

Abstract
Nonribosomal peptide synthetases are large, multi-domain enzymes that produce peptide molecules with important biological activity such as antibiotic, antiviral, anti-tumor, siderophore and immunosuppressant action. The adenylation (A) domain catalyzes two reactions in the biosynthetic pathway. In the first reaction, it activates the substrate amino acid by adenylation and in the second reaction it transfers the amino acid onto the phosphopantetheine arm of the adjacent peptide carrier protein (PCP) domain. The conformation of the A domain differs significantly depending on which of these two reactions it is catalyzing. Recently, several structures of A–PCP di-domains have been solved using mechanism-based inhibitors to trap the PCP domain in the A domain active site. Here, we present an alternative strategy to stall the A–PCP di-domain, by engineering a disulfide bond between the native amino acid substrate and the A domain. Size exclusion studies showed a significant shift in apparent size when the mutant A–PCP was provided with cross-linking reagents, and this shift was reversible in the presence of high concentrations of reducing agent. The cross-linked protein crystallized readily in several of the conditions screened and the best crystals diffracted to ≈8 Å.
Keywords: crystallization, disulfide cross-link, nonribosomal peptide synthetase, protein engineering, thioesterification
Introduction
Nonribosomal peptide synthetases (NRPSs) are large macromolecular enzymes that catalyze peptide bond formation between monomeric subunits to produce nonribosomal peptide molecules (NRPs) with important biological and therapeutic activity (Konz and Marahiel, 1999; Schwarzer et al., 2003; Fischbach and Walsh, 2006; Felnagle et al., 2008). The substrates used by NRPSs to make NRPs include the 20 ‘normal’ amino acids, but in total over 500 monomers are known to be utilized, including d-amino, aryl amino, keto, hydroxyl and fatty acids (Caboche et al., 2008). Two examples of the thousands of NRPs are the well-known antibiotic penicillin (van Liempt et al., 1989) and the immunosuppressant cyclosporin A (Zocher et al., 1986), each of which completely transformed medicine in the 20th century (Tedesco and Haragsim, 2012; Zaffiri et al., 2012).
NRPSs are organized into repeating modules, with each module responsible for adding one substrate to the growing NRP, in an assembly-line manner (Konz and Marahiel, 1999). The basic architecture of a module includes a condensation (C) domain, an adenylation (A) domain and a peptide carrier protein (PCP) domain (Fig. 1A), with each domain performing specific roles in peptide synthesis (Weber and Marahiel, 2001). The first two reactions of the synthetic cycle are performed by the A domain (Fig. 1B). The A domain first selects the appropriate substrate using a well-characterized binding site (Conti et al., 1997; Stachelhaus et al., 1999), binds ATP and catalyzes adenylation to produce an activated aminoacyl adenylate. Next, the A domain catalyzes the transfer of the amino acyl moiety to the thiol group of prosthetic phosphopantetheine (PPE) arm attached to the PCP domain (Sundlov et al., 2012). The amino acyl-PCP domain then moves to the C domain, where, through peptide bond formation, it accepts and elongates the peptidyl group from the peptidyl–PCP domain of the upstream module. The elongated peptidyl–PCP then donates the peptidyl group to the downstream amino acyl-PCP through a peptidyl transferase reaction at the downstream C domain, continuing peptide elongation and freeing the PCP for another round of its synthetic cycle.
Fig. 1.

Schematic diagrams for ACV synthetase NRPS and products. (A) Schematic diagram showing the domain organization of P. chrysogenum ACV synthetase (E: epimerization domain, TE: thioesterase domain). (B) Schematic diagrams showing the two reactions performed by the A domain. (C) Ribbon diagram showing the 140° rotation of the A subdomain (labeled Asub) between the adenylation (orange) and thioesterification (yellow) conformations. A domains of PheA (PDBID:1AMU, orange) and PA1221 (PDBID:4DG9, yellow) were superimposed using the main body of the A domain (labeled A). PheA substrates are shown in gray spheres to mark the active site. (D) The chemical structures of (i) the tri-peptide product of ACV synthetase and (ii) penicillin G.
Normally, the number and order of the modules correspond to the length and sequence of amino acids in the peptide product (Fischbach and Walsh, 2006). NRPS assembly lines can consist of a single huge polypeptide of between 2 and 18 modules, with a mass of ∼220 kDa–2.2 MDa, or be split over multiple proteins which assemble through noncovalent interactions.
