

Abstract
Salvia miltiorrhiza Bunge (Labiatae) is an emerging model plant for traditional medicine, and tanshinones are among the pharmacologically active constituents of this plant. Although extensive chemical and pharmaceutical studies of these compounds have been performed, studies on the basic helix-loop-helix (bHLH) transcription factors that regulate tanshinone biosynthesis are limited. In our study, 127 bHLH transcription factor genes were identified in the genome of S. miltiorrhiza, and phylogenetic analysis indicated that these SmbHLHs could be classified into 25 subfamilies. A total of 19 sequencing libraries were constructed for expression pattern analyses using RNA-Seq. Based on gene-specific expression patterns and up-regulated expression patterns in response to MeJA treatment, 7 bHLH genes were revealed as potentially involved in the regulation of tanshinone biosynthesis. Among them, the gene expression of SmbHLH37, SmbHLH74 and SmbHLH92 perfectly matches the accumulation pattern of tanshinone biosynthesis in S. miltiorrhiza. Our results provide a foundation for understanding the molecular basis and regulatory mechanisms of bHLH transcription factors in S. miltiorrhiza.
The basic helix-loop-helix (bHLH) transcription factor family is one of the largest transcription factor families in both animals and plants. More than 630 bHLH transcription factors have been identified in several important food crops1, and based on genome-wide analyses, 167, 177, 190 and at least 191 bHLH transcription factors have been predicted in the Arabidopsis thaliana L., Oryza sativa L., Nicotiana tabacum L. and Vitis vinifera L. genomes, respectively1,2,3. Interestingly, more bHLH transcription factors have been identified in the A. thaliana genome than in most animal species4. The completion of the Salvia miltiorrhiza Bunge genome sequence has facilitated studies on bHLH transcription factors in S. miltiorrhiza, a plant belonging to the Labiatae family that is an emerging model plant for traditional medicine5. For thousands of years, S. miltiorrhiza has been widely used for the treatment of cardiovascular disease, amenorrhea and dysmenorrhea6. Tanshinones, a major group of pharmacologically active constituents of this plant, mainly accumulate in the root periderm, and their biosynthesis is regulated by MeJA7,8,9. Although numerous chemical and pharmaceutical studies of S. miltiorrhiza have been conducted, investigations of transcription factors are limited, especially with regard to the regulation of transcription factors involved in tanshinone biosynthesis. Sequencing data show that are 15 members of the SPL transcription factor family in the S. miltiorrhiza genome10, and genome-wide characterisation of the R2R3-MYB transcription factor subfamily has also been performed11. Furthermore, the repressor role of SmMYB39 in the rosmarinic acid pathway has been characterised through overexpression and RNAi-mediated silencing12. An analysis of the S. miltiorrhiza transcriptome revealed 1,341 unigenes belonging to 46 transcription factor families, including 76 unigenes of bHLH transcription factors13. Additionally, upon treatment with yeast extract and Ag+14, 412 transcription factors (40 bHLH genes) were identified among the differentially expressed genes in S. miltiorrhiza hairy roots.
Nonetheless, transcriptome sequencing is not likely to identify all bHLH genes simultaneously, especially those genes expressed under specific conditions. Thus, to systematically elucidate the regulatory role of bHLH transcription factors in S. miltiorrhiza, a genome-wide analysis of bHLH transcription factors was performed. Based on various expression patterns and the results of MeJA treatment and qPCR analyses, 3 bHLH genes that likely regulate tanshinone biosynthesis were identified. The findings from this study provide a foundation for understanding the molecular basis and regulatory mechanisms of bHLH transcription factors in S. miltiorrhiza.
Results
Gene prediction, sequence features and phylogenetic analysis
A total of 127 SmbHLHs containing ORF sequences were identified and named SmbHLH1-SmbHLH127 (Table S1). The number of bHLH genes in S. miltiorrhiza is similar to the number of bHLH genes in A. thaliana, O. sativa, N. tabacum and V. vinifera. The gene length of SmbHLHs was found to vary from 459 bp (SmbHLH58) to 8,663 bp (SmbHLH61) (Table S2), and the length of SmbHLH cDNAs varied from 255 bp (SmbHLH71) to 2,154 bp (SmbHLH81) (Table S2). The molecular weights of the predicted proteins range from 9,533.9 Da (SmbHLH71) to 79,156.6 Da (SmbHLH81) (Table S2), and the theoretical isoelectric points are predicted to range from 4.8 (SmbHLH69) to 9.9 (SmbHLH126 and SmbHLH127) (Table S2).
A neighbour-joining tree constructed among the 127 bHLH members identified in S. miltiorrhiza indicated a total of 25 distinct subfamilies (designated A to Y) (Fig. 1), with subfamily A having the largest number of members (20 SmbHLHs) and subfamilies C, L, Q and X the fewest (1 bHLH). An un-rooted phylogenetic tree was constructed using the bHLH gene family in S. miltiorrhiza and 5 other bHLH genes (AtTT8, NtMYC2a, NtMYC2b, CjbHLH1, and CrMYC2) that regulate secondary metabolism15,16,17,18. Interestingly, AtTT8, NtMYC2a, NtMYC2b, and CrMYC2 cluster in subfamily R, and CjbHLH1 clusters in subfamily O. According to the alignment, the transcription factors in subfamily R (9 genes) are putative regulators of tanshinone biosynthesis.
