

Abstract
This article reviews the zinc(II)-dependent photophysical properties of arylvinylbipyridines (AVBs), a class of fluoroionophores in which 2,2′-bipyridyl and an aryl moiety are electronically conjugated. Zinc(II) binding of an AVB may lead to an emission bathochromic shift of the fluoroionophore without diminishing its fluorescence quantum yield. This observation can be explained using the excited state model of electron donor–π bridge–electron acceptor “push–pull” fluorophores, in which the bipy moiety acts as an electron acceptor, and zinc(II)-coordination strengthens its electron affinity. The spectral sensitivity of bipy-containing fluoroionophores, such as AVBs, to zinc(II) can be exploited to prepare fluorescent indicators for this ion. In several cases, AVB moieties are incorporated in fluorescent heteroditopic ligands, so that the variation of zinc(II) concentration over a relatively large range can be correlated to fluorescence changes in either intensity or color. AVB fluoroionophores are also used to introduce an intramolecular Förster resonance energy transfer (FRET) strategy for creating zinc(II) indicators with high photostability and a narrow emission band, two desired characteristics of dyes used in fluorescence microscopy.
Keywords: Bipy, Solvent effect, Internal charge transfer, Zinc, Photoinduced electron transfer, Förster resonance energy transfer, Fluorescence microscopy
1. Brief background on 2,2′-bipyridine/metal complexes
2–2′-Bipyridine (bipy) is a bidentate metal coordination ligand for both early and late transition metal ions [1,2]. The metal/bipy complex formation solubilizes the metal ions in organic phases, which leads to methods of (a) metal extraction from aqueous media [3,4], and (b) homogenous catalysis (e.g., [Cu(bipy)2Cl] in Fig. 1, left) in organic phases [5–9]. Metal complexes of bipy and its analogs often bear intense colors (e.g., [Fe(bipy)3]SO4 in Fig. 1, middle), which has been applied in colorimetric analysis of metal ions [10,11], and as sun light-absorbing dyes (e.g., N3 dye in Fig. 1, right) in sensitized solar cells [12–14]. The colors of these complexes are attributed to metal-to-ligand-charge-transfer (MLCT, or d–π*) absorption transition [15–17], which means that in the lowest excited state of the complex, the metal center is oxidized by the bipy ligand. In order for this to happen, the bipy ligand has to have a low-lying lowest-unoccupied molecular orbital (LUMO) to receive an electron from the photo-excited metal center. By installing electron-withdrawing groups such as carboxylate on bipy further lowers its LUMO energy [18], thus enhancing the MLCT transition.
Fig. 1.
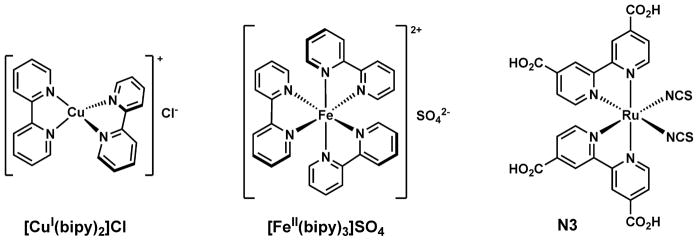
Left: a copper(I)/bipy complex [5] used as an early catalyst in atom transfer radical polymerization (ATRP); middle: an iron(II)/bipy complex that is intensely red; right: a ruthenium(II) complex [14] with a bipy derivative, which is an effective dye in dye-sensitized solar cells. Of the octahedral complexes only one stereoisomer is shown.
In addition to an oxidizable metal center such as ruthenium(II) or iron(II), an excited organic fluorophore could also offer an electron to the LUMO of bipy, when permitted by the frontier molecular orbital (FMO) energetics and molecular geometries. Similar to the strategy of facilitating MLCT transitions in ruthenium (II) dyes in sensitized solar cells, in which the LUMO of bipy is lowered by the electron-withdrawing carboxyl substituents, the electron transfer from an excited organic fluorophore to bipy can also be favored by lowering the LUMO of bipy. This can be done either via covalent bipy functionalization with electron-withdrawing groups, or, more relevant to the topic of this article, via metal coordination.