The synthesis cycle performed by NRPSs requires high mobility, in both domain movement and intra-domain conformational changes. The PCP domain moves to interact with the A and C domains in its own modules and the C domain of the downstream module, as well as with any optional tailoring domains that may be present in the module (Walsh et al., 2001; Samel et al., 2007; Frueh et al., 2008; Tanovic et al., 2008; Sundlov et al., 2012; Sundlov and Gulick, 2013). Intra-domain conformational changes have been observed with all core NRPS domains. The PCP domain is known to adopt different conformations depending not only on its functional state (Koglin et al., 2006), but also upon interactions with other NRPS domains (Frueh et al., 2008). The two N and C terminal halves of the C domain can move relative to each other (Samel et al., 2007; Bloudoff et al., 2013). X-ray crystallographic structures show that the A domain adopts distinct conformations for performing the adenylation and thioesterification reactions, with its C-terminal subdomain (Asub) undergoing a rotation of ∼140° between the two catalytic states (Fig. 1C) (Conti et al., 1997; May et al., 2002; Reger et al., 2008; Tanovic et al., 2008; Yonus et al., 2008; Drake et al., 2010; Mitchell et al., 2012; Sundlov et al., 2012).
Recent studies have used mechanism-based inhibitors to produce A–PCP di-domain samples locked with A and PCP in the catalytic state for thioesterification (Mitchell et al., 2012; Sundlov et al., 2012; Sundlov and Gulick, 2013). These vinyl sulfonamide adenylate analogs mimic the aminoacyl adenylates formed in the A domain. The A domain will catalyze the nucleophilic attack of the PPE arm thiol on the vinyl sulfonamide analog, but the adenine moiety is not released in the reaction. This produces a dead-end substrate analog, noncovalently bound at the A site and covalently linked to the PCP domain (Qiao et al., 2007). These compounds enabled structure determination of A–PCP di-domains which was not possible without such a stalling mechanism, providing a wealth of information about the thioesterification step of NRP synthesis. In this study, we present an alternative strategy for stalling NRPSs with the PCP domain locked to the A domain.
Our experiments are performed with the second module of δ-(l-α-aminoadipyl)-l-cysteinyl-d-valine (ACV) synthetase from Penicillium chrysogenum (van Liempt et al., 1989). ACV synthetase is a tri-modular NRPS which is responsible for synthesizing the NRP ACV (Fig. 1D(i)), the core structure of the β-lactam antibiotics, including the well-known penicillin and cephalosporin groups of antibiotics. ACV is modified by isopenicillin N synthase and transaminated by isopenicillin N N-acyltransferase to produce mature penicillin G (the compound commonly referred to as ‘penicillin’) (Fig. 1D(ii)). Penicillin acts by inhibiting peptidoglycan cell-wall synthesis in bacteria. Penicillin itself is on the World Health Organization's List of Essential Medicines (World Health Organization, 2013) and has saved millions of lives worldwide (Zaffiri et al., 2012), while dozens of other β-lactams are in common use in modern clinics.
Here, we report a strategy to constrict the mobile PCP domain of NRPSs using an engineered disulfide cross-link between the amino acyl-PCP and the A domain. For this strategy, we cloned and expressed the A–PCP di-domain from the second module of ACV synthetase of P. chrysogenum, with a cysteine mutation engineered into the A domain active site. Under cross-linking conditions, we observed a significant shift in apparent size of the protein by size exclusion chromatography, which was reversible in the presence of high-reducing agent. The cross-linked protein was able to be crystallized in several different conditions.
Materials and methods
Cloning
The A–PCP constructs were designed by aligning the sequence of the second A and PCP domains of ACV synthetase from P. chrysogenum to A and PCP domains of known structure (Conti et al., 1997; Samel et al., 2007; Tanovic et al., 2008; Yonus et al., 2008). The gene sequence was amplified by PCR from plasmid pESC-npgA-pcbAB (Siewers et al., 2009) using the following oligonucleotides: GATCACATGTATACCACGCTTCATGAGATG (ACVSA-PCP2For, PciI site underlined) and ATATGTCGACATCGTTCAGGATCAAGT (ACVSA_PCP2Rev, SalI site underlined). After amplification, the PCR product was digested with PciI and SalI restriction enzymes and ligated using T4 DNA ligase (NEB) into the vector pET28a (Novagen), which had been digested with the restriction enzymes NcoI and XhoI to create compatible sticky ends. The ligated vector was transformed into DH5α competent cells, and clones from the resulting colonies were verified by sequencing. Ligation of the compatible PciI and NcoI sticky ends caused the mutation of a single base in the desired coding sequence from a G to T, resulting in the substitution of an aspartate to a tyrosine in the protein sequence. This was corrected by standard site-directed mutagenesis methods using the following primer pair: AAGAAGGAGATATACCATGGGCAGCCTGGAGTATCTC (A-PCP2QKFor, mutated base underlined) and GAGATACTCCAGGCTGCCCATGGTATATCTCCTTCTT (A-PCP2QKRev, mutated base underlined) to generate the plasmid pA–PCP2, comprising the A and PCP domains of the second module of ACV synthetase with a C terminal hexahistidine tag. The valine to cysteine mutation at residue number 296 (pA–PCP2 numbering) was introduced by standard site-directed mutagenesis methods using the following primer pair: CGCCGTGTGGACTGCTGCGGGGAGGCGTTCAGC (A-PCP2V296CQKFor, mutated bases underlined) and GCTGAACGCCTCCCCGCAGCAGTCCACACGGCG (A-PCP2V296CQKRev, mutated bases underlined) to generate the plasmid pA–PCP2V296C.