Figure 1. An un-rooted phylogenetic tree of the bHLH gene family in S. miltiorrhiza.

The amino acid sequences were aligned using Clustal W, and the phylogenetic tree was constructed using neighbour-joining criteria. The letters (A-Y) represent the main subfamilies. The putative bHLH genes involved in the regulation of tanshinone biosynthesis are SmbHLH37 (subfamily R), SmbHLH51 (subfamily R), SmbHLH53 (subfamily R), SmbHLH60 (subfamily W), SmbHLH74 (subfamily H), SmbHLH92 (subfamily P), and SmbHLH103 (subfamily P).
Structure analysis
Structure analyses of all of the bHLH genes revealed that the number of exons varies from 1 to 12; a total of 6 genes are intronless. The genes in the 25 subfamilies have an average exon number per gene ranging from 1 (subfamily U) to 9 (subfamily D), and the average exon number in subfamilies G, H and L is 5. The same pattern was observed in subfamilies C, K and M but with an average number of 4. The 6 intronless genes are distributed across 3 subfamilies, particularly subfamily U, in which all 4 genes are intronless. The remaining 2 intronless genes belong to subfamily R and subfamily T. Although these subfamilies contain only 1 gene, all of the other genes in subfamilies K and N contain 4 and 2 exons, respectively. The structural features of each bHLH gene in subfamily A are listed in Fig. 2, and those of the others are listed in Figure S1.
Figure 2. The structural features of each bHLH gene in subfamily A.
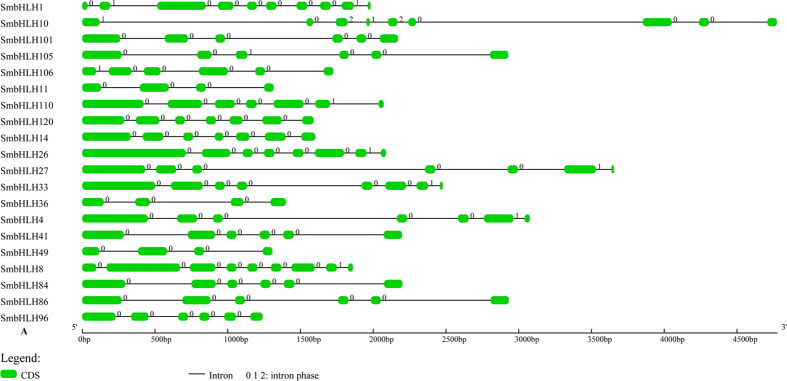
The exons are represented by green round-cornered rectangles. The black lines connecting two exons represent introns. The numbers above the line represent the intron phase.
Exons with the same splicing phase at both ends are called symmetric exons, and an excess of symmetric exons and phase 0 introns is likely to facilitate exon shuffling, recombinational fusion and protein domain exchange19,20. According to the 575 exons analysed herein, 230 exons are symmetric with phase 0 introns, 2 exons are symmetric with phase 1 introns, and 11 exons are symmetric with phase 2 introns. Among the 448 introns of the bHLH genes, 363 are phase 0, 41 are phase 1 and 44 are phase 2. Therefore, our analysis of the bHLH gene structures strongly indicates a great bHLH transcription factor family diversity in S. miltiorrhiza.
Conserved motif analysis
A total of 22 conserved motifs were characterised (motifs 1–22) (Table S3). The SmbHLHs in subfamily A contain the highest number of motifs (11 types), whereas the SmbHLHs in subfamilies C, T, U and X contain the smallest number of motifs (2 types). Additionally, the average motif number per sequence varies across subfamilies, ranging from 2 (subfamilies C, N, T and U) to 7 (subfamily V).
In most cases, a motif is repeated only once, but a few special cases were found. Motif 2 is repeated twice in SmbHLH72 and SmbHLH107. Motif 4 and motif 21 are repeated twice in SmbHLH94 and SmbHLH20, respectively. The highest number of motif repetitions occurs in SmbHLH54, in which motifs 2, 6 and 10 are each repeated twice. Furthermore, certain conserved motifs are nested in specific subfamilies in the un-rooted phylogenetic tree (Fig. 1). For example, motif 16 is shared by 2 members (SmbHLH89 and SmbHLH121) in subfamily D; motif 8 is shared by 2 members (SmbHLH90 and SmbHLH109) in subfamily M; and motif 21 is shared by 5 members (SmbHLH20, SmbHLH23, SmbHLH25, SmbHLH90, and SmbHLH109) in subfamily M. Motifs 12, 13 and 18 are only distributed in subfamily A. Similarly, motifs 6, 10, 11, 17 and 22 are specific to subfamily V. The distribution of conserved motifs of each bHLH gene is listed in Fig. 3.
Figure 3. The distribution of conserved motifs in each bHLH gene.
