

Summary
PICK1 is a neuronal scaffolding protein containing a PDZ domain and an autoinhibited BAR domain. BAR domains are membrane-sculpting protein modules generating membrane curvature and promoting membrane fission. Previous data suggest that BAR domains are organized in lattice-like arrangements when stabilizing membranes but little is known about structural organization of BAR domains in solution. Through a small-angle X-ray scattering (SAXS) analysis, we determine the structure of dimeric and tetrameric complexes of PICK1 in solution. SAXS and biochemical data reveal a strong propensity of PICK1 to form higher order structures and SAXS analysis suggest an offset, parallel mode of BAR-BAR oligomerization. Furthermore, unlike accessory domains in other BAR domain proteins, the positioning of the PDZ domains is flexible, enabling PICK1 to perform long-range, dynamic scaffolding of membrane-associated proteins. Together with functional data, these structural findings are compatible with a model where oligomerization governs auto-inhibition of BAR domain function.
Introduction
PICK1 (Protein Interacting with C Kinase) is a scaffolding protein localized to neurons as well as to muscle and endocrine cells (Cao et al., 2007; Jansen et al., 2009; Staudinger et al., 1995). The protein serves distinct roles in scaffolding of kinases (Perez et al., 2001; Staudinger et al., 1997), direct modulation of membrane protein function (Sogaard et al., 2013), regulation of membrane protein trafficking (Citri et al., 2010; Madsen et al., 2012) and membrane remodeling during biogenesis of dense core vesicles (Cao et al., 2013; Holst et al., 2013). At the N-terminus, PICK1 harbors a PSD-95/Discs-Large/ZO-1 homology (PDZ) domain that mediates protein-protein interactions with a number of transmembrane proteins including receptors, ion channels and transporters, as well as with PKC α (Xu and Xia, 2006). A central Bin/amphiphysin/Rvs (BAR) domain facilitates PICK1 dimerization, which is important for both its scaffolding function of PICK1 and for its ability to bind to and remodel the cell membrane (Citri et al., 2010; Jin et al., 2006; Lu and Ziff, 2005; Madsen et al., 2012).
PICK1 is central in regulating trafficking of AMPA-type glutamate receptors (AMPARs) during synaptic plasticity. PICK1 binds the intracellular C-terminus of the AMPAR subunit GluA2 (Xia et al., 1999) and regulates its plasma membrane localization in an activity dependent manner (Anggono and Huganir, 2012). This function, as well as the synaptic localization of PICK1, depends on the membrane binding BAR domain (Jin et al., 2006; Steinberg et al., 2006). Similar to endophilin and the F-BAR protein syndapin-1 (Meinecke et al., 2013; Rao et al., 2010), the membrane and protein binding function of the PICK1 BAR domain is auto-inhibited (Jin et al., 2006; Lu and Ziff, 2005; Madsen et al., 2008; Rocca et al., 2008). This auto-inhibition of the PICK1 BAR domain is believed to involve the N-terminal PDZ domain (Lu and Ziff, 2005; Madsen et al., 2008) and as well as the unstructured C-terminus (Jin et al., 2006). A steric hindrance model suggesting direct interaction of the PDZ and BAR domains has been proposed for the PICK1 auto-inhibition (Hanley, 2008; Lu and Ziff, 2005).
For both endophilin and syndapin, auto-inhibition is relieved by dynamin binding to the SH3 domain (Meinecke et al., 2013; Rao et al., 2010) and similarly peptide binding in the PDZ domain was suggested to relieve PICK1 auto-inhibition (Lu and Ziff, 2005; Rocca et al., 2008). In addition, Ca2+-binding was proposed to regulate the auto-inhibition (Citri et al., 2010), whereas our previous work suggests that membrane recruitment is key to BAR domain activation (Madsen et al., 2008).
Crystal structures of numerous BAR domains have been solved, but only few studies have provided insight into the structural organization of individual domains relative to one another in full-length BAR domain proteins. For sorting nexin 9 (Snx9), endophilin, APPL1 and syndapin-1, their respective accessory domains were all associated with either the side or the tip of the BAR domain (Li et al., 2007; Pylypenko et al., 2007; Rao et al., 2010; Wang et al., 2008; Zhu et al., 2007). To investigate the structural interdomain arrangement in PICK1, which is believed to underlie the auto-inhibition mechanism, we engaged in small-angle X-ray scattering (SAXS) studies of full-length PICK1 in solution. We demonstrate that PICK1 is highly prone to oligomerization, even in absence of lipid membrane, and we obtain the first solution based structural information of BAR domain oligomerization using rigid body modelling in combination with Ensemble Optimization Method (EOM) analysis. The data suggest an elongated tetrameric conformation with individual dimers overlapping along one third of the length of the dimer - i.e. an elongated, overlapping mode of BAR-BAR oligomerization. Using a non-oligomerizing mutant of PICK1, together with a novel method for analytical decomposition of the scattering data followed by EOM, we furthermore show that the PDZ domains are flexible and far apart and consequently enable PICK1 to perform highly flexible, long range scaffolding of membrane associated protein complexes.
Results
Biochemical characterization of PICK1WT and PICK1LKV
Wild type PICK1 (PICK1WT) was expressed in E. Coli and purified as described (Madsen et al., 2005). The size of purified PICK1WT was verified by SDS-PAGE (Figure 1A) and subsequently evaluated by size exclusion chromatography (SEC) revealing two peaks for PICK1WT. The early peak, eluting at ~8.5 ml (peak 1), most likely represented large oligomeric states (Figure 1B). These large oligomers were almost completely removed by ultracentrifugation (Figure 1B, dashed). The main peak (peak 2, ~12 ml) was composed of at least two overlapping peaks presumably representing the dimeric form of PICK1WT as well as higher order oligomeric forms (Figure 1B, dashed). This suggests very little monomeric protein in our samples. To test whether we could indeed detect the monomer, we increased the Triton X-100 (TX-100) concentration to 0.1%. Under these conditions the main peak eluted later (~13 ml), suggesting separation into monomers (Figure S1A). This was further substantiated by crosslinking experiments (Figure S1B). Analytical ultra-centrifugation (AUC) of peak 2 obtained in 0.01% TX-100 supported PICK1 oligomerization by revealing several species (Figure 1C), likely corresponding to dimers (~3S [Svedberg], b)), tetramers (~4S, c) and octamers (~7S, e), as well as a featureless signal at high S representing even higher order oligomers. Moreover, a small peak was seen (~1S), that might be the degradation product seen as a faint band on the SDS-PAGE (Figure 1A).
Figure 1. PICK1 exists in multiple oligomeric states and oligomerization is reduced in the self-binding mutant PICK1LKV.