In this article, the photophysical properties of selected fluorescent compounds that incorporate bipy as the receptor for zinc(II) ions are reviewed. The utilities of certain compounds as fluorescent indicators for illuminating zinc(II) ions in living mammalian cells using fluorescence microscopy are described. The purpose of this article is to summarize a few lessons on both fundamental photophysics and fluorescence microscopy fronts that we have learned by studying bipy-containing fluorophores. Hopefully these lessons may guide the future studies of fluorescent compounds and their possible utilities as indicators for interesting and important analyte molecules. In addition to the references cited earlier, several other research and review articles are listed here for the readers to explore the broader bipy chemistry that is beyond the scope of this article [19–39]. 2,2′:6′,2″-Terpyridine (terpy) and phenanthroline (phen) have similar metal coordination properties and electron-withdrawing effects to those of bipy. Therefore, a few examples containing terpy or phen are also mentioned.
2. Zinc(II)-dependent emission of bipy-appended fluorophores—early reports
In this section, a few studies on bipy-containing fluorescent compounds that inspired our own work are described. Compound 1 (Fig. 2a), which was reported by de Silva et al. in the metal-free form exhibits anthracene fluorescence [40]. When zinc(II) coordinates the bipy moiety, the LUMO level of bipy drops to the extent that electron transfer from the excited anthryl group to bipy is feasible and rapid. This electron transfer process, sometimes called donor-excited photoinduced electron transfer (d-PeT) because the excited fluorophore is the electron donor [43], quenches the anthryl emission in the solvents that were applied in this study [40]. Sohna et al. reported bipy-containing compounds 2 [41] and 3 [42] (Fig. 2b); the pyrene emission of both compounds is similarly diminished upon zinc(II) binding.
Fig. 2.
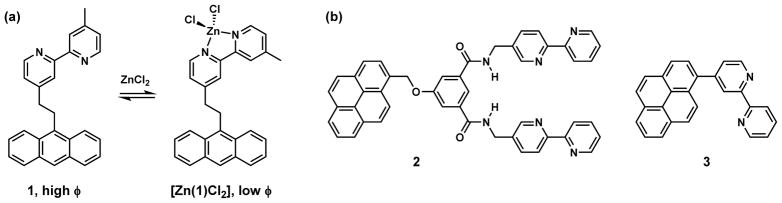
(a) Zinc(II) coordination quenches the emission of compound 1 through photoinduced electron transfer (PET) from the excited anthryl to zinc(II)-bound bipy [40]. φ: fluorescence quantum yield. (b) Similarly, zinc(II) coordination also decreases the fluorescence quantum yields of compounds 2 and 3 [41,42].
Leroy et al. also reported compounds 4 and 5 (Fig. 3), in which the bipy moieties are conjugated with pyrene fluorophore via π bridges [45]. The emission of both compounds exhibits bathochromic shifts, but undiminished emission quantum yields upon binding ZnCl2 in THF and toluene. Similar observations were made in selective organic solvents by Wasielewski, Schmehl, and their coworkers, on bipy-included conjugate polymer [46] and arylvinylterpy ligands [47], respectively. Ajayaghosh et al. reported compound 6 (Fig. 3), the fluorescence of which turns from green/ yellow to orange/red upon zinc(II) complex formation in acetonitrile [44]. Highly fluorescent analogs of 6 have since been reported to also have zinc(II)-sensitive fluorescence properties [48–52]. Similar to 4, the bipy moiety in 6 is in conjugation with N-methylpyrrole caps via vinyl bridges. The free ligand has a fluorescence quantum yield (φ) of ~0.4 in acetonitrile. The φ value of the zinc(II) complex was not reported. Nonetheless, the striking zinc(II)-effected color change of emission drew us to investigate the photophysical mechanism of the zinc(II)-dependent emission of similar compounds, and to find ways to incorporate arylvinylbipy (AVB) moiety in fluorescent indicators for zinc(II) ions that do not undergo fluorescence quenching upon zinc(II) binding.
Fig. 3.
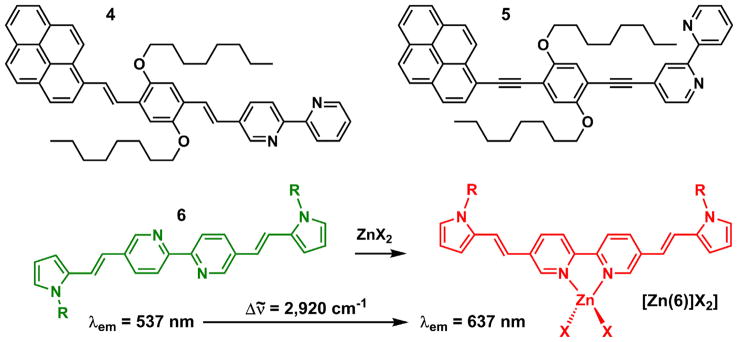
Structures of compounds 4–6, and the emission color change of 6 upon zinc(II) binding in acetonitrile [44].