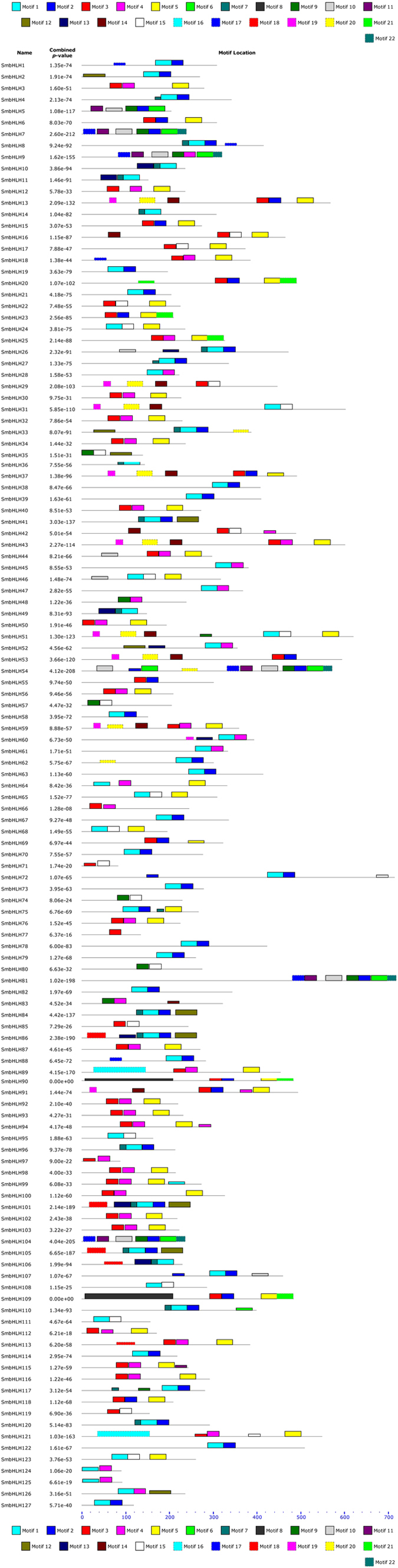
The relative positions of each conserved motif within the bHLH protein are shown in colour.
Differential expression of bHLH genes in various organs
After mapping these clean reads to the S. miltiorrhiza genome database, the expression levels were estimated according to RPKM (reads per kilobase per million) values (Table S4). Among the 127 bHLH genes, the expression of 117 bHLH genes, comprising 24 subfamilies, was detected in at least one of the four organs tested. The other 10 genes, distributed across 8 subfamilies, were undetected (RPKM values of 0). Among the 117 detected genes, 99 were detected with expression in the flower, 93 in the leaf, 93 in the root and 99 in the stem.
To understand the spatial transcriptional patterns of bHLH genes among various organs, hierarchy clustering was performed using the 117 genes detected. As illustrated in Fig. 4, 45 genes showed relatively high expression (RPKM values higher than 1) among the four organs. In addition, most of the bHLH genes exhibited diverse expression profiles. For example, 8 genes (SmbHLH17, SmbHLH23, SmbHLH58, SmbHLH79, SmbHLH104, SmbHLH106, SmbHLH117 and SmbHLH118) were specifically expressed in the flower; 2 genes (SmbHLH32 and SmbHLH114) were specifically expressed in the leaf; 3 genes (SmbHLH54, SmbHLH92 and SmbHLH124) were specifically expressed in the root; and 3 genes (SmbHLH55, SmbHLH69 and SmbHLH125) were specifically expressed in the stem (Fig. 1). Additionally, 17 genes exhibited higher expression levels in flowers than in vegetative organs (at least 2-fold higher than other organs, with RPKM values greater than 1). Furthermore, 22 genes exhibited higher expression levels in the root than in the other three organs, and this group included 2 genes (SmbHLH92 and SmbHLH124) that were specifically expressed in the root (Fig. 1); however, the RPKM value of the other gene (SmbHLH54) specifically expressed in the root was 0.075. Two of the 22 genes (SmbHLH51 and SmbHLH53) belong to subfamily R; 7 of the 22 genes belong to subfamily P; and 3 of the 22 genes belong to subfamily W.
Figure 4. Heatmaps representing the expression profiles of S. miltiorrhiza bHLH genes in the flower, leaf, root and stem.

The numbers to the right of the figure indicate the bHLH gene name. The colour scale is shown at the top. Higher expression levels are shown in green. The genes with RPKM values of 0 in the four tested organs are not included in the figure.
Differential expression of bHLH genes in various tissues
The expression of 101 bHLH genes, comprising 23 subfamilies, was detected in at least one of the three root tissues (periderm, phloem and xylem) tested (Table S5). A total of 26 genes, distributed among 12 subfamilies, were undetected (RPKM values of 0). Compared to the differential expression data for the four plant organs, 20 of these 26 genes were not detected in the root; 4 of the remaining 6 genes were detected in the flower, stem and leaf, with 1 of the 6 genes detected in the flower and stem, whereas 1 of the 6 genes was specifically expressed in the root. Of the 101 detected genes, the expression of 90 was detected in the periderm; 94 genes were found to be expressed in the phloem, with 97 expressed in the xylem. Eight of the 10 genes had an RPKM value of 0 in all four tested organs, and the RPKM values were 0 in the three root tissues for all 8 genes, except SmbHLH10 and SmbHLH77. The RPKM values of SmbHLH10 and SmbHLH77 in the three tissues were less than 1 under normal conditions. A total of 52 bHLH genes with relatively high expression (RPKM values higher than 1) were detected across the three tissues (Fig. 1). Compared with the RPKM values of the four organs, 48 of the 52 genes (all, except SmbHLH5, SmbHLH12, SmbHLH66 and SmbHLH123) exhibited relatively high expression levels in the root.