(A) Representative SDS-PAGE (n=5) showing PICK1WT (left) and PICK1LKV (right). PICK1WT elutes at 46 kD (monomer). A weak putative degradation product is also seen. PICK1LKV displays only a band at 46 kD. (B) Size exclusion chromatography (SEC) of PICK1WT (solid line), ultracentrifuged PICK1WT (dashed line) and PICK1LKV (dotted line). PICK1WT (solid line) shows two major peaks. Peak 1 is removed by ultracentrifugation and is almost absent in PICK1LKV. (C) Analytical ultracentrifugation (AUC) of fractions corresponding to peak 2 in the SEC. PICK1WT (solid line) displays distinct peaks. PICK1LKV displays reduced size of peaks corresponding to larger sizes. (D) Representative western blot showing lysates from COS7 cells transiently expressing PICK1WT with or without pretreatment with the membrane permeable crosslinking agent DSS (n=3) (E) Representative confocal micrographs showing YFP-PICK1WT (left) and YFP-PICK1LKV (right) transiently expressed in COS7 cells (n=3). Arrows indicate clusters. Scale bar 10 μM. (F) Quantification of liposome vesiculation capacity of endophilin, PICK1WT and PICK1LKV in arbitrary units. Fluorescently labeled liposomes were incubated with indicated proteins before ultracentrifugation. Fluorescent lipids in the supernatant indicate scission from liposomes, which are pelleted (means ± sem, n=3, *p < 0.05). See also Figure S1.
We also investigated the oligomeric status of PICK1 in living cells. COS-7 cells transfected with PICK1WT (N-terminal cmyc-tag) were treated with the cell permeable crosslinking agent disuccinimidyl suberate (DSS, 1 mM). Western blotting of the cell lysates revealed that crosslinking not only increased the dimeric fraction (100 kD), but also produced several distinct larger species suggesting the presence of dimers and higher order oligomers in living cells as well (Figure 1D).
The PICK1 PDZ domain alone has previously been stabilized for structural studies by fusion of a C-terminal sequence that docks into the peptide binding groove in the PDZ domain (Elkins et al., 2007). Using a similar approach for the full-length protein, we substituted the three C-terminal residues of PICK1 with the three C-terminal residues of the dopamine transporter - the minimal binding sequence of the PICK1 PDZ domain (CDS->LKV; PICK1LKV)). Purified PICK1LKV remained stable without precipitating at high concentrations (data not shown) and no degradation was visible on SDS-PAGE (Figure 1A). Interestingly, SEC of purified PICK1LKV showed almost no higher order oligomers or aggregates (peak 1) (Figure 1B). In addition, the second peak (peak 2) was more distinct and almost without the shoulder seen for PICK1WT (Figure 1B). AUC of peak 2 from the SEC revealed a more distinct peak and a larger fraction of dimer (~3S) than seen for PICK1WT (Figure 1C). Consistently, the tetrameric peak (~4S) (c) was markedly reduced compared to that of PICK1WT and at higher S values we only observed two very shallow peaks (Figure 1C). We did not observe the small peak (~1S) for PICK1LKV, in agreement with the lack of degradation on the SDS-PAGE (Figure 1C). In conclusion, PICK1LKV appeared more stable and had lower tendency to form higher order oligomers and aggregates as compared to PICK1WT.
PICK1LKV alleviates the auto-inhibition of PICK1
Next, we expressed YFP-PICK1WT and -PICK1LKV in COS7 cells. Confocal microscopy showed diffuse localization of YFP-PICK1WT throughout the cytosol albeit with numerous weak clusters of the protein (Figure 1E). This partial clustering was previously suggested to reflect the membrane binding capacity of the protein (Jin et al., 2006). Somewhat counterintuitively in light of the biochemical data, YFP-PICK1LKV demonstrated massive clustering indicating stronger membrane binding capacity (Figure 1E). One possible explanation could be that the auto-inhibition of the BAR domain was alleviated in YFP-PICK1LKV.
To address this in more detail, we tested the membrane vesiculation capacity of PICK1WT and PICK1LKV by adopting a modified version of a vesiculation assay that recently was used to demonstrate a critical role in membrane scission of amphipathic helices in the N-BAR proteins endophilin A1 and amphiphysin (Boucrot et al., 2012). Indeed, our previous data have indicated that an amphipathic helix adjacent to the BAR domain is critical for the membrane-deforming capacity of PICK1 (Holst et al., 2013). The assay is based on the ability of N-BAR domains to promote formation of nanovesicles by scission/fission when incubated with liposomes. If formed, the nanovesicles will be present in the supernatant after ultracentrifugation in contrast to the liposomes that will be present in the pellet (Boucrot et al., 2012). We incubated fluorescently labeled liposomes with or without purified PICK1WT, PICK1LKV or endophilin1A followed by ultra-centrifugation. Consistent with the ability of endophilin1A to promote formation of nanovesicles, endophilin markedly increased fluorescence in the supernatant compared to control (no protein added) (Figure 1F). PICK1WT also increased fluorescence and the increase was significantly higher for PICK1LKV (Figure 1F). This demonstrates the membrane vesiculation capacity of PICK1 and further supports that the auto-inhibition of the PICK1 BAR domain is at least partially alleviated in PICK1LKV.
SAXS analysis of PICK1WT and PICK1LKV - Visual inspection
SAXS profiles of ultra-centrifuged PICK1WT and PICK1LKV both showed a monotonously decaying scattering intensity as a function of the scattering vector q (Figure 2A, B). In both systems the forward scattering, I(0), of the concentration normalized data increases with concentration (Figure 2A and 2B, see insets). This is a clear indication of a concentration-dependent oligomerization (see table in Figure 2). The effect was more pronounced for PICK1WT than for PICK1LKV.
Figure 2. Small-angle X-ray scattering (SAXS) experiments demonstrated concentration dependent oligomerization PICK1WT and PICK1LKV.

Experimental SAXS data measured on five concentrations of PICK1WT ranging from 0.9 - 7.5 mg/ml (A), and four concentrations of PICK1LKV ranging from 1.2 - 8.8 mg/ml (B), scaled by concentration and on absolute scale. C-D) Pair distance distribution functions, p(r), calculated by IFT of data shown in A and B, respectively. (Tables) Model-independent parameters derived by IFT, i.e. maximal internal distance, Dmax, Radius of Gyration, Rg and forward scattering, I(0). Note the concentration dependent shift to larger Dmax values. See also Figure S2.
The pair distance distribution functions, p(r), functions for PICK1WT (Figure 2C) and PICK1LKV (Figure 2D), calculated by indirect Fourier transformation (IFT) (Glatter., 1977; Pedersen et al., 1994), all revealed skewed shapes with a prominent tail at large distances, akin to the p(r)'s previously found for endophilin and Snx9 (Wang et al., 2008), and consistent with an overall elongated shape. The Dmax and Rg values also exhibited a concentration-dependent increase consistent with that observed in the I(0)-data and showed that, at similar concentrations, larger structures are formed by PICK1WT than by PICK1LKV (see table in Figure 2).