Protein expression and purification
The pA–PCP2 or pA–PCP2V296C plasmid was transformed into BL21 (DE3) cells and plated onto kanamycin selective plates at 37°C. Single colonies were used to inoculate LB starter cultures containing kanamycin, which were grown at 37°C for 16 h and then used in a 1:100 dilution to inoculate 1 l of TB supplemented with kanamycin. Cultures were grown at 37°C until the OD600 reached 1.0 at which time the temperature was reduced to 16°C. After a further hour, cultures were induced with 0.1 mM IPTG. Cells were grown for 16 h before harvesting by centrifugation and either used immediately or stored at −80°C for later use.
Cell pellets were homogenized in 2 ml buffer A (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4, 5 mM imidazole, 1 mM PMSF and 0.5 mM β-mercaptoethanol (βME)) per gram of cells, supplemented with several crystals of DNase (BioShop) and one cOmplete EDTA-free protease inhibitor tablet (Roche). Cells were lysed by two passages through an EmulsiFlex-C3 homogenizer (Avestin) at 15 000 psi operating pressure. Cellular debris and unlysed cells were removed by centrifugation at 18 000 g for 30 min at 4°C. The supernatant was incubated with 2 ml Ni2+ charged Profinity IMAC resin (BioRad) for several hours at 4°C. After centrifugation at 500 g, the supernatant was removed and the resin was incubated with 50 ml buffer A containing 25 mM imidazole for 10 min. The sample was then centrifuged again and the supernatant was removed again. Finally, the resin was incubated for 10 min with 50 ml buffer B (as buffer A, but 250 mM imidazole) to release protein from the resin. Sample was centrifuged and the supernatant taken as partially purified protein. To increase yields, these steps were repeated iteratively with the sample that did not bind resin in the initial incubation.
The partially purified protein was loaded onto a 5 ml Q-sepharose HP column (GE Healthcare) equilibrated in buffer C (20 mM HEPES, pH 7.4, 50 mM NaCl, 0.5 mM βME). The column was then washed with 50 ml of 45% buffer D (as buffer C but 500 mM NaCl) and bound protein was eluted on a 50 ml gradient to 60% buffer D.
The eluted fractions were diluted 1:1 with buffer E (20 mM HEPES, pH 7.4, 200 mM NaCl, 0.2 mM βME) containing 2 M ammonium sulfate and loaded on a 1 ml PhenylHP column (GE Healthcare), equilibrated in buffer E containing 1 M ammonium sulfate. Bound protein was eluted on a 100 ml gradient to buffer E.
Size exclusion chromatography
Concentrated protein was applied to a Superdex-200 10/300 column (GE Healthcare) equilibrated in buffer E and eluted in the same buffer at 0.4 ml/min. Blue dextran (2 MDa), thyroglobulin (669 kDa), apoferritin (443 kDa), alcohol dehydrogenase (200 kDa) and bovine serum albumin (66 kDa) were used as molecular weight standards.
Cross-linking
A–PCP2V296C in buffer E was incubated with 12.5 mM MgCl2, 1 mM ATP and 2 mM cysteine at 37°C for 30 min. Cysteine and ATP were removed from the reaction mixture by serial dilution and concentration using a 30 kDa molecular weight cutoff Amicon Ultra concentrator (Millipore). Phosphopantetheinylation was then performed using 10 nM Sfp, 12.5 mM MgCl2 and 1 mM CoA for 1 h at 22°C, prior to subjecting the sample to size exclusion chromatography.
Crystallization
Sparse matrix screening was performed to identify crystallization conditions using the sitting drop, vapor diffusion method. Drops were set up in 96-well trays with commercially available screens using a Crystal Phoenix crystallization robot (Art Robbins Instruments) by combining 0.1 µl protein solution with 0.1 µl reservoir solution. Drops were equilibrated against 50 µl reservoir solution at 22°C.