3. Arylvinylbipyridines (AVBs)—solvent effect on spectral shape and frequency
A series of 5-arylvinyl-substituted bipy ligands (7–15) were prepared in our laboratory [53]. Most of these compounds have intermediate to large fluorescence quantum yields (φ > 0.2) and short lifetimes (τ <2 ns) in the vast majority of the tested organic solvents. Depending on the aryl group and the solvent, the emission color of these compounds spans from purple (e.g., 14 in hexanes) to green/yellow (e.g., 7 in acetonitrile). A positive correlation between the electron donor strength of the aryl ring and emission wavelength as well as the Stokes shift in acetonitrile is apparent (Fig. 4). This trend is expected for edonor–πbridge–eacceptor (D–π–A, interchangeable with “push–pull”) type dyes, in which enhancing either the donor (arylvinyl) or the acceptor (bipy) strength increases the charge transfer character of the excited state (as measured by the excited state dipole moment). The enlarged excited state dipole leads to substantial stabilization in a polar solvent such as acetonitrile, hence a large Stokes shift [54].
Fig. 4.
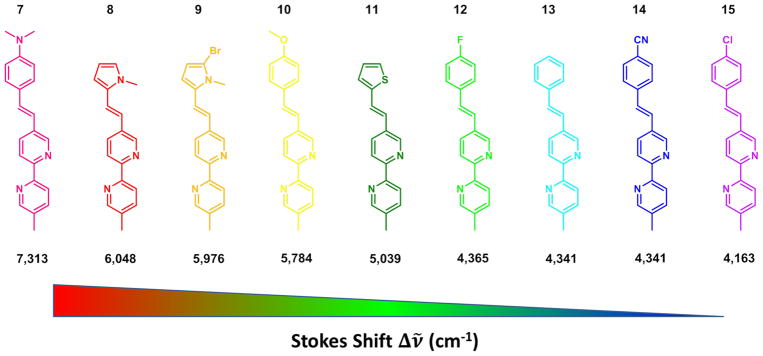
The Stokes shift (listed below the structures in cm−1) of AVB measured in acetonitrile decreases (from left to right) as the aryl group becomes more electron-withdrawing [53].
Solvent effect on emission often reveals important aspects of structure and dynamics of excited fluorophores [54–57]. The normalized emission spectra of 7 and 14 are shown in Fig. 5. Compound 7 contains an electron-rich N,N-dimethylamino substituent, which leads to a sensitive positive solvatochromic emission shift. The emission of the electron-poor cyano-bearing compound 14, on the other hand, experiences a small solvent effect. Vestiges of vibronic structure are apparent in the spectra of 14 in all five solvents, while the emission of 7 in relatively polar solvents is broad and structureless. The latter is a result of significant inhomogeneous spectral broadening, which is usually caused by the thermal disorder of solvent molecules that strongly interact with a large excited state dipole [55,58].
Fig. 5.

Normalized fluorescence spectra of 7 (upper) and 14 (lower) in different solvents. Methylcyclohexane (7)/hexanes (14): purple; benzene: blue; dioxane: cyan; chloroform: green; acetonitrile: dark green; DMF: yellow; DMSO: orange; methanol: red. The samples were excited at the maxima of their respective absorption spectra. Adapted with permission from Ref. [53]. Copyright (2009) American Chemical Society. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The emission of compound 14 may be considered as from a “locally excited” (LE) state of a small dipole moment, because it carries a vibronic structure and little solvent dependence. The structured emission of compound 7 in methylcyclohexane can also be referred as the LE emission by analogy. The structureless, low energy emission of 7 in highly polar solvents, therefore, can be regarded as from a “charge transfer” (CT) excited state. In order to explain the solvent polarity-dependent transition of LE to CT emission of 7, we shall first clarify the physical meanings of the terms “LE” and “CT” in the context of excited state solvation.
As opposed to “charge transfer” (CT), “locally excited” (LE) shall literally refer to the excited state resulting directly from the excitation of either the e-donor or the e-acceptor ground state. Therefore, “LE” carries a clear physical meaning in the model of exciplex formation, in which either donor or acceptor is excited to their respective excited states before the exciplex (i.e., the CT excited) state forms. Over time, the use of “LE” (sometimes also referred as the π–π* state) and “CT” (also known as internal charge transfer (ICT) or intraligand charge transfer (ILCT)) has been extended to describe electronically excited species under many different circumstances, including cases in which donor and acceptor are electronically conjugated in the ground state, and their physical meanings have become ambiguous [59–63]. Herein, we make an attempt to define the LE and CT emission (assuming that we will continue to use these terms in the future) of D–π–A fluorophores similar to AVB compounds in Fig. 4.