Neither calcium nor peptide binding induce significant conformational changes in PICK1
Ca2+ or PDZ ligand binding was previously suggested to regulate auto-inhibition of the PICK1 BAR domain (Citri et al., 2010; Lu and Ziff, 2005; Rocca et al., 2008). SAXS data and p(r) functions for PICK1WT in absence and presence of 50 μM free Ca2+, 100 μM GluA2 or both, showed no significant differences (Figure S2), strongly suggesting no significant change in tertiary or quaternary structure upon binding of Ca2+ and/or GluA2 PDZ-ligand. This is in agreement with our previous findings that peptide binding did not increase lipid binding and neither peptide binding or Ca2+ were sufficient to induce cellular clustering (Madsen et al., 2008).
Decomposition of the PICK1LKV SAXS signal into dimer and tetramer form factors
With the concentration-induced oligomerization of the samples it was clear that each sample contained more than one structural species. To circumvent this problem, the PICK1LKV SAXS data were decomposed into their underlying dimeric and tetrameric form factors by combining data from samples with different protein concentrations and hence different dimer/tretamer ratios. This was done through an approach that is a special case of singular value decomposition that assumes that only two species are present in the sample (details in SI). Assuming that the four PICK1LKV samples only contained dimers and tetramers, the four PICK1LKV SAXS data-set were combined in six different ways providing six different versions of the decomposed dimer and tetramer form factors (Figure 3). Direct comparison of the form factors and their p(r) functions (Figure 3B, C) revealed a sufficiently strong similarity between the results obtained by different combinations to implicitly validate the decomposition method and its preassumption of only two species in the case of PICK1LKV. The final average PICK1LKV dimer and tetramer form factors and p(r)'s are plotted in Figure 3D. For the PICK1LKV dimer, Dmax was consistently 200 Å and Rg was 61 +/−1 Å. This is slightly larger than reported values for the related BAR domain proteins Snx9full-length (Dmax 172.5 Å, Rg 51.7 +/− 0.5 Å) and endophilinfull-length (Dmax 167.5 Å, Rg 48.0 +/− 0.5 Å) (Wang et al., 2008). The tetramer form factor was slightly less well defined and gave a Dmax of ~250 Å and an Rg of 75 +/− 3 Å. In the PICK1WT system, a similar decomposition was not possible due to the presence of higher order oligomeric species.
Figure 3. Decomposition of polydisperse PICK1LKV SAXS data into dimer and tetramer scattering signals.

(A) Concentration series of PICK1LKV (same as Figure 2B and D). Left: SAXS data. Right: corresponding p(r) functions. (B) Six dimer SAXS form factors (left) and corresponding p(r) functions (right) resulting from decomposition through pairwise combination of samples at different concentrations. (C) Six tetramer SAXS form factors (left) and p(r) functions (right). (D) Direct comparison of the mean dimer and tetramer form factors and p(r) functions. Model-independent parameters are listed in Table S1.
Definition of structural modules composing PICK1
To enable rigid body modelling the structure of the individual protein domains was defined. For the PDZ domain, the NMR-structure (PDB 2LUI) (Figure 4A) (Erlendsson et al., 2014) was used. For the BAR domain dimer, we generated a homology model of the monomers by threading PICK1 residues on the crystal structure of the arfaptin-2 BAR domain. Different dimer interfaces giving rise to varying length and curvature of the BAR domain were initially tested in rigid body modeling against the SAXS data, however the interface from the arfaptin-2 crystal structure clearly gave the best result. We subsequently used molecular dynamics (MD) simulations to identify the structure of its dimeric assembly and assess the quality of the model before performing final all atom MD simulation to obtain a stable BAR domain dimer model (Figure 4B, see Figure S3, methods and SI for details). Unfortunately, oligomerization and inability for substitute endogenous cysteines hampered experimental validation of the BAR domain dimer interface by e.g. cross-linking or FRET. Finally, the N- and C-termini, as well as the linker between the PDZ domain and the BAR domain, were considered to have a random coil structure, which was supported by backbone chemical shifts from NMR experiments (see Figure 4C and S3 as well as SI for details).
Figure 4. Structural modules in PICK1.
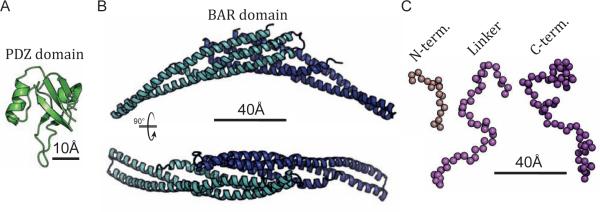
(A) PDZ domain of PICK1 from NMR structure (PDB 2LUI) (Erlendsson et al., 2014). (B) Side and top view of the PICK1 BAR domain modeled by AA-MD simulation based on the crystal structure of the homologous Arfaptin 2. (C) Representative structures of the unstructured N-terminus, the linker between the PDZ and BAR domain, and the C-terminus. See also Figure S3.
Structural Analysis of PICK1LKV dimers using the Ensemble Optimisation Method (EOM)
Classical Bio-SAXS approach to the data analysis (Koch et al., 2003), based on bead and/or rigid-body modeling of a single monodisperse structure to the decomposed experimental dimer form factor did not provide good or meaningful fits. This is an indirect sign of structural dispersion of the dimer structure arising from e.g. the flexible linker between the BAR and PDZ domains as well as the N- and C-termini. Consequently, to extract structural information from the SAXS data, the refinement of the dimer structure against the SAXS dimer form factor was carried out using an approach that combined rigid body modelling and EOM (SI for details) (Bernadó et al. 2007, Petoukhov, et al. 2012). A large number of potential dimeric structures were generated using the above described structural modules. The linker between the PDZ and BAR domains, as well as the N- and C-termini, were defined as flexible, whereas the PDZ domains and the BAR domain dimer were kept conformationally rigid. This yielded a population of dimers with varying positions of the PDZ domains, within the constraints given by the length of the linker and steric avoidance. The theoretical scattering from each model was calculated (with RANCH), and an ensemble of structures was optimized using the genetic algorithm (GAJOE) in EOM to fit the theoretical scattering to the SAXS data. Through this approach, a model for the PICK1LKV dimer was obtained by refinement against the above described decomposed dimer form factor (Figure 5A). The wide distributions of Dmax and Rg selected by EOM, show that the PICK1LKV dimer indeed has a very flexible structure (Figure 5A). The best ensemble, containing 4 different poses, is visualized in Figure 5B.
Figure 5. Interdomain arrangement in PICK1LKV dimer.