The highest percentage of lipid-soluble tanshinones is found in the periderm of the root. In the current study, 12 bHLH genes exhibited higher expression levels in the periderm (at least 2-fold higher than in other tissues, with RPKM values greater than 1) than in the two other tissues tested (Fig. 1). These genes were SmbHLH6, SmbHLH12, SmbHLH30, SmbHLH55, SmbHLH74, SmbHLH92, SmbHLH102, SmbHLH103, SmbHLH115, SmbHLH117, SmbHLH123 and SmbHLH125. Seven of the 12 genes (all, except SmbHLH12, SmbHLH55, SmbHLH117, SmbHLH123 and SmbHLH125) showed relatively high expression levels in the root.
Up-regulated bHLH genes in response to methyl jasmonate
A total of 10 bHLH genes were up-regulated by MeJA treatment of S. miltiorrhiza leaves, SmbHLH4, SmbHLH8, SmbHLH21, SmbHLH37, SmbHLH51, SmbHLH53, SmbHLH60, SmbHLH74, SmbHLH90, and SmbHLH109 (Fig. 1), 3 of which (SmbHLH37, SmbHLH51, and SmbHLH53) are in subfamily R. In addition, 4 of the 10 genes (SmbHLH51, SmbHLH53, SmbHLH60, and SmbHLH74) exhibited higher expression levels in the root than in the other three organs. These 4 genes are most likely involved in the regulation of tanshinone biosynthesis in S. miltiorrhiza and are distributed among 3 subfamilies (subfamilies H, R and W). More interestingly, SmbHLH74 (subfamily H) exhibited high expression levels not only in the root but also in the periderm. In addition, the expression level of SmbHLH74 was up-regulated upon MeJA treatment.
qPCR analysis
The expression levels of 7 bHLH genes (Fig. 1) identified as putative regulators of tanshinone biosynthesis were analysed using qPCR. The DXS2 gene exhibited relatively high expression in the root and root periderm. Additionally, the expression level of DXS2 was up-regulated upon MeJA treatment (Fig. 5). The expression patterns of 3 (SmbHLH37, SmbHLH74, and SmbHLH92) of the 7 genes in various organs or tissues or in response to MeJA treatment were similar to the results obtained from RNA-Seq (Fig. 5). However, the expression patterns of SmbHLH51, SmbHLH53, SmbHLH60 and SmbHLH103 in the four organs did not match those obtained by RNA-Seq (Fig. 5).
Figure 5. Expression patterns of 7 putative bHLH genes and the DXS2 gene.

Fold changes in gene expression levels in S. miltiorrhiza R (root), S (stem), L (leaf), F (flower), R1 (periderm), R2 (phloem), R3 (xylem), T0 (treatment with carrier solution for 12 h), and T12 (treatment with MeJA for 12 h) are shown. The transcript levels in the leaf after treatment with the carrier solution for 12 h were normalised to 1.
Promoter sequence analysis of enzyme-coding genes in tanshinone biosynthesis pathways
As the biosynthesis of tanshinones depends on cross-talk between the MEP and MVA pathways21, the promoter sequences of 72 enzyme-coding genes (19 enzymes) in tanshinone biosynthesis pathways were analysed (Table S6). Interestingly, 70 enzyme-coding genes were predicted based on bHLH binding sites (E-box), including 4 functionally characterised genes (HMGR2, CPS1, KSL1 and CYP76AH1)22,23,24. The E-box was predicted in the promoter sequences of 6 enzyme-coding genes (DXS, DXR, MCT, CMK, MDS and HDR, but not HDS) in the MEP pathway. Additionally, the E-box was predicted in the promoter sequences of genes encoding different types of enzymes (AACT, HMGS, HMGR, MK, PMK and MDC) in the MVA pathway. These results indicated that bHLH transcription factors may play a role in regulating tanshinone biosynthesis via the modulation of the MEP and MVA pathways.
Discussion
Although the bHLH family has been broadly studied in animals and a diverse range of plants, the present study is the first to report the identification and characterisation of bHLH transcription factors based on the entire genome sequence of S. miltiorrhiza. According to their evolutionary origin, sequence relatedness, functional activity and DNA-binding specificity, bHLH proteins are classified into 6 main groups (designated A to F) in animal systems25,26,27; however, the classification of bHLH transcription factors in plants is on-going28. In this study, 127 bHLH members in S. miltiorrhiza were classified into 25 subfamilies, consistent with other plants4,28,29,30,31. Previous studies have suggested that the classification of bHLH transcription factors in plants might be different from that in animals. Based on a phylogenetic tree, bHLHs in S. miltiorrhiza and A. thaliana can be classified into 52 subfamilies (Figure S2); 23 of these 52 subfamilies include proteins from S. miltiorrhiza and A. thaliana, with the other 10 subfamilies (18 genes) specific to S. miltiorrhiza and 19 (48 genes) specific to A. thaliana. These results indicate that certain bHLHs play strongly conserved roles in S. miltiorrhiza and A. thaliana, whereas other bHLHs may have specific functions. Most of the bHLH genes specific to S. miltiorrhiza were found to cluster in subfamilies R and P. Additionally, 4 of the 7 putative genes regulating tanshinone biosynthesis are specific to S. miltiorrhiza (Fig. 1).