Prior to data collection, crystals were cryo-protected by dipping the looped crystal in 4.5 M sodium formate. Diffraction images were collected using a Rigaku micromax rotating copper-anode generator fitted with varimax HF optics and a Saturn 944+ CCD (Rigaku, Tokyo, Japan) at the Centre for Structural Biology at McGill University, Montreal, Canada, and indexed with HKL2000 (Otwinowski and Minor, 1997).
Results
Rationale for engineered disulfide approach
At the time we initiated this project, the catalytic conformation adopted by the A and PCP domains for the thioesterification reaction was not known. We reasoned that restricting an A–PCP di-domain construct to a single conformation would greatly increase the chance of crystallization, which has proven correct, as illustrated by A–PCP di-domain structures locked with vinyl sulfonamide adenylate analogs (Mitchell et al., 2012; Sundlov et al., 2012; Sundlov and Gulick, 2013). We devised a different approach to lock the A and PCP domains together: engineering a disulfide cross-link between the amino acyl moiety in amino acyl-ACP and the amino acid binding site in the A domain. For proof of principle, we chose to use the second module of ACV synthetase from P. chrysogenum. Module 2 of ACV synthetase has cysteine as its substrate, meaning the native substrate could be used in combination with an A–PCP construct with a single point mutation to form the desired disulfide cross-link, making it an ideal candidate for this study.
To select a target residue for introduction of the cysteine mutation, a homology model of A–PCP2 with bound substrate was made with the server SWISS-MODEL (Biasini et al., 2014) using the co-complex crystal structure of the first A domain of gramicidin S synthetase I from Bacillus brevis with its substrate phenylalanine (Conti et al., 1997). Valine 296 was identified as the most promising residue to mutate to cysteine to allow disulfide bond formation (Fig. 2A). This mutation places the two thiols in close proximity (2.3 Å), does not cause steric disruption in the active site, and does not alter the domain specificity away from cysteine as assessed by the NRPSpredictor2 webserver (Rottig et al., 2011).
Fig. 2.
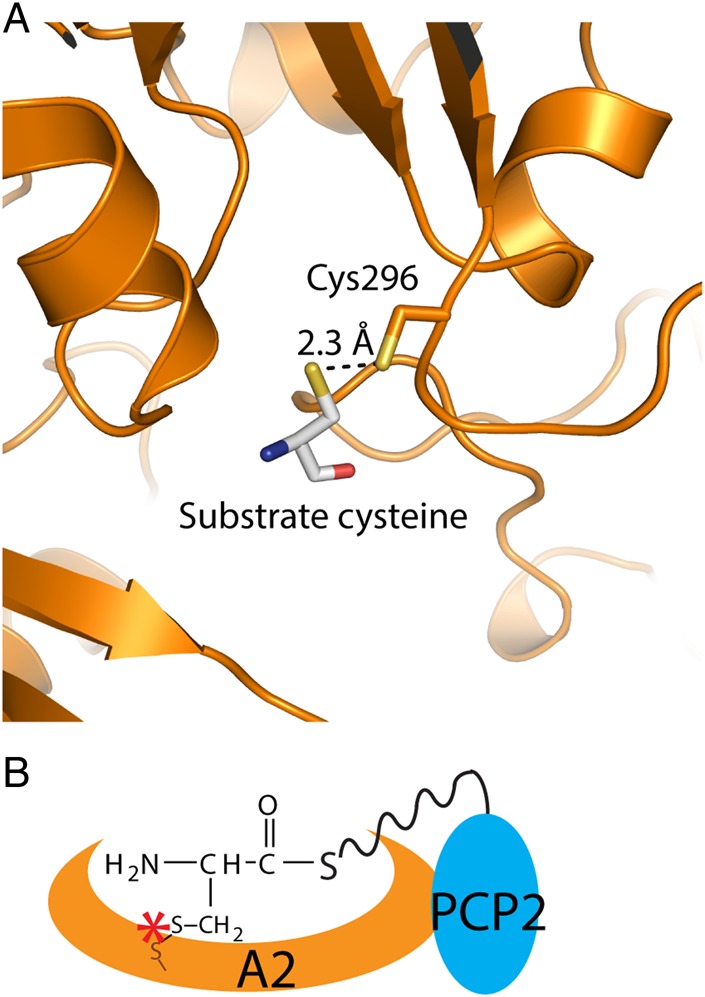
Engineered disulfide cross-linking strategy to stall the A–PCP di-domain. (A) Homology model of A–PCP2V296C showing cysteine 296 and the substrate cysteine. (B) Schematic diagram of the A–PCP2V296C trapped engineered disulfide cross-link. The dark orange sulfur atom represents the sulfur of the cysteine introduced into the amino acid binding site of the A domain by site-directed mutagenesis.