(A) EOM on decomposed dimer form factor of PICK1LKV. Fit of optimal ensemble to combination of PICK1LKV samples at 2.3 and 3.5 mg/ml. Top panels show Dmax and middle panel Rg of generated pools in blue and selected pools in green. Bottom panel shows fit to SAXS form factor. (B) Illustration of flexibility of PICK1LKV. The rigid BAR domain is shown in cyan/blue. The PDZ domain is green. Transparency reflects frequency of the conformation in the optimal ensemble. The linker region is in purple. The C-terminal is removed for visual clarity. (C) Histograms of sampled distances between the PDZ domain and the position of the linker on the BAR domain (cyan), the tip of the BAR domain (blue), and the center of the BAR domain (green), and between the PDZ domains (orange) (100 ensembles, bin size 20 Å). (D) Quantification of cellular FRET of N- and C-terminal fusions of PICK1. CFP-YFP is a direct fusion of the two fluorescent proteins. Means ± sem, n=4, *p < 0.05, **p < 0.01.
The PDZ domains of PICK1 dimers are flexible with respect to the BAR domain
Analysis of the structural models from 100 optimized ensembles provided distributions of distances between PDZ domains (center of mass) and specific points on the BAR domain dimer (Figure 5C). The most frequent structures in the ensemble displayed distances from the PDZ domains to the attachment point on the BAR domain of 20-60 Å in (random coil ~50 Å for 40 amino acids (Tanford et al., 1966)). Moreover, the distance from the PDZ domains to the tip of the BAR domain (blue) is generally shorter than to the center of the BAR domain (green) (20-40 Å vs. 60-100 Å, respectively). The distance between PDZ domains (orange) were mostly between 120 Å and 180 Å, often representing the largest distances in the protein. To experimentally test this model, we performed cellular FRET measurements of N- and C-terminal fusion of fluorescently labeled PICK1. Whereas the NFRET for PICK1-eYFP + PICK1-eCFP was significantly higher than for eYFP + eCFP (0.096 ± 0.01 vs 0.018 ± 0.01, P<0.01) there was no significant difference between eYFP-PICK1 + eCFP-PICK1 and eYFP + eCFP (0.024 ± 0.004 vs 0.018 ± 0.01) (n=4, one-way ANOVA with Bonferroni's post test). Indeed, this supports the EOM model suggesting that the N-terminal PDZ domain are spaced far apart. In conclusion, our findings suggest that the PDZ domains in the PICK dimer, in contrast to accessory domains in other BAR domain proteins, are separated from the BAR domain, staying far from each other and freely exploring the space around the tips of the BAR domain.
EOM investigation of the PICK1 oligomerisation mode
An EOM analysis against the experimental SAXS data clearly underlined that neither PICK1WT nor PICK1LKV could be described by an ensemble of dimers only. This prompted us to include tetramers into the EOM pool, where the conformation of the BAR-domain of the PICKWT was assumed similar to that determined for PICK1LKV. By translating and rotating the BAR domain dimer, several putative tetrameric arrangements of the BAR domains were tested (Figure 6). The modes termed ‘offset’ and ‘side’ (Figure 6A and D) mimicks the BAR-BAR interactions observed for PinkBAR and the F-BAR protein FBP17/CIP4, with different extent of overlap (Frost et al., 2008; Pykalainen et al., 2011). The ‘helix’ mode (Figure 6B) mimics the BAR-BAR interaction observed for endophilin, whereas the ‘tip-tip’ interactions (Figure 6C) have been observed for endophilin, FBP17/CIP4 and PACSIN (Bai and Zheng, 2013; Frost et al., 2008; Mim et al., 2012). A hypothetical configuration resembling a circle was included as control (Figure 6E). These tetrameric arrangements were kept fixed while generating random positions of the linker regions (and consequently the PDZ domains) and the N- and C-termini, as described above for the dimeric structures.
Figure 6. Dimers of dimers of PICK1LKV via an ‘offset’ BAR-BAR arrangement.

EOM fitting of five putative tetrameric structures [(A) off-set, (B) helix, (C) tip-tip, (D) side and (E) circle] to PICK1LKV SAXS data at 8.8 mg/ml. Dimers are included in the analysis. Middle panels show Dmax and Rg distributions of the randomly generated pools (blue) and selected pools (green) by EOM. Bottom panels show SAXS data and fit of the optimal ensemble. χ2-values for the fits are shown.
Each tetramer pool, in combination with the dimer pool (from Figure 5), was evaluated with EOM against SAXS data of PICK1LKV, which consisted mainly of dimers and tetramers as suggested by the biochemical data (see Figure 1). Figure 6 shows the EOM analysis of the putative modes of oligomerization for PICK1LKV. Comparing the fits and chi-values, we found that the ‘offset’ mode provided the best fit to the data (Figure 6A). Consequently, this mode was used in the subsequent analysis. The ‘helix’ mode based on the arrangement of endophilin BAR domains on tubules (Mim et al., 2012) also yielded good fits to the experimental data, although inferior to the fits from the ‘offset’ mode (Figure 6B). On the other hand, the ‘tip-to-tip’, side-by side’ and ‘circle’ mode produced significantly worse fits (Figure 6C-E), including systematic underestimation of the scattering intensities at low q region (Figure 6D-E), suggesting that these modes are too compact to fit the scattering data well.
Structural investigation and flexibility of PICK1LKV tetramers
Based on the information from the dimers and the mode of oligomerisation, the structure and flexibility of the PICK1LKV tetramers were investigated. EOM selected distributions of Dmax and Rg that were narrower than for the dimer, suggesting that the tetramer is less flexible (Figure 7A). The best ensemble, again containing 4 poses, is shown in Figure 7B and the distance distributions are shown in Figure 7C. The distance distribution histograms reveal that the distal PDZ domains localize closer to the center of the BAR domain, whereas the central PDZ domains stretch out compared to the dimeric structure. The distance between PDZ domains from the same dimer (100-140 Å) (orange) is slightly shorter than the corresponding distance in the dimer model. The distance between distal PDZ domains (red) was mostly between 200 and 260 Å, indicating that the maximal distance is represented by the BAR domain tetramer (270 Å). Finally, the distance between the central PDZ domains (yellow) display a broad distribution (40-120 Å) indicating flexibility but also that they occasionally may come in contact. In conclusion, despite minor changes in the interdomain arrangements between the dimeric and tetrameric species, the PDZ domains in the tetrameric structure are also separated from the BAR domain with highly flexible interconnecting linkers.
Figure 7. Interdomain arrangement in PICK1LKV tetramer.