As shown in Fig. 6, a D–π–A (or “push–pull”) molecule, such as 7, is vertically excited to the Franck–Condon S1 state [D–π–A]FC, or a higher electronic state Sn, which could be a true “LE” state if the electronic perturbation is localized in the donor portion. The relaxation follows rapidly via vibrational relaxation (VR), or initially via internal conversion (IC) if from Sn (n > 1), to the ground vibrational state of the first excited state—S1(0) [64]. The VR step of a molecular fluorophore takes place regardless of solvation, which results in the Stokes shift between emission and excitation [65]. The magnitude of Stokes shift increases as the vibrational quantum number difference between ground and the Franck–Condon state (a function of nuclear displacement between the excited and ground states) grows. In the absence of strong solvation, if the ground and excited states share similar spacing of vibrational energy levels as most small organic fluorophores do, a mirror-image relationship between the S1(0) emission and excitation spectra is expected. That should be the case for compound 7 in methylcyclohexane (Fig. 5a, purple) and compound 14 in all solvents (Fig. 5b), of which the structured low-wavelength band is referred in this article as the LE emission. Hence, the so-called LE emission originates from the S1(0) state without significant solvation, regardless whether the relevant FMOs are restricted within the donor (i.e., the true “local”) portion of the molecule, or extended beyond it.
Fig. 6.
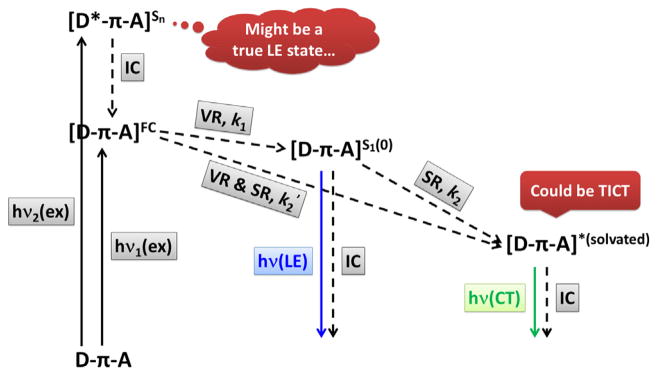
A solvation model for “edonor–πbridge–eacceptor” (D–π–A) fluorophores. ex = excitation; IC = internal conversion; VR = vibrational relaxation; SR = solvent relaxation; LE = locally excited; CT = charge transfer; TICT = twisted intramolecular charge transfer; k1, k2, and k2′ are the rates of respective processes.
When a D–π–A molecule such as 7 is excited in a polar solvent, solvent relaxation (SR, Fig. 6), which would involve the reorientation of solvent dipoles around the large excited state dipole of the solute, will occur to make things complicated. First, the emission band of the solvent-relaxed state is often broad with no vibronic structure (Fig. 5a, except purple). This “inhomogeneous spectral broadening” is the result of the thermal disorder of the reoriented polar solvent molecules around polar excited solute molecules. Often times the broad band of a D–π–A fluorophore observed in a polar solvent is referred as the charge transfer (CT) emission from a CT excited state, even if the excited state dipole moment is not necessarily much larger than the excited state formed in the low polarity solvent that displays a structured “LE” emission.
The second complication is in the kinetic profiles from the Franck–Condon to the solvated states. If SR is slower than VR (k2 < k1, Fig. 6), S1(0) could be populated. The other possibility is that SR is on the same time scale with or faster than VR (k2 ≥ k1), especially when the vibrational energy of the Franck–Condon state is so high due to the large nuclear displacement between excited and ground states, that the large amount of dissipated energy drives (i.e., accelerates) solvent reorientation. In that case, SR might bypass S1(0) so that the Franck–Condon excited state relaxes directly to a solvated state [D–π–A]*solvated with rate k2′. The solvent relaxation process of compound 10 (Fig. 4) in acetonitrile was captured by femtosecond time-resolved transient absorption spectroscopy with a time constant of 53 ps [66], which is shortened as the solvent polarity grows. Regardless how the solvated state is reached from the Franck–Condon state, as long as the excited dipole moment remains unchanged during solvation (i.e., solute polarizability can be neglected) and no specific solvent–solute interaction (e.g., hydrogen bonding) is involved, the Lippert–Mataga equation based on a thermodynamic model could predict the change of Stokes shift as a function of solvent polarity to a moderate accuracy [54,56,67–69]. The slope of this linear equation is the change of molecular dipole of the fluorophore upon photo-excitation. In the former case (k2 < k1) in which S1(0) is a local minimum on the excited state potential energy surface, dual emission from the LE and CT states might be observed (more on this later).