Cloning, expression and purification of A–PCP2 and A–PCP2V296C
The gene construct of A–PCP2 was designed based on domain boundaries, established using alignments to previously determined A and PCP structures to create the A–PCP ‘wild-type’ construct. Site-directed mutagenesis was used to introduce the V296C point mutation to create the PCP2V296C construct.
A–PCP2 protein was heterologously expressed in large scale using BL21 (DE3) Escherichia coli and TB media. A purification protocol of nickel affinity, anion exchange and hydrophobic interaction chromatography, and size exclusion chromatography was established to produce highly pure protein sample (Fig. 3A, inset). A–PCP2 eluted from the gel filtration column at approximately the same volume as the 232 kDa molecular weight standard (Fig. 3A). The molecular weight of A–PCP2 is 68 kDa, suggesting the protein likely forms a trimer or tetramer. The final yield of A–PCP2 was <0.5 mg protein/l growth culture due to low expression. A–PCP2V296C was expressed and purified by the same protocol with the same yield and also eluted with the same molecular weight as the wild-type protein (Fig. 3A), indicating that the mutation did not destabilize the protein.
Fig. 3.

Specific cross-linking alters the elution profile of the A–PCP2 di-domain in size exclusion chromatography. (A) Elution profiles of the wild type and A–PCP2V296C proteins show no shift in apparent molecular weight. The elution volume and weight (kDa) of protein standards are shown. Inset: SDS-PAGE of purified A–PCP2V296C, with molecular weight makers (kDa) given to the left. (B) Elution profiles of cross-linked A–PCP2V296C before and after incubation with 10 mM βME show the shift is dependent on oxidizing conditions. (C) Elution profiles of A–PCP2V296C after undergoing the cross-linking protocol with and without the PPTase Sfp show the shift is dependent on the phosphopantetheinyl arm. (D) Elution profiles of A–PCP2V296C after undergoing the cross-linking protocol with and without the cysteine substrate show the shift is dependent on cysteine. (E) The wild-type A–PCP2 protein eluted at the same volume under cross-linking and non-cross-linking conditions. A newer size exclusion column was used in panel E.
A–PCP2 cross-linking
The disulfide cross-link was formed by incubating cysteine with A–PCP2V296C in the presence of ATP, which covalently attaches the substrate and resulting adenylate to the A domain binding site. The promiscuous PPE transferase Sfp (Quadri et al., 1998) was then used to load the PPE group from coenzyme A, which allows attack of the PPE arm on the adenylate, producing cysteinyl-PCP covalently attached to the A domain amino acid binding site (Fig. 2B).
Cross-linked A–PCP2V296C was observed to shift elution volume by the size exclusion chromatography, eluting slightly after the 443 kDa molecular weight standard (Fig. 3). To demonstrate that it was the cysteine cross-linking that was responsible for this change in the protein, the purified, cross-linked sample was incubated with 10 mM βME to break the cross-link and then reapplied to the size exclusion column and eluted in buffer containing 5 mM βME. The protein now eluted with a molecular weight corresponding to that of uncross-linked protein (Fig. 3B). Furthermore, the observed shift was dependent on both cysteine and PPE, as control experiments in which either the cysteine or Sfp was omitted from the reactions did not show the shift (Fig. 3C and D), indicating that both Sfp and cysteine are essential for cross-linking to occur. No substantial difference was observed in the elution position of the wild-type protein under cross-linking and non-cross-linking conditions (Fig. 3E).
Crystallography of A–PCP2V296C
Cross-linked A–PCP2V296C was subjected to sparse matrix screening at a protein concentration of 4 mg/ml using commercially available screens JCSG+, Classics I and Classics II (Qiagen). Crystals appeared in multiple conditions after several days (Fig. 4A). The best crystal, grown with a precipitant of 3.5 M sodium formate, produced diffraction to ∼8 Å (Fig. 4B). Further optimization of growth conditions to improve diffraction was not successful. The diffraction pattern was indexed to a primitive hexagonal unit cell with cell edge lengths of a,b = 153.0 Å, c = 239.5 Å. Matthews probabilities and solvent content calculations indicate it is likely to belong to space group P6, with a 58% solvent content and four copies of A–PCP2V296C per asymmetric unit. Wild-type A–PCP2 also crystallized in some conditions, but never produced a crystal with the size or diffraction potential of this A–PCP2V296C crystal.