(A) EOM on decomposed tetramer form factor of PICK1LKV. Fit of optimal ensemble to combination of PICK1LKV samples at 2.3 and 3.5 mg/ml. Top panels show Dmax and middle panel Rg of generated pools in blue and selected pools in green. Bottom panel shows fit to SAXS form factor. (B) Illustration of flexibility of PICK1LKV. The BAR domain is shown in cyan/blue. PDZ domain is green and the transparency reflects frequency of conformation in optimal ensemble. Linker region is in purple. The C-terminal is removed for visual clarity. (C) Histograms of sampled distances between the PDZ domain and the position of the linker on the BAR domain (light blues), the tip of the BAR domain (dark blues), and the center of the BAR domain (greens) and between the PDZ domains within the dimer (orange), as well as between the two distal PDZ domains (red) and the central PDZ domains (yellow) (100 ensembles, bin size 20 Å).
Concentration dependence of the PICK1 oligomerization
The biochemical analysis, the model independent SAXS parameters and the EOM analysis consistently suggested concentration dependent oligomerization of PICK1. To further characterize the concentration dependence of the oligomerization process, we performed EOM analysis for the different concentrations using the dimer pool combined with the ‘offset’ mode tetramer pool. For the PICK1LKV data, we observed a concentration dependent shift from predominantly dimers (at the lowest concentration), to almost exclusively tetramers (at the highest concentration) (Figure 8). Similar analysis of PICK1WT, using only dimers and tetramers, allowed reasonable fits at low concentrations (up to 3.2 mg/ml), but at higher concentrations the fits systematically underestimated the scattering intensity, particularly at lower scattering angles, suggesting the presence of higher order species (Figure S4) in aggrement with the AUC experiments.
Figure 8. Concentration dependent tetramerization of PICK1LKV.

EOM on PICK1LKV at increasing protein concentrations; (A) 1.2 mg/ml, (B) 3.2 mg/ml, (C) 4.2 mg/ml, and (D) 8.8 mg/ml. Top panels and middle panels show Dmax and Rg of the generated pools in blue and selected pools in green. Left pool (160-230 Å) represents dimers, right pool (260-340 Å) represent tetramers. Bottom panels show SAXS data and fit of optimal ensemble. (E) Quantification of the fraction of dimers and tetramers at different concentrations of PICK1LKV. For analysis of PICK1WT with dimers and tetramers see Figure S4.
Hexamers and octamers were generated in RANCH following the same principles as for the tetramer formation by duplicating the BAR dimer. For PICK1WT, the EOM did not select any structures from the hexamer pool, but included a significant octamer pool, allowing a good fit to the data (Figure 9). Since the fitting procedure at this level includes a number of random parameters, a reliable conclusion regarding the presence or absence of hexamers is not possible. Rather, it suffices to state that PICK1WT has a pronounced tendency to form higher oligomers in solution, which can be fitted to our SAXS data by introducing a combination of dimers, tetramers and octamers. For PICK1LKV, the inclusion of higher oligomers in the random pools did not improve the fit (Figure S5). This confirms that there is no significant proportion of higher oligomers present in PICK1LKV.
Figure 9. Concentration dependent oligomerization of PICK1WT.

EOM on PICK1WT at increasing protein concentrations; (A) 0.9 mg/ml (B) 2.3 mg/ml (C) 3.2 mg/ml (D) 5.3 mg/ml and (E) 7.5 mg/ml, including dimers, tetramers, hexamers and octamers in the generated pool. Top panels show Dmax and Rg of generated pools in blue and selected pools in green. Bottom panels show SAXS data and fit of optimal ensemble. (F) Quantification of the fraction of dimers, tetramers, hexamers and octamers at different concentrations of PICK1WT. (G) Quantification of the fraction of dimers, tetramers, hexamers and octamers at different concentrations of PICK1LKV from analysis repeated with all species (see Figure S5 for analysis).
Discussion
BAR domains are membrane binding and sculpting protein modules involved in a wide variety of different cellular processes, including e.g. budding and fission during endocytosis from the plasma membrane and during production of dense core vesicles at the trans-Golgi network. The BAR domain is composed of two subunits and is believed to bind membranes as dimers. The curvature sensing capacity, as well as the scission capacity of the domains, largely reside in amphipathic helixes flanking the BAR domains (Bhatia et al., 2009; Boucrot et al., 2012) and recently a lattice-like arrangement of BAR domains on membrane tubules has been suggested to stabilize and shape tubule structure (Frost et al., 2008; Mim et al., 2012).
Higher order oligomers have also been observed in solution in absence of lipid membrane for the F-BAR domain CDC15 (Roberts-Galbraith et al., 2010), the F-BAR domain protein PACSIN1 (Halbach et al., 2007) and the I-BAR protein PinkBAR (Pykalainen et al., 2011). Here, we demonstrate that the BAR domain protein PICK1 forms higher order structures in the absence of lipids in solution at micromolar concentrations. The oligomerization was reversible and supported both by SEC and AUC. Moreover, EOM analysis of the SAXS data suggests a mix of dimers, tetramers and higher oligomers (likely octamers). The absolute distribution between the different oligomeric forms, however, is not easily compared between AUC and SAXS data, due to differences in preparation (FPLC peak 2 for AUC vs ultracentrifuged samples for SAXS), and equilibration times. Neither AUC nor EOM analysis of the SAXS data indicated any monomers in our samples suggesting high affinity for the PICK1 BAR domain dimer.
The use of EOM analysis in this study allowed us to obtain the first direct structural information on the oligomerization mode in solution for a BAR domain protein. The data can only be adequately fitted using an elongated oligomerization mode involving an offset between the individual BAR domain bearing more resemblance to the crystal structure of PinkBAR (Pykalainen et al., 2011), than the side-by-side arrangement in the crystal structure of PACSIN2 (Bai and Zheng, 2013). The resolution of the SAXS/EOM procedure does not allow us to investigate the oligomerization interface in detail, but based on the model of the BAR domain from MD simulations, we cannot identify any protruding hydrophobic residues similar to Trp141, which is important for Pinkbar oligomerization (Pykalainen et al., 2011). This lateral oligomeric assembly mode also somewhat resembles the one reported for the F-BAR domains FBP17/CIP4 on lipid tubules (Frost et al., 2008), although with a smaller overlap. The tip-to-tip interaction, seen for FBP17/CIP4, and also for endophilin A1 and PACSIN 2 on membrane tubules (Bai and Zheng, 2013; Mim et al., 2012), however, was not supported by the EOM analysis for PICK1 in solution. Although our best model suggests a direct interaction between the BAR-BAR dimers, the data are almost equally compatible with the helix-helix interaction mode observed for endophilin A1 on membrane tubules (Mim et al., 2012), in which the N-terminal amphipathic helices embedded in the membrane served as a perpendicular spacer of individual dimers. Detailed molecular insight into interplay between membrane binding, oligomerization and folding/shielding of the amphipathic helix is likely to shed light on the functional regulation of the protein. Of note, the PDZ domains from two separate dimers are in proximity within this oligomeric arrangement, suggesting that the reported dimerization of the PICK1 PDZ domain (Pan et al., 2007) could be formed between separate dimers. The EOM analysis, however, does not support a stable PDZ-PDZ interaction.