The regulation of growth and development, stress resistance, and signal transduction by bHLH transcription factors has been reported in plants27,30,32,33,34,35,36,37,38. To date, at least 43 bHLH transcription factors have been identified as regulators of secondary metabolism in at least 21 distinct plants (Table S7), and these active components include flavonoids, alkaloids, and terpenoids. bHLH transcription factor genes are expressed in stimulus-responsive, constitutive or organ-specific manners39,40,41, and organ-specific and MeJA-responsive expression patterns of bHLHs were observed in our study. In addition, available functional information from Zea mays L., A. thaliana and Matthiola incana R. Br. suggests that bHLH transcription factors can participate in flavonoid biosynthesis in both a positive and negative fashion42,43,44,45. In Catharanthus roseus (L.) G. Don, the bHLH transcription factor CrMYC2 regulates ORCA gene expression, which in turn regulates alkaloid biosynthesis genes17. Additionally, a regulatory role for Coptis japonica Makino CjbHLH1 in the transcription of isoquinoline alkaloid (IQA) biosynthesis genes has been reported16. The main objectives of the present research were to identify potential bHLH transcription factors involved in tanshinone biosynthesis.
Genes expressed at a higher level in the root and root periderm or in MeJA-treated leaves might regulate tanshinone biosynthesis in S. miltiorrhiza. After mapping all of the bHLH genes (genes up-regulated in response to MeJA, genes specifically expressed in the root, genes more highly expressed in the root than in the other three organs, genes exhibiting a relatively high expression level in the three tissues, genes more highly expressed in the periderm than in the other two tissues, and genes specific to S. miltiorrhiza compared to A. thaliana) to the phylogenetic tree of the bHLH gene family in S. miltiorrhiza, most genes were found to cluster in subfamilies H, P, R and W (Fig. 1). The regulation of sesquiterpene biosynthesis gene expression by Arabidopsis MYC2 (bHLH transcription factor) has been reported46, and tanshinones and sesquiterpenes are both terpenoids. In addition, upon construction of a neighbour-joining phylogenetic tree using MEGA 5.1, MYC2 was found in the same subfamily as SmbHLH37, SmbHLH51 and SmbHLH53. Based on gene-specific expression patterns, up-regulated expression patterns in response to MeJA treatment and qPCR analyses, the gene expression of 3 bHLH genes (SmbHLH37, SmbHLH74, and SmbHLH92) perfectly matches the accumulation pattern of tanshinone biosynthesis. Previous studies have shown that roots contain the highest concentration of tanshinones, though the concentration in leaves is very low47, and some key enzyme-coding genes (SmGGPPS2 and SmCPS1) have been reported to be relatively highly expressed in roots48. These results indicated that tanshinone biosynthesis was more active in roots than in other organs, further suggesting that tanshinones are originally produced in the roots of S. miltiorrhiza. These results provide the foundation for understanding the role of bHLH transcription factors in the regulation of tanshinone biosynthesis in S. miltiorrhiza.
Materials and Methods
Plant materials and treatment conditions
Salvia miltiorrhiza Bunge (line 99–3) was grown in a field at the Institute of Medicinal Plant Development in Beijing. When the plants were blooming in May, the flowers, leaves, stems and roots of S. miltiorrhiza were collected, treated with liquid nitrogen and stored at −80 °C until subsequent analysis. In addition, some roots were separated into the periderm, phloem and xylem immediately after collection. Leaves were treated with MeJA (200 μM) and collected as described in a previous study49. At least two biological duplicate samples of each organ or tissue were used.
RNA-Seq and bioinformatics analysis
Total mRNA was isolated using the RNeasy Plus Mini kit (Qiagen, Hilden, Germany); 1% agarose gel electrophoresis was used to confirm the integrity and quality of the total RNA. An accurate RNA concentration was determined using a NanoDrop 2000 spectrophotometer (Thermo, Waltham, MA, USA) and Qubit (Invitrogen, Carlsbad, CA, USA). An Agilent 2100 (Agilent, California, USA) was used to identify the integrity of the total RNA in more detail and to confirm accuracy before sequencing. A total of 19 sequencing libraries were constructed, including 8 for the 4 organs, 9 for the 3 root tissues and 2 for the MeJA treatment experiment49. The libraries were sequenced using the Illumina HiSeq 2500 platform to produce 100 bp paired-end reads. All produced reads were mapped to the genome of S. miltiorrhiza using Tophat 2.0.1150. Differential gene expression was analysed using Cufflinks 2.2.151. To visualise the global transcription profile of bHLH genes, hierarchical clustering was performed using R52.