Fig. 4.

Crystallography of cross-linked A–PCP2V296C. (A) Crystals of A–PCP2V296C grown in 3.5 M sodium formate. The biggest crystals had dimensions of ∼100 µm × 20 µm × 20 µm and appeared after several days. (B) X-ray diffraction image of a crystal of A–PCP2V296C showing a diffraction limit of ∼8 Å.
Discussion
NRPSs are dynamic enzymes that must undergo many conformational changes during the synthesis of their small peptide products. Developing methods to stall NRPSs at specific stages in the reaction cycle is vital to understanding the precise mechanisms of the pathway. In this study, we present a novel strategy to isolate and crystallize an A–PCP di-domain in a defined conformation.
The A domain catalyzes two reactions in the NRPS cycle. Almost two decades ago, Conti et al. determined the structure of the A domain of gramicidin S synthetase I with its substrate phenylalanine and an adenosine nucleotide (Conti et al., 1997). This seminal structure helped to explain the mechanism of adenylation and led to the excellent understanding of substrate selection that allows robust substrate prediction from gene sequence (Stachelhaus et al., 1999; Challis et al., 2000; Rausch et al., 2005; Rottig et al., 2011).
Visualizing and understanding the second reaction catalyzed by the A domain was achieved only recently. This required both the A domain and the phosphopantetheinyled PCP domain, which is the acceptor substrate in the thioesterification reaction. Crystallographic trials with unmodified A–PCP di-domain did not produce the desired structures; density was visible only for the A domain and not the PCP domain, likely because of the transient nature of the interaction between the domains (Mitchell et al., 2012). However, with the use of dead-end mechanism-based inhibitor vinyl sulfonamide adenylate analogs to promote the interaction between the A and PCP domains (Fig. 5), both domains were visible and observed in a productive conformation (Mitchell et al., 2012; Sundlov et al., 2012; Sundlov and Gulick, 2013). These structures showed the dramatic 140° rotation of the Asub domain required to catalyze the thioesterification reaction (Fig. 1C) (Zettler and Mootz, 2010; Mitchell et al., 2012; Sundlov et al., 2012; Sundlov and Gulick, 2013).
Fig. 5.

Comparison of two techniques to stall A–PCP di-domains. (A) The native thioesterification reaction catalyzed by the A domain. (B) How the thioesterification reaction is stalled by vinyl sulfonamide adenylate analogs. A cysteinyl sulfonamide adenylate analog is shown in this example. (C) How the thioesterization reaction is stalled by the engineered disulfide cross-link approach. The dark orange sulfur atom represents the sulfur of the cysteine introduced into the amino acid binding site of the A domain by site-directed mutagenesis.
We have described an alternate approach to the use of vinyl sulfonamide adenylate analogs, an engineered disulfide cross-link to isolate the thioesterification complex of the PCP domain bound to the A domain (Fig. 5C). In this method, cysteine is introduced into the amino acid binding site of the A domain by point mutation and can form a disulfide bond with a thiol in the amino acid substrate. This approach avoids several disadvantages of using vinyl sulfonamide adenylates. The vinyl sulfonamide adenylates have notoriously low affinities for A domains (apparent Ki ∼ 0.1–0.3 mM), orders of magnitude lower than noncovalent intermediate analogs of the thioesterification reaction (apparent Ki ∼ 3 nM–3 μM) (Qiao et al., 2007). The kinetics of the dead-end reaction are very slow, because the electrophile in the reaction has been changed from a ketone group to an alkene group. Together, this necessitates large excesses of inhibitor and multi-day incubation times, during which the NRPS sample can become unstable and precipitate (data not shown). Also, although in the published cases the analog was sufficient to promote the interaction of the A and PCP domains (Mitchell et al., 2012; Sundlov et al., 2012), the vinyl sulfonamide adenylate is covalently attached only to the PCP domain and not to the A domain, so it is possible vinyl sulfonamide adenyl-PCP can reposition away from the A domain. Indeed, stand-alone A and vinyl sulfonamide adenyl-PCP domains have been reported not to form a stable complex (Sundlov et al., 2012). Furthermore, specific vinyl sulfonamide adenylates must be synthesized for every A domain substrate, requiring a nontrivial synthesis of at least eight total steps (Qiao et al., 2007; Mitchell et al., 2012; Sundlov et al., 2012).