We were unable to fit the SAXS data using classical rigid body modeling even with a number of putative dimer interfaces producing different length and curvature of the BAR domain. Consequently, we turned to EOM analysis of the PICK1LKV dimer, which suggests that the PDZ domains are clearly separated from the BAR domain, and cellular FRET studies extended these findings to the WT protein in a cellular context. This is different from what has been observed for the PX/PH domains of Snx9, endophilin, and APPL1 that were all associated with the BAR domain either at the side or the tip. Such a flexible interdomain arrangement has been observed from computational studies as well (He et al., 2011), concieveably enables PICK1 to perform highly flexible scaffolding of membrane associated protein complexes and potentially flexible supramolecular scaffolding for the oligomers.
As for the auto-inhibition, it could be speculated that the C-terminal self-association in PICK1LVK restricts the PDZ domain from entering the space under the BAR domain. Steric hinderance by such an interdomain arrangement has been suggested to underlie the auto-inhibition of the PICK1 BAR domain (Hanley, 2008; Lu and Ziff, 2005), and fits with the alleviation of auto-inhibition seen for PICK1LKV. There is, however, no evidence of reduced flexibility of PICK1WT compared to PICK1LKV, and the selected dimer pools showed size and Rg distributions similar to PICK1LKV. Furthermore, the PICK1WT EOM analysis does not sample conformations with PDZ domains close to the BAR domain. Alternatively, the reduced cellular clustering of PICK1LKV might result from tethering of the C-terminus, which has been demonstrated to counteract cellular clustering and lipid binding (Jin et al., 2006), however the low density of the unstructured C-terminus prevents us from addressing this flexibility reliably in SAXS measurements. Finally, if the oligomerization indeed involves the amphipathic helix preceeding the BAR domain, an alternative scenario is that oligomerization could directly compete with membrane interaction and consequently mediate the auto-inhibition of PICK1.
The possible relevance of our structural data was supported by cell-based experiments providing evidence based on crosslinking that PICK1 indeed forms higher order structures also in living cells. Our cellular data moreover showed a striking redistribution of eYFP-PICK1LKV compared to eYFP-PICK1WT. Whereas eYFP-PICK1WT exhibited diffuse localization, eYFP-PICK1LKV displayed massive clustering, that likely reflects enhanced membrane binding. This, together with the enhanced vesiculation capacity of eYFP-PICK1LKV, supports alleviated auto-inhibition in this mutant. The clustering also suggest that the combined ability of PICK1 to bind and deform lipid membranes can lead to dramatically high local concentrations of PICK1 when unrestrained. Altogether, we therefore suggest a model where oligomerization in solution governs auto-inhibition of the BAR domain and thereby unrestrained activity, which in turn allows for specific and reversible cellular functions of PICK1 that otherwise would be impossible.
Experimental Procedures
Cloning, protein purification and biochemical characterization
WT PICK1 and PICK1-LKV were cloned and expressed in E. coli as described previously (Madsen 2005) (see also SI).
Cellular cross-linking
COS-7 cells were grown as described (Madsen et al., 2008) and seeded on poly ornithine coated Labtek chambers (Thermo Scientific) and transfected using Lipofectamine2000 (Invitrogen) (0.5μg DNA/300,000 cells). For crosslinking in cells, COS7 cells were transfected with pCMVmycPICK1 (Madsen et al., 2008) and grown in a 40 mm petridish. Crosslinking was performed 48 h after transfection with 1mM DSS (disuccinimidyl suberate) (Thermo Scientific) in PBS, pH 7.4 on ice for 30 minutes. The reaction was stopped by adding TBS with 1 vol% TX-100, 1 mM DTT, DNAse and protease inhibitor cocktail (Pierce). Cell lysates were analyzed by 10% SDS-PAGE followed by western blotting using rabbit αPICK1 ab (Abcam).
Liposome vesiculation assay
The vesiculation capacity was measured as modified from (Boucrot et al., 2012). Liposomes produced from Folch brain liposomes and 1% fluorescently conjugated lipid DiD (1,1′-Dioctadecyl - 3,3,3′,3′-tetramethylindodicarbocyanine iodide) were extruded through 200 nm filters at a ‘concentration’ of 0.25 mg/ml and incubated with 10 μM protein for 1 h, before spinning at 200,000 × G for 15 minutes in a final buffer of TBS + 0.0025 vol% TX-100 + 0.25 mM DTT. Supernatants were collected immediately and pellet was resuspended in buffer. Vesiculation was measured in the supernatants as the absorbance at 648 nm reflecting the amount of DiD present and thereby the degree of liposome disruption.
Molecular modeling and dynamics simulation
A molecular model of the PICK1-BAR domain monomer was built with homology modeling using arfaptin 2 as a template (PDB ID: 1I49). This model was used in extensive coarse-grained (CG) molecular dynamics (MD) simulations to determine the structure of its dimeric assembly (See also SI). Additionally, three alternative models for the PICK1-BAR dimers were constructed with homology modeling using, arfaptin, amphiphysin and endophilin BAR dimers as templates. The relative quality of these three models together with the best dimer model obtained from the CG-MD (CG-model) was assessed with all-atom (AA) MD simulations (See also SI) using several different criteria, such as their stability (RMSD), solvent-accessible surface area (SASA) of hydrophobic residues, and surface area of hydrophobic residues buried in the dimer interface. The CG-model performed best and was further equilibrated in subsequent AA-MD and used as our final model of the PICK1-BAR dimer (see SI for details).
SAXS data acquisition and analysis
Acquisition
The scattering experiments were carried out at the EMBL X33 beamline (C. E. Blanchet, 2012) at the DORIS storage ring (DESY, Hamburg) following standard procedures (see also SI).
Ensemble Optimization Method (EOM)
Populations of 10.000 structures each were generated with Ranch (Bernado et al., 2007; Petoukhov and Svergun, 2012). Scattering curves were calculated for each structure from [0.0001 to 0.5Å−1] using 15 spherical harmonics. Optimal ensembles were fitted to experimental data with the program Gajoe (Bernado et al., 2007; Petoukhov and Svergun, 2012), with 2000 generations and 100 repeats (see also SI).
Analytical decomposition of SAXS data
The small-angle scattering from oligomers, in the absence of structure factors, are simply the sum the scattering from the single components: This implies that the scattering from two samples with different fractions of the same two components, can be decomposed to obtain the form factors of the single components by solving two equations with two unknowns. This is exploited in order to determine the solution structure of the dimer and the tetramer (see also SI).
Supplementary Material
Highlights.