If a specific solvent–solute interaction (e.g., solvent forms hydrogen-bonds with the excited state dipole) influences the energy of the excited state, and/or the excited state dipole changes as the solvent relaxation proceeds, a poor fit to the Lippert–Mataga equation is to be expected [70]. This is the case for compound 7 [48], because its emission energy is at the lowest in methanol, which has the largest hydrogen bond acidity, not DMSO, which has the largest dielectric constant. In many cases, solvation might lead to an increase of the excited state dipole and even conformational changes if the lifetime of the excited state is long enough. The extreme scenario would be the formation of the “twisted intramolecular charge transfer” (TICT) state [59,71], which has been extensively studied as a model of dual emission (“LE” vs “CT”).
Based on the above discussions, we hereby define the LE and CT states of AVB fluorophores. An LE state is the vibrationally relaxed first excited state (S1(0)) without significant solvation, regardless of the magnitude of the excited state dipole moment. The LE emission band shall exhibit a vibronic structure and mirror-image relationship with its excitation spectrum. A CT state is the solvated excited state with a large dipole moment in a polar solvent, which affords a broad, structureless emission band of a lower energy than its S1(0) state. Based on this definition, an LE state could be achieved by any fluorophore, regardless of the size of its excited state dipole, in an environment lacking a strong solvent–solute interaction (i.e., either solvent or solute dipole is small, or both). A CT state requires both a large excited state dipole and a polar solvent. Compound 14 therefore exhibits LE emission in all solvents, because its excited state dipole moment is not large enough to drive significant solvent reorientation. For compound 7, an LE state is assigned to the emission in methylcyclohexane, while in more polar solvents the emission is assigned to a CT state. Many reported push–pull dyes exhibit similar “LE” →”CT” emission spectral evolution in shape and frequency as the solvent polarity increases. Examples 16–22 are shown in Fig. 7 that are grouped based on the structures of π-bridges.
Fig. 7.

Examples of D–π–A (push–pull) dyes similar to AVBs. Different colors represent different π bridges: blue—(poly) vinyl bridges in compounds 16 [54], 17 [72], and 18 [73]; yellow: fused aromatics in 20 [74] and 21 [62]; yellow and blue: both fused aromatic and vinyl in 19 [75]; red: single ring aromatics in 22 [76]. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4. Zinc(II)-dependent emission of AVB in the context of fluorescence imaging
Compound 8 carries an electron-rich N-methylpyrrole group (Fig. 8) [53]. Upon photo-excitation, an increase in molecular dipole moment is expected regardless of solvent, during which the electron density shifts from the N-methylpyrrole side to the bipy moiety. In the non-polar solvent hexanes, an emission band of the LE character was observed, while in polar solvents (e.g., acetonitrile) a broad, structureless, and solvent-sensitive CT band appeared. When zinc(II) coordinates at the bipy position, the cationic zinc(II) interacts with the negative end of the excited state dipole to strongly stabilize the excited state. Therefore, a large zinc (II)-enabled spectral shift results (4277 cm−1 in acetonitrile), which is somewhat insensitive to solvent polarity, because the strong interaction between the cationic zinc(II) and the dipolar excited state largely overrides the solvent effect.
Fig. 8.
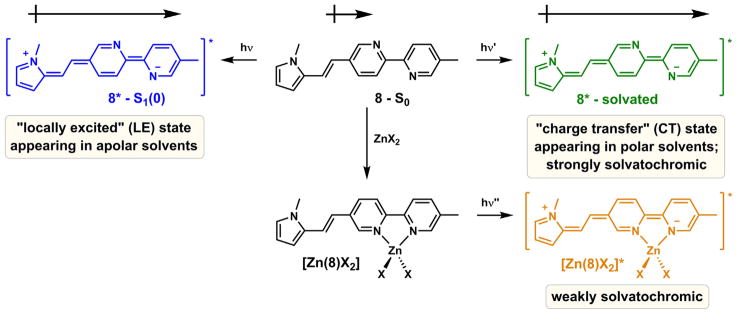
The dependency of the emission of 8 on solvent and zinc(II) binding. X: a monodentate counter ion.