With the engineered disulfide approach, there is a covalent link between the thiol-aminoacyl-PCP and the A domain to physically lock the two domains together, physically limiting the conformational flexibility. There is no alteration in the reactive atoms of the A domain-catalyzed reactions, allowing full locking within an hour. In addition, this approach is much easier to export to new NRPS systems. Instead of a lengthy and involved synthesis of novel vinyl sulfonamide adenylates with the appropriate amino acid side chain, all that is required is a single point mutation and a thiol-containing amino acid. The many published A domain structures available (Conti et al., 1997; May et al., 2002; Samel et al., 2007; Tanovic et al., 2008; Drake et al., 2010; Mitchell et al., 2012; Sundlov et al., 2012; Sundlov and Gulick, 2013) and automated modeling servers (Lambert et al., 2002; Ashworth and Baker, 2009; Biasini et al., 2014) allow facile generation of a homology model of the desired A domain to inform point mutation design, and many amino acids with thiol substitutions are commercially available. For example, available thiol-substituted analogs include, for valine: penicillamine and aminomercaptobutanoic acid; for phenylalanine: 2- 3- or 4-mercaptophenylalanine; for alanine: cysteine (SciFinder, American Chemical Society). It should also be noted that the two approaches target close, but different stages in the reaction cycle—the vinyl sulfonamide adenylates stall the reaction in an intermediate-like state, whereas the disulfide cross-link traps the product state (Fig. 5).
We used homology modeling of the A domain of domain 2 of ACV synthetase to produce an A–PCP2V296C di-domain construct that could be trapped by a disulfide, covalently linking the PCP domain to the A domain active site. We observed a significant shift in the apparent molecular weight only for A–PCP2V296C and not for wild-type A–PCP2. The shift was dependent on both cysteine and the phosphopantetheinyl arm, and was reversed by reducing agent. Shifting to a higher apparent molecular weight initially seems unexpected, as a restricted A–PCP structure might be expected to have a more compact conformation than an unrestricted structure. We propose that the apparent size increase is likely due to locking of a PCP to the A domain of a neighboring A–PCP2V296C di-domain in the oligomeric assembly, as was observed in the crystal structure of the EntBE A–PCP di-domain (Sundlov et al., 2012). Likewise, that our wild-type A–PCP2 protein was able to form crystals, albeit never to the size or diffraction potential of A–PCP2V296C, is consistent with reports of Mitchell et al. that an uninhibited A–PCP di-domain construct of protein PA1221 could form crystals, but the PCP was visible only with stalling with vinyl sulfonamide adenylates (Mitchell et al., 2012).
NRPSs are fascinating enzymes with interesting and complicated synthetic cycle and immense potential for production of novel therapeutics. A complete structural characterization is essential to understand and exploit NRPSs, and techniques to lock them in particular conformations are required to potentiate successful structural study. Our A–PCP disulfide cross-linking approach could be a very useful tool to allow successful structure determination of di-domain constructs or indeed entire multimodular NRPS megaenzymes.
Funding
This research was supported by the Canadian Institutes of Health Research (CIHR) Operating Grant No. MOP 106615 awarded to T.M.S., a Human Frontiers Science Program Organization Career Development Award to T.M.S., a Tier 2 Canada Research Chair in Macromolecular Machines held by T.M.S. and a CIHR Postdoctoral Fellowship to M.J.T.
Acknowledgements
We thank Janice Reimer for purification of Sfp, Martin Aloise for efforts with protein purification and members of the Schmeing laboratory for helpful discussion. Plasmid pESC-npg-pcbAB was a kind gift from Professor Jens Nielsen, Center for Microbial Biotechnology, Department of Systems Biology, Technical University of Denmark.