The PDZ domains are far apart and flexible with respect to the BAR domain.
PICK1 forms higher oligomeric species in solution.
The N-BAR domain is directly involved in the parallel, offset oligomerization mode.
Oligomerization is likely to regulate BAR domain auto-inhibition.
Acknowledgments
The work was supported by the National Institute of Health Grants P01 DA 12408 (UG), the Danish Medical Research Council (UG, BV), University of Copenhagen BioScaRT Program of Excellence (UG, KLM), the Lundbeck Foundation Center for Biomembranes in Nanomedicine (UG,), the Drug Research Academy (XT), Novo Nordisk A/S (XT), DANSCATT (MLC, LA) and the UNIK Center for Synthetic Biology (UG, MLC, KLM). Computational resources of XSEDE, and of the David A. Cofrin Center for Biomedical Information in the HRH Prince Alwaleed Bin Talal Bin Abdulaziz Alsaud Institute for Computational Biomedicine are gratefully acknowledged.
Biography
Karlsen et al. present structures of PICK1 demonstrating that the protein-binding PDZ domains are flexible with respect to the membrane binding BAR domain enabling long range scaffolding interactions. Furthermore, PICK1 oligomerization involves an offset, parallel arrangement of the BAR domains, possibly governing BAR domain auto-inhibition.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
TT purified, characterized and prepared protein for SAXS. MLK performed and developed SAXS analysis.
References
- Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. 2012 doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Zheng X. Tip-to-tip interaction in the crystal packing of PACSIN 2 is important in regulating tubulation activity. Protein Cell. 2013 doi: 10.1007/s13238-013-3041-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- Bhatia VK, Madsen KL, Bolinger PY, Kunding A, Hedegard P, Gether U, Stamou D. Amphipathic motifs in BAR domains are essential for membrane curvature sensing. The EMBO journal. 2009;28:3303–3314. doi: 10.1038/emboj.2009.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucrot E, Pick A, Camdere G, Liska N, Evergren E, McMahon HT, Kozlov MM. Membrane fission is promoted by insertion of amphipathic helices and is restricted by crescent BAR domains. Cell. 2012;149:124–136. doi: 10.1016/j.cell.2012.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet CE, A.V.Z., Kikhney AG, Franke D, Konarev PV, Shang W, Klaering R, Ro brahn B, Hermes C, Cipriani F, Svergun DI, Roessle M. Instrumental setup for high-throughput small-and wide-angle solution scattering at the X33 beamline of EMBL Hamburg. Journal of Applied Crystallography. 2012;45 [Google Scholar]
- Cao M, Mao Z, Kam C, Xiao N, Cao X, Shen C, Cheng KK, Xu A, Lee KM, Jiang L, et al. PICK1 and ICA69 control insulin granule trafficking and their deficiencies lead to impaired glucose tolerance. PLoS biology. 2013;11:e1001541. doi: 10.1371/journal.pbio.1001541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, Xu J, Shen C, Kam C, Huganir RL, Xia J. PICK1-ICA69 heteromeric BAR domain complex regulates synaptic targeting and surface expression of AMPA receptors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:12945–12956. doi: 10.1523/JNEUROSCI.2040-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Bhattacharyya S, Ma C, Morishita W, Fang S, Rizo J, Malenka RC. Calcium binding to PICK1 is essential for the intracellular retention of AMPA receptors underlying long-term depression. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:16437–16452. doi: 10.1523/JNEUROSCI.4478-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins JM, Papagrigoriou E, Berridge G, Yang X, Phillips C, Gileadi C, Savitsky P, Doyle DA. Structure of PICK1 and other PDZ domains obtained with the help of self-binding C-terminal extensions. Protein Sci. 2007;16:683–694. doi: 10.1110/ps.062657507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlendsson S, Rathje M, Heidarsson PO, Poulsen FM, Madsen KL, Teilum K, Gether U. PICK1 (protein interacting with C-kinase 1) binding promiscuity relies on unconventional PDZ (PSD-95/Discs-large/ZO-1 homology) binding modes for non-class II PDZ ligands. The Journal of biological chemistry. 2014 doi: 10.1074/jbc.M114.548743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost A, Perera R, Roux A, Spasov K, Destaing O, Egelman EH, De CP, Unger VM. Structural basis of membrane invagination by F-BAR domains. Cell. 2008;132:807–817. doi: 10.1016/j.cell.2007.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatter. O. new method for the evaluation of small-angle scattering data. Journal of Applied Crystallography. 1977:10. [Google Scholar]
- Halbach A, Morgelin M, Baumgarten M, Milbrandt M, Paulsson M, Plomann M. PACSIN 1 forms tetramers via its N-terminal F-BAR domain. FEBS J. 2007;274:773–782. doi: 10.1111/j.1742-4658.2006.05622.x. [DOI] [PubMed] [Google Scholar]
- Hanley JG. PICK1: a multi-talented modulator of AMPA receptor trafficking. Pharmacol Ther. 2008;118:152–160. doi: 10.1016/j.pharmthera.2008.02.002. [DOI] [PubMed] [Google Scholar]
- He Y, Liwo A, Weinstein H, Scheraga HA. PDZ binding to the BAR domain of PICK1 is elucidated by coarse-grained molecular dynamics. Journal of molecular biology. 2011;405:298–314. doi: 10.1016/j.jmb.2010.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst B, Madsen KL, Jansen AM, Jin C, Rickhag M, Lund VK, Jensen M, Bhatia V, Sorensen G, Madsen AN, et al. PICK1 deficiency impairs secretory vesicle biogenesis and leads to growth retardation and decreased glucose tolerance. PLoS biology. 2013;11:e1001542. doi: 10.1371/journal.pbio.1001542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen AM, Nassel DR, Madsen KL, Jung AG, Gether U, Kjaerulff O. PICK1 expression in the Drosophila central nervous system primarily occurs in the neuroendocrine system. J Comp Neurol. 2009;517:313–332. doi: 10.1002/cne.22155. [DOI] [PubMed] [Google Scholar]
- Jin W, Ge WP, Xu J, Cao M, Peng L, Yung W, Liao D, Duan S, Zhang M, Xia J. Lipid binding regulates synaptic targeting of PICK1, AMPA receptor trafficking, and synaptic plasticity. Journal of Neuroscience. 2006;26:2380–2390. doi: 10.1523/JNEUROSCI.3503-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MH, Vachette P, Svergun DI. Small-angle scattering: a view on the properties, structures and structural changes of biological macromolecules in solution. Quarterly reviews of biophysics. 2003;36:147–227. doi: 10.1017/s0033583503003871. [DOI] [PubMed] [Google Scholar]