Ajayaghosh et al. postulated that zinc(II) coordination planarizes the bipy-containing ligand 6 (Fig. 3), which results in a larger aromatic system with a lower HOMO–LUMO gap, and consequently leads to a bathochromic shift in emission [44]. However, considering the fact that unlike biphenyl, the transoid co-planar conformation of 2,2′-bipyridine [77] is the most stable in the ground state [78], the stabilization of the polarized excited state via metal coordination could be a more dominant factor in contributing to the emission bathochromic shift than forcing coplanarity. To gauge the relative contributions of fluorophore planarization and excited state dipole stabilization to the zinc(II) coordination-effected emission bathochromic shifts in AVBs, the emission data of compounds 11, 23, and 24 are compared (Fig. 9).
Fig. 9.
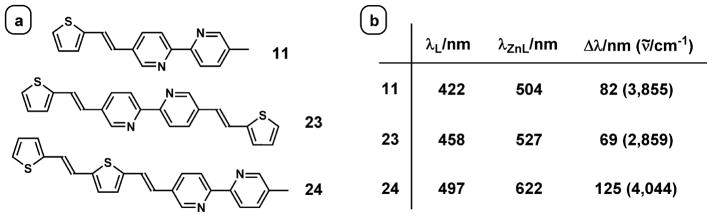
(a) The structures of compounds 11, 23, and 24, and (b) the emission maxima of the compounds and their zinc(II) complexes, and the zinc(II)-effected emission shifts.
Zinc(II) binding results in an emission shift of 3855 cm−1 for 11 in acetonitrile [53]. If fluorophore planarization is the dominant factor leading to the bathochromic shift, compound 23, which would have provided a bigger planar conjugated system upon zinc (II) complexation, should have given a larger zinc(II)-induced shift under the same conditions. To the contrary, zinc(II)-coordination of 23 affords a smaller shift of 2859 cm−1. When both thienylvinyl groups locate on the same side of bipy, as in 24, the largest shift of 4044 cm−1 upon zinc(II) binding was observed [79]. The comparison of zinc(II)-effected emission shifts of these three compounds points to the stabilization of the excited state dipoles (or the CT excited states) as the major factor in determining the magnitude of the shifts. Phen-containing fluorophores, which have rigid planar conformations to begin with, also undergo substantial emission bathochromic shifts when binding zinc(II) in organic solvents [80], attributable to the stabilization of CT excited states.
| (1) |
| (2) |
Zinc(II) complexation increases the fluorescence lifetimes (τ) of compounds 8–15 in acetonitrile (zinc(II) completely quenches the emission of 7) [48]. The fluorescence quantum yields (φ) of the zinc (II)-bound 8–15 are also larger than those of the metal-free forms, although to lesser extents comparing to the changes in τ. Similar zinc(II)-dependent effect on τ and φ of bipy-containing conjugate polymers was also observed by Chen et al. [46]. The rates of radiative (kr) and nonradiative (knr) decays were calculated from Eqs. (1) and (2). Both rates for 8–15 decrease upon zinc(II) coordination. Because zinc(II) coordination of the ligands enhances the CT character of the excited state, the diminishing orbital overlap between the excited and ground states shall reduce the rate of radiative decay (kr). This argument has been used to explain the low fluorescence quantum yields of exciplexes [81] and TICT [71] excited states, two limiting cases with complete excited state charge transfer. The rate of non-radiative decay (knr) of an AVB also decreases upon zinc(II) coordination [53]. This observation is not wholly expected because it runs counter to the energy gap law [82,83], which predicts an increase in knr when the emission frequency drops upon zinc(II) binding. Likely the restriction of the single bond rotation in the zinc(II)-bound bipy moiety reduces the probability of non-radiative decay. Therefore, the τ value increases because both kr and knr decrease upon zinc(II) binding (Eq. (1)), while the φ value of the ligand (Eq. (2)) is spared from the quenching effect of a substantially CT excited state of the zinc(II)-bound complex by a lowered knr.