Identification of bHLH genes and sequence feature analysis
All SmbHLH genes were identified by BLAST analysis of the bHLH domain against the S. miltiorrhiza genome. All of the predicted SmbHLH proteins contain the conserved bHLH domain, as determined using sequence search in Pfam53. The SmbHLH genes were manually corrected using a protein BLAST algorithm (http://blast.ncbi.nlm.nih.gov/Blast.cgi). In addition, the Compute pI/Mw tool on the ExPASy server (http://web.expasy.org/compute_pi/) was used to predict the theoretical isoelectric point (pI) and the molecular weight (Mw) of the SmbHLH proteins.
Phylogenetic, gene structure and MEME motif analyses
A Clustal W analysis was performed, and an un-rooted neighbour-joining tree was constructed in MEGA 5.1, with 1000 bootstrap replicates based on the amino acid sequences of the bHLH domain of SmbHLH proteins (Table S8)54. The online Gene Structure Display Server (GSDS 2.0) (http://gsds.cbi.pku.edu.cn/index.php) was used to investigate the gene structure based on each coding sequence (CDS) and corresponding genomic sequence. Conserved motifs in S. miltiorrhiza bHLH transcription factors were identified using MEME (Suite version 4.9.1) with the following criteria: expected E-values less than 2 × 10−30, any number of repetitions of a motif, and an optimum width of 10–200 amino acids55,56.
Quantitative real-time reverse transcription PCR (qPCR) and promoter sequences analysis
Total RNA was reverse-transcribed using a FastQuant RT kit (TIANGEN, China). The qPCR reactions were performed according to the manufacturer’s protocol with the SYBR premix Ex Taq kit (TaKaRa, China) and conducted in triplicate using the Applied Biosystems 7500 Real-Time PCR system (Life Technologies, USA). The primers were designed using Primer Premier 6 (Table S9), with an amplicon size ranging from 130 bp to 180 bp and an optimal Tm of 54 ± 1 °C. The specificity of the primers was assessed by 2% agarose gel electrophoresis and a dissociation curve. SmActin was used as the reference gene in the current study. In addition, the expression level of the DXS2 (1-deoxy-D-xylulose 5-phosphate synthase) gene, which encodes the enzyme for the first step in the MEP pathway, was analysed as a positive control48. The promoter sequences (1500 bp) of enzyme-coding genes in the tanshinone biosynthesis pathways were used to predicted cis-elements in the PLACE database (http://www.dna.affrc.go.jp/PLACE/signalscan.html).
Accession codes
Illumina HiSeq and genome sequencing data have been submitted to Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) under accessions SRP051564, SRP028388, SRR1640458 and SRP051524.
Additional Information
How to cite this article: Zhang, X. et al. Genome-wide characterisation and analysis of bHLH transcription factors related to tanshinone biosynthesis in Salvia miltiorrhiza. Sci. Rep. 5, 11244; doi: 10.1038/srep11244 (2015).
Supplementary Material
Acknowledgments
The authors thank Professor Xian’en Li (Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College) for providing the Salvia miltiorrhiza Bunge material (line 99-3) used in this work. This work was supported by the National Key Technology R&D Program (grant no. 2012BAI29B01) and the Program for Innovative Research Team at the Institute of Medicinal Plant Development (grant no. IT1304).
Footnotes
Author Contributions X.Z. contributed to the sample collection, RNA extraction, and real-time PCR performance; analysed the data; and drafted the manuscript. H.M.L. participated in the design of the study. Z.C.X. contributed to analysis of the data and RNA extraction. Y.J.Z. contributed to analysis of the data. A.J.J. participated in the discussion of the results and revision of the manuscript. This work was conducted in the laboratory of J.Y.S. and S.L.C., who contributed to the evaluation and discussion of the results and revision of the manuscript.