- Li J, Mao X, Dong LQ, Liu F, Tong L. Crystal structures of the BAR-PH and PTB domains of human APPL1. Structure. 2007;15:525–533. doi: 10.1016/j.str.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Lu W, Ziff EB. PICK1 interacts with ABP/GRIP to regulate AMPA receptor trafficking. Neuron. 2005;47:407–421. doi: 10.1016/j.neuron.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Madsen KL, Beuming T, Niv MY, Chang CW, Dev KK, Weinstein H, Gether U. Molecular determinants for the complex binding specificity of the PDZ domain in PICK1. Journal of Biological Chemistry. 2005;280:20539–20548. doi: 10.1074/jbc.M500577200. [DOI] [PubMed] [Google Scholar]
- Madsen KL, Eriksen J, Milan-Lobo L, Han DS, Niv MY, mmendrup-Johnsen I, Henriksen U, Bhatia VK, Stamou D, Sitte HH, et al. Membrane localization is critical for activation of the PICK1 BAR domain. Traffic. 2008;9:1327–1343. doi: 10.1111/j.1600-0854.2008.00761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen KL, Thorsen TS, Rahbek-Clemmensen T, Eriksen J, Gether U. Protein Interacting with C Kinase 1 (PICK1) Reduces Reinsertion Rates of Interaction Partners Sorted to Rab11-dependent Slow Recycling Pathway. The Journal of biological chemistry. 2012;287:12293–12308. doi: 10.1074/jbc.M111.294702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinecke M, Boucrot E, Camdere G, Hon WC, Mittal R, McMahon HT. Cooperative recruitment of dynamin and BIN/amphiphysin/Rvs (BAR) domain-containing proteins leads to GTP-dependent membrane scission. J Biol Chem. 2013;288:6651–6661. doi: 10.1074/jbc.M112.444869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mim C, Cui H, Gawronski-Salerno JA, Frost A, Lyman E, Voth GA, Unger VM. Structural basis of membrane bending by the N-BAR protein endophilin. Cell. 2012;149:137–145. doi: 10.1016/j.cell.2012.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L, Wu H, Shen C, Shi Y, Jin W, Xia J, Zhang M. Clustering and synaptic targeting of PICK1 requires direct interaction between the PDZ domain and lipid membranes. The EMBO journal. 2007;26:4576–4587. doi: 10.1038/sj.emboj.7601860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen JS, Hansen S, Bauer R. The aggregation behavior of zinc-free insulin studied by small-angle neutron scattering. Eur Biophys J. 1994;22:379–389. doi: 10.1007/BF00180159. [DOI] [PubMed] [Google Scholar]
- Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB. PICK1 Targets Activated Protein Kinase C{alpha} to AMPA Receptor Clusters in Spines of Hippocampal Neurons and Reduces Surface Levels of the AMPA-Type Glutamate Receptor Subunit 2. Journal of Neuroscience. 2001;21:5417–5428. doi: 10.1523/JNEUROSCI.21-15-05417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov MV, Svergun DI. Applications of small-angle X-ray scattering to biomacromolecular solutions. Int J Biochem Cell Biol. 2012;45:429–437. doi: 10.1016/j.biocel.2012.10.017. [DOI] [PubMed] [Google Scholar]
- Pykalainen A, Boczkowska M, Zhao H, Saarikangas J, Rebowski G, Jansen M, Hakanen J, Koskela EV, Peranen J, Vihinen H, et al. Pinkbar is an epithelial-specific BAR domain protein that generates planar membrane structures. Nature structural & molecular biology. 2011;18:902–907. doi: 10.1038/nsmb.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylypenko O, Lundmark R, Rasmuson E, Carlsson SR, Rak A. The PX-BAR membrane-remodeling unit of sorting nexin 9. EMBO J. 2007;26:4788–4800. doi: 10.1038/sj.emboj.7601889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Y, Ma Q, Vahedi-Faridi A, Sundborger A, Pechstein A, Puchkov D, Luo L, Shupliakov O, Saenger W, Haucke V. Molecular basis for SH3 domain regulation of F-BAR-mediated membrane deformation. Proc Natl Acad Sci U S A. 2010;107:8213–8218. doi: 10.1073/pnas.1003478107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts-Galbraith RH, Ohi MD, Ballif BA, Chen JS, McLeod I, McDonald WH, Gygi SP, Yates JR, 3rd, Gould KL. Dephosphorylation of F-BAR protein Cdc15 modulates its conformation and stimulates its scaffolding activity at the cell division site. Molecular cell. 2010;39:86–99. doi: 10.1016/j.molcel.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca DL, Martin S, Jenkins EL, Hanley JG. Inhibition of Arp2/3-mediated actin polymerization by PICK1 regulates neuronal morphology and AMPA receptor endocytosis. Nature cell biology. 2008;10:259–271. doi: 10.1038/ncb1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogaard R, Borre L, Braunstein TH, Madsen KL, MacAulay N. Functional modulation of the glutamate transporter variant GLT1b by the PDZ domain protein PICK1. The Journal of biological chemistry. 2013;288:20195–20207. doi: 10.1074/jbc.M113.471128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudinger J, Lu J, Olson EN. Specific interaction of the PDZ domain protein PICK1 with the COOH terminus of protein kinase C-alpha. Journal of Biological Chemistry. 1997;272:32019–32024. doi: 10.1074/jbc.272.51.32019. [DOI] [PubMed] [Google Scholar]
- Staudinger J, Zhou J, Burgess R, Elleedge SJ, Olson EN. PICK1: A Perinuclear Binding Protein and Substrate for Protein Kinase C Isolated by the Yeast Two-hybrid System. Journal of Cell Biology. 1995;128:263–271. doi: 10.1083/jcb.128.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, Jin W, Thomas GM, Linden DJ, Huganir RL. Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron. 2006;49:845–860. doi: 10.1016/j.neuron.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Tanford C, Kawahara K, Lapanje S. Proteins in 6-M guanidine hydrochloride. Demonstration of random coil behavior. The Journal of biological chemistry. 1966;241:1921–1923. [PubMed] [Google Scholar]
- Wang Q, Kaan HY, Hooda RN, Goh SL, Sondermann H. Structure and plasticity of Endophilin and Sorting Nexin 9. Structure. 2008;16:1574–1587. doi: 10.1016/j.str.2008.07.016. [DOI] [PubMed] [Google Scholar]
- Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 1999;22:179–187. doi: 10.1016/s0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]
- Xu J, Xia J. Structure and function of PICK1. Neurosignals. 2006;15:190–201. doi: 10.1159/000098482. [DOI] [PubMed] [Google Scholar]
- Zhu G, Chen J, Liu J, Brunzelle JS, Huang B, Wakeham N, Terzyan S, Li X, Rao Z, Li G, et al. Structure of the APPL1 BAR-PH domain and characterization of its interaction with Rab5. EMBO J. 2007;26:3484–3493. doi: 10.1038/sj.emboj.7601771. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.