Compound 10 is an example of AVBs that could be used in fluorescence imaging of intracellular zinc(II) ions [84]. Zinc(II) complex formation raises the φ of 10 in an neutral aqueous solution from 0.05 to 0.31 with an emission maximum at 513 nm. The dissociation constant of [Zn(10)]2+ was estimated at ~1 μM using a 1:1 binding model. Bipy-derived compounds selectively bind transition metal ions of intermediate hardness over alkali and alkaline earth metal ions such as sodium(I) or magnesium(II). The selectivity of 10 for transition metal ions follows the Irving–Williams order and hard/soft acids and bases (HSAB) theory, which means that bipy does not show a particular affinity to zinc(II) over other transition metal ions. Nonetheless, the bipy moiety is considered to have adequate selectivity in biological settings, in which the major interference would have come from sodium(I), potassium(I), magnesium(II), and calcium(II). Compound 10 was taken up by mammalian cells readily upon incubation at a low concentration (~2 μM). It did not exhibit any sign of cytotoxicity over the course of the experiment [85]. Therefore, the brightness, zinc(II) sensitivity, selectivity, and uptake/toxicity of 10 are all suitable as an indicator for biological zinc(II) in a matching concentration range. One major weakness of bipy as a receptor for zinc(II) is the uncertainty in binding stoichiometry in differently buffered environments. Bipy is known to form ZnL, ZnL2, and ZnL3 complexes [86], depending on counter ion and solvent. The variability of the binding stoichiometry complicates quantitative analysis of zinc(II).
Selected bipy- or bipy analog-containing, zinc(II) sensitive-fluorophores 25–33 are shown in Fig. 10. Considering bipy as an electron acceptor, the zinc(II)-dependent fluorescence bathochromic shifts of 25–27 can also be explained using the D–π–A model [95] that is applied to compound 8 (Fig. 8). Similar to that of 25, the zinc(II)-sensitive emission of 4-methoxy-2,6-dimethylphenyl-substituted bipy ligands was reported by Loren and Siegel [96]. Four bulky mesityl groups are included in compound 27, which biases toward a 1:1 binding stoichiometry with zinc(II) [89]. Although compound 28 lacks a D–A type structure, zinc(II) binding nonetheless leads to an emission red shift [90]. More experimental data is needed to establish a photophysical model of 28 to account for this observation. The zinc(II)-dependent spectral change of phen- (29) and terpy- (30–33) included fluorophores can also be explained using the D–π–A model. However, for terpy-functionalized fluorophores, fluorescence quenching usually accompanies the bathochromic shift upon zinc(II) coordination. In these cases, a terpy/zinc(II) complex acts as an excellent electron acceptor to possibly lead to the formation of fully charge separated excited states, which tend to decay predominantly via non-radiative processes.
Fig. 10.

Selected fluorescent ligands containing bipy or its analogs: 25 [87], 26 [88], 27 [89], 28 [90], 29 [80], 30 [91], 31 [92], 32 [93], and 33 [94].
5. AVBs in fluorescent heteroditopic ligands
A fluorescent heterotopic ligand has two different metal coordination sites (Fig. 11) [97]. When a metal ion encounters the ligand, it associates with the high-affinity binding site (the rectangle in Fig. 11) to afford a mononuclear complex. Further increase of the metal ion concentration shall lead to the formation of the dinuclear complex. If these three species – metal-free ligand, mononuclear, and dinuclear complexes – have distinct fluorescence properties as characterized by their emission wavelengths (λ) and fluorescence quantum yields (φ), the fluorescence changes could be correlated to the metal ion fluctuation over a large concentration range. These compounds could be particularly useful as fluorescent indicators for zinc(II), a biologically significant ion that has a broad physiological concentration range [98,99] in its strictly regulated “free” forms [100]. Moreover, studies on the interplay between ligand-centered photophysics and zinc(II) coordination chemistry in a heteroditopic architecture are challenging, but on the other hand are rewarded with new insights on the fundamental knowledge on both fronts. In this section, we describe four fluorescent heteroditopic ligands that contain the AVB components. The subsections are titled by the photophysical switches [101] that are turned either “on” or “off” by sequential binding of zinc(II). A brief tutorial on how these switches work and how they are characterized is included in a recent review from our group [102].
Fig. 11.
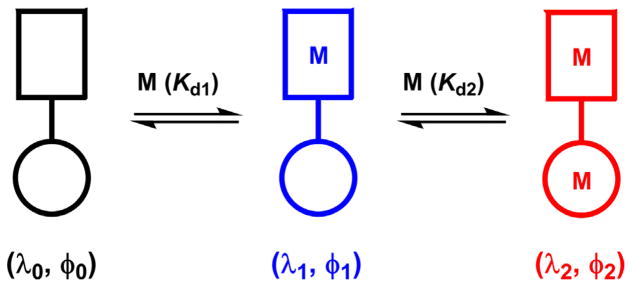
An illustration of a sequential metal coordination process of a fluorescent heteroditopic ligand. Rectangle: high-affinity binding site; circle: low-affinity binding site. Kd: dissociation constant (Kd1 < Kd2).