References
- Carretero-Paulet L. et al. Genome-wide classification and evolutionary analysis of the bHLH family of transcription factors in Arabidopsis, poplar, rice, moss, and algae. Plant Physiol. 153, 1398–1412 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007). [DOI] [PubMed] [Google Scholar]
- Rushton P. J. et al. Tobacco transcription factors: novel insights into transcriptional regulation in the Solanaceae. Plant Physiol. 147, 280–295 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires N. & Dolan L. Origin and diversification of basic-helix-loop-helix proteins in plants. Mol. Biol. Evol 27, 862–874 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao F. & Lu S. Genome-wide identification, molecular cloning, expression profiling and posttranscriptional regulation analysis of the Argonaute gene family in Salvia miltiorrhiza, an emerging model medicinal plant. BMC Genomics 14, 512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T. O. Danshen: a popular chinese cardiac herbal drug. J. Am. Coll. Cardiol. 47, 1498–1500 (2006). [DOI] [PubMed] [Google Scholar]
- Hao G. et al. Cloning, molecular characterization and functional analysis of 1-hydroxy-2-methyl-2-(E)-butenyl-4-diphosphate reductase (HDR) gene for diterpenoid tanshinone biosynthesis in Salvia miltiorrhiza Bge. f. alba. Plant Physiol. Biochem. 70, 21–32 (2013). [DOI] [PubMed] [Google Scholar]
- Hao X. et al. Effects of methyl jasmonate and salicylic acid on tanshinone production and biosynthetic gene expression in transgenic Salvia miltiorrhiza hairy roots. Biotechnol. Appl. Biochem. 10.1002/bab.1236 (2014). [DOI] [PubMed] [Google Scholar]
- Yang D. et al. PEG and ABA trigger methyl jasmonate accumulation to induce the MEP pathway and increase tanshinone production in Salvia miltiorrhiza hairy roots. Physiol. Plant. 146, 173–183 (2012). [DOI] [PubMed] [Google Scholar]
- Zhang L. et al. Genome-wide analysis and molecular dissection of the SPL gene family in Salvia miltiorrhiza. J. Integr. Plant Biol. 56, 38–50 (2014). [DOI] [PubMed] [Google Scholar]
- Li C. & Lu S. Genome-wide characterization and comparative analysis of R2R3-MYB transcription factors shows the complexity of MYB-associated regulatory networks in Salvia miltiorrhiza. BMC Genomics 15, 277 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. et al. Cloning and characterization of a putative R2R3 MYB transcriptional repressor of the rosmarinic acid biosynthetic pathway from Salvia miltiorrhiza. PloS One 8, e73259 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua W., Zhang Y., Song J., Zhao L. & Wang Z. De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics 98, 272–279 (2011). [DOI] [PubMed] [Google Scholar]
- Gao W. et al. Combining metabolomics and transcriptomics to characterize tanshinone biosynthesis in Salvia miltiorrhiza. BMC Genomics 15, 73 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesi N. et al. The TT8 gene encodes a basic helix-loop-helix domain protein required for expression of DFR and BAN genes in Arabidopsis siliques. The Plant Cell 12, 1863–1878 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y. et al. Isoquinoline alkaloid biosynthesis is regulated by a unique bHLH-type transcription factor in Coptis japonica. Plant Cell Physiol. 52, 1131–1141 (2011). [DOI] [PubMed] [Google Scholar]
- Zhang H. et al. The basic helix-loop-helix transcription factor CrMYC2 controls the jasmonate-responsive expression of the ORCA genes that regulate alkaloid biosynthesis in Catharanthus roseus. The Plant J. 67, 61–71 (2011). [DOI] [PubMed] [Google Scholar]
- Zhang H. B., Bokowiec M. T., Rushton P. J., Han S. C. & Timko M. P. Tobacco transcription factors NtMYC2a and NtMYC2b form nuclear complexes with the NtJAZ1 repressor and regulate multiple jasmonate-inducible steps in nicotine biosynthesis. Mol. Plant 5, 73–84 (2012). [DOI] [PubMed] [Google Scholar]
- Gilbert W. The exon theory of genes. Cold Spring Harb. Symp. Quant. Biol. 52, 901–905 (1987). [DOI] [PubMed] [Google Scholar]
- Patthy L. Intron-dependent evolution: preferred types of exons and introns. FEBS Lett. 214, 1–7 (1987). [DOI] [PubMed] [Google Scholar]
- Laule O. et al. Crosstalk between cytosolic and plastidial pathways of isoprenoid biosynthesis in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U. S. A. 100, 6866–6871 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. et al. CYP76AH1 catalyzes turnover of miltiradiene in tanshinones biosynthesis and enables heterologous production of ferruginol in yeasts. Proc. Natl. Acad. Sci. U. S. A. 100, 12108–12113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z. et al. Cloning and characterization of a novel 3-hydroxy-3-methylglutaryl coenzyme a reductase gene from Salvia miltiorrhiza involved in diterpenoid tanshinone accumulation. J. Plant. Physiol. 168, 148–157 (2011). [DOI] [PubMed] [Google Scholar]
- Zhou Y. et al. Modular pathway engineering of diterpenoid synthases and the mevalonic acid pathway for miltiradiene production. J. Am. Chem. Soc. 134, 3234–3241 (2012). [DOI] [PubMed] [Google Scholar]
- Atchley W. R. & Fitch W. M. A natural classification of the basic helix-loop-helix class of transcription factors. Proc. Natl. Acad. Sci. U. S. A. 94, 5172–5176 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang C. V., Dolde C., Gillison M. L. & Kato G. J. Discrimination between related DNA sites by a single amino acid residue of Myc-related basic-helix-loop-helix proteins. Proc. Natl. Acad. Sci. U. S. A. 89, 599–602 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledent V. & Vervoort M. The basic helix-loop-helix protein family: comparative genomics and phylogenetic analysis. Genome Res. 11, 754–770 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. et al. Genome-wide analysis of basic/helix-loop-helix transcription factor family in rice and Arabidopsis. Plant Physiol. 141, 1167–1184 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck M. J. & Atchley W. R. Phylogenetic analysis of plant basic helix-loop-helix proteins. J. Mol. Evol. 56, 742–750 (2003). [DOI] [PubMed] [Google Scholar]