References
- Ashworth J., Baker D. (2009) Nucleic Acids Res., 37, e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini M., Bienert S., Waterhouse A., et al. (2014) Nucleic Acids Res., 42, W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloudoff K., Rodionov D., Schmeing T.M. (2013) J. Mol. Biol., 425, 3137–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caboche S., Pupin M., Leclere V., Fontaine A., Jacques P., Kucherov G. (2008) Nucleic Acids Res., 36, D326–D331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis G.L., Ravel J., Townsend C.A. (2000) Chem. Biol., 7, 211–224. [DOI] [PubMed] [Google Scholar]
- Conti E., Stachelhaus T., Marahiel M.A., Brick P. (1997) EMBO J., 16, 4174–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake E.J., Duckworth B.P., Neres J., Aldrich C.C., Gulick A.M. (2010) Biochemistry, 49, 9292–9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felnagle E.A., Jackson E.E., Chan Y.A., Podevels A.M., Berti A.D., McMahon M.D., Thomas M.G. (2008) Mol. Pharm., 5, 191–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach M.A., Walsh C.T. (2006) Chem. Rev., 106, 3468–3496. [DOI] [PubMed] [Google Scholar]
- Frueh D.P., Arthanari H., Koglin A., Vosburg D.A., Bennett A.E., Walsh C.T., Wagner G. (2008) Nature, 454, 903–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koglin A., Mofid M.R., Lohr F., et al. (2006) Science, 312, 273–276. [DOI] [PubMed] [Google Scholar]
- Konz D., Marahiel M.A. (1999) Chem. Biol., 6, R39–R48. [DOI] [PubMed] [Google Scholar]
- Lambert C., Leonard N., De Bolle X., Depiereux E. (2002) Bioinformatics, 18, 1250–1256. [DOI] [PubMed] [Google Scholar]
- May J.J., Kessler N., Marahiel M.A., Stubbs M.T. (2002) Proc. Natl Acad. Sci., 99, 12120–12125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell C.A., Shi C., Aldrich C.C., Gulick A.M. (2012) Biochemistry, 51, 3252–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z., Minor W. (1997) Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Qiao C., Wilson D.J., Bennett E.M., Aldrich C.C. (2007) J. Am. Chem. Soc., 129, 6350–6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri L.E.N., Weinreb P.H., Lei M., Nakano M.M., Zuber P., Walsh C.T. (1998) Biochemistry, 37, 1585–1595. [DOI] [PubMed] [Google Scholar]
- Rausch C., Weber T., Kohlbacher O., Wohlleben W., Huson D.H. (2005) Nucleic Acids Res., 33, 5799–5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reger A.S., Wu R., Dunaway-Mariano D., Gulick A.M. (2008) Biochemistry, 47, 8016–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottig M., Medema M.H., Blin K., Weber T., Rausch C., Kohlbacher O. (2011) Nucleic Acids Res., 39, W362–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samel S.A., Schoenafinger G., Knappe T.A., Marahiel M.A., Essen L.O. (2007) Structure, 15, 781–792. [DOI] [PubMed] [Google Scholar]
- Schwarzer D., Finking R., Marahiel M.A. (2003) Nat. Prod. Rep., 20, 275–287. [DOI] [PubMed] [Google Scholar]
- Siewers V., Chen X., Huang L., Zhang J., Nielsen J. (2009) Metab. Eng., 11, 391–397. [DOI] [PubMed] [Google Scholar]
- Stachelhaus T., Mootz H.D., Marahiel M.A. (1999) Chem. Biol., 6, 493–505. [DOI] [PubMed] [Google Scholar]
- Sundlov J.A., Gulick A.M. (2013) Acta Crystallogr. D Biol. Crystallogr., 69, 1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundlov J.A., Shi C., Wilson D.J., Aldrich C.C., Gulick A.M. (2012) Chem. Biol., 19, 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanovic A., Samel S.A., Essen L.O., Marahiel M.A. (2008) Science, 321, 659–663. [DOI] [PubMed] [Google Scholar]
- Tedesco D., Haragsim L. (2012) J. Transplant., 2012, 230386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Liempt H., von Döhren H., Kleinkauf H. (1989) J. Biol. Chem., 264, 3680–3684. [PubMed] [Google Scholar]
- Walsh C.T., Chen H., Keating T.A., Hubbard B.K., Losey H.C., Luo L., Marshall C.G., Miller D.A., Patel H.M. (2001) Curr. Opin. Chem. Biol., 5, 525–534. [DOI] [PubMed] [Google Scholar]
- Weber T., Marahiel M.A. (2001) Structure, 9, R3–R9. [DOI] [PubMed] [Google Scholar]
- World Health Organization. (2013) WHO Model List of Essential Medicines. http://apps.who.int/iris/bitstream/10665/93142/10661/EML_10618_eng.pdf.
- Yonus H., Neumann P., Zimmermann S., May J.J., Marahiel M.A., Stubbs M.T. (2008) J. Biol. Chem., 283, 32484–32491. [DOI] [PubMed] [Google Scholar]
- Zaffiri L., Gardner J., Toledo-Pereyra L. (2012) J. Invest. Surg., 25, 67–77. [DOI] [PubMed] [Google Scholar]
- Zettler J., Mootz H.D. (2010) FEBS J., 277, 1159–1171. [DOI] [PubMed] [Google Scholar]
- Zocher R., Nihira T., Paul E., Madry N., Peeters H., Kleinkauf H., Keller U. (1986) Biochemistry, 25, 550–553. [DOI] [PubMed] [Google Scholar]