- Heim M. A. et al. The basic helix-loop-helix transcription factor family in plants: a genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 20, 735–747 (2003). [DOI] [PubMed] [Google Scholar]
- Toledo-Ortiz G., Huq E. & Quail P. H. The Arabidopsis basic/helix-loop-helix transcription factor family. The Plant Cell 15, 1749–1770 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A., Zhao M., Leavitt J. M. & Lloyd A. M. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. The Plant J. 53, 814–827 (2008). [DOI] [PubMed] [Google Scholar]
- Khanna R. et al. The basic helix-loop-helix transcription factor PIF5 acts on ethylene biosynthesis and phytochrome signaling by distinct mechanisms. The Plant Cell 19, 3915–3929 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. H. et al. SOMNUS, a CCCH-type zinc finger protein in Arabidopsis, negatively regulates light-dependent seed germination downstream of PIL5. The Plant Cell 20, 1260–1277 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa Y., Yamashino T. & Mizuno T. The circadian clock regulates the photoperiodic response of hypocotyl elongation through a coincidence mechanism in Arabidopsis thaliana. Plant Cell Physiol. 50, 838–854 (2009). [DOI] [PubMed] [Google Scholar]
- Sonnenfeld M. J., Delvecchio C. & Sun X. Analysis of the transcriptional activation domain of the Drosophila tango bHLH-PAS transcription factor. Dev. Genes. Evol. 215, 221–229 (2005). [DOI] [PubMed] [Google Scholar]
- Xu J. et al. The aborted microspores regulatory network is required for postmeiotic male reproductive development in Arabidopsis thaliana. The Plant Cell 22, 91–107, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi K., Menand B., Bell E. & Dolan L. A basic helix-loop-helix transcription factor controls cell growth and size in root hairs. Nat. Genet. 42, 264–267, (2010). [DOI] [PubMed] [Google Scholar]
- Hichri I. et al. The basic helix-loop-helix transcription factor MYC1 is involved in the regulation of the flavonoid biosynthesis pathway in grapevine. Mol. Plant. 3, 509–523 (2010). [DOI] [PubMed] [Google Scholar]
- Quattrocchio F., Wing J. F., van der Woude K., Mol J. N. & Koes R. Analysis of bHLH and MYB domain proteins: species-specific regulatory differences are caused by divergent evolution of target anthocyanin genes. The Plant J. 13, 475–488 (1998). [DOI] [PubMed] [Google Scholar]
- Steyn W. J., Wand S. J., Jacobs G., Rosecrance R. C. & Roberts S. C. Evidence for a photoprotective function of low-temperature-induced anthocyanin accumulation in apple and pear peel. Physiol. Plant. 136, 461–472, (2009). [DOI] [PubMed] [Google Scholar]
- Ludwig S. R., Habera L. F., Dellaporta S. L. & Wessler S. R. Lc, a member of the maize R gene family responsible for tissue-specific anthocyanin production, encodes a protein similar to transcriptional activators and contains the myc-homology region. Proc. Natl. Acad. Sci. U. S. A. 86, 7092–7096 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne C. T., Zhang F. & Lloyd A. M. GL3 encodes a bHLH protein that regulates trichome development in arabidopsis through interaction with GL1 and TTG1. Genetics 156, 1349–1362 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay N. A., Walker A. R., Mooney M. & Gray J. C. Two basic-helix-loop-helix genes (MYC-146 and GL3) from Arabidopsis can activate anthocyanin biosynthesis in a white-flowered Matthiola incana mutant. Plant Mol. Biol. 52, 679–688 (2003). [DOI] [PubMed] [Google Scholar]
- Sasaki-Sekimoto Y. et al. Basic helix-loop-helix transcription factors jasmonate-associated MYC2-LIKE1 (JAM1), JAM2, and JAM3 are negative regulators of jasmonate responses in Arabidopsis. Plant Physiol. 163, 291–304 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong G. J., Xue X. Y., Mao Y. B., Wang L. J. & Chen X. Y. Arabidopsis MYC2 interacts with DELLA proteins in regulating sesquiterpene synthase gene expression. The Plant Cell 24, 2635–2648 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matkowski A. et al. Antioxidant activity of extracts from leaves and roots of Salvia miltiorrhiza Bunge, S. przewalskii Maxim., and S. verticillata L. Bioresour. Technol. 99, 7892–7896 (2008). [DOI] [PubMed] [Google Scholar]
- Ma Y. et al. Genome-wide identification and characterization of novel genes involved in terpenoid biosynthesis in Salvia miltiorrhiza. J. Exp. Bot. 63, 2809–2823 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H. et al. Transcriptional data mining of Salvia miltiorrhiza in response to methyl jasmonate to examine the mechanism of bioactive compound biosynthesis and regulation. Physiol. Plant. 152, 241–255 (2014). [DOI] [PubMed] [Google Scholar]
- Trapnell C., Pachter L. & Salzberg S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M. et al. The Pfam protein families database. Nucleic Acids Res. 40, D290–301 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T. L., Williams N., Misleh C. & Li W. W. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34, W369–373 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Z. X. et al. Genome-wide identification, classification, and analysis of two-component signal system genes in maize. Genet. Mol. Res. 10, 3316–3330 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.