5.1. PET/ICT
Compound 34 contains a di(2-picolyl) amino (DPA) group and a phenylvinylbipy fluorophore (Fig. 12a) [103]. In its metal-free form in acetonitrile, 34 is weakly fluorescent because the excited state fluorophore is quenched via photoinduced electron transfer (PET) from the tertiary amino group on DPA. In this medium, DPA has a higher affinity for zinc(II) than bipy. Therefore, DPA first captures a zinc(II) ion to form the mononuclear [Zn(34)Cl2]. The coordinative bond between the tertiary amino and zinc(II) renders the PET less likely to occur, which results in the increase of fluorescence quantum yield. The fluorescence enhancement via turning off a “PET switch” is a widely applied strategy in developing turn-on fluorescent indicators [101,104–106]. The binding of the second zinc(II) ion necessarily occurs at the bipy site, which stabilizes the CT excited state to lead to a bathochromic shift of emission. Therefore, the sequential zinc(II) binding of 34 leads to two distinct segments of fluorescence changes—fluorescence enhancement (φ1/φ0) followed by emission bathochromic shift (Δλ = λ2 – λ1). The extents of these two changes are the benchmarks for evaluating the effectiveness of heteroditopic ligands of the PET/ ICT type.
Fig. 12.

(a) The sequential binding of zinc(II) of compound 34. φ1/φ0 = 18, Δλ = 60 nm in acetonitrile; (b) the dinuclear zinc(II) complex of compound 35. φ1/φ0 = 5.4, Δλ = 28 nm in an aqueous solution at neutral pH.
In order for a heteroditopic ligand to be useful as a fluorescent indicator for illuminating biological zinc(II) ions, the zinc(II) coordination-modulated fluorescence changes need to be replicated under physiological conditions. It is not the case for compound 34 because the affinities of DPA and bipy are too close at neutral pH in water for a discrete mononuclear complex to populate. To rectify this situation, the tridentate DPA is replaced by a pentadentate ligand that contains two fluorinated pyridyl groups in compound 35 (Fig. 12b) [107]. A clear-cut two-segment fluorescence change of 35 was observed in an aqueous solution buffered at neutrality. The monozinc(II) complexation leads to a 5.4-fold amplification of φ, while the secondary binding at bipy results in an emission shift of 28 nm.
The 28-nm zinc(II)-dependent emission band separation of 35 jeopardizes the potential utility of fluorescent heteroditopic ligands in fluorescence microscopy [108], while the two emission bands need to be resolved using different filter sets. It is noted that in acetonitrile, the zinc(II)-induced shift of compound 34 reaches 60 nm [103]. The dependence of zinc(II)-induced emission shift on solvent is a consequence of the differing solvatochromic effects of the metal-free and zinc(II)-bound forms. A metal-free push–pull AVB has a much larger degree of positive emission solvatochromism than its zinc(II) complex (e.g., see compound 8 in Fig. 8). Therefore, as the solvent polarity increases when going from acetonitrile to water, the free ligand emission moves to the red while that of the zinc(II) complex barely changes. Consequently, the emission gap between zinc(II)-free and zinc-bound forms is shrinking.
The small emission band separation between zinc(II)-free and zinc-bound forms in water (or highly polar solvents) of an AVB ligand is the manifestation of the inherent property of a push–pull fluoroionophore. To improve the emission shift (Δλ), which is a benchmark for evaluating fluorescent heteroditopic ligands, a molecular design that utilizes entirely different zinc(II)-modulated photophysical processes is called upon, which is represented in 36 (Fig. 13).
Fig. 13.

Sequential zinc(II) binding of compound 36, and corresponding fluorescence change. φ1/φ0 = 7.5; Δλ = 100 nm in acetonitrile.
5.2. PET/ICT/FRET
Compound 36 contains two fluorophores [109]. The higher energy fluorophore anthryl is not solvatochromic. It is connected to a PET-switching DPA unit, which quenches the anthryl emission in the metal-free form. Thienylvinylbipy is the second fluorophore that also contains the secondary (lower-affinity) binding site bipy. The photo-excitation of the anthryl group only leads to a weak emission of the metal-free 36 because the excited anthryl non-radiatively relaxes via the PET pathway. Zinc(II) acts on the PET switch in the mononuclear complex to restore the anthryl emission. The following bipy/zinc(II) complexation leads to an absorption bathochromic shift of the thienylvinylbipy fluorophore, which increases the spectral overlap with the anthryl emission. Consequently, Förster resonance energy transfer (FRET) occurs to result in the emission of the zinc(II)-bound thienylvinylbipy. Both anthryl and zinc(II)-bound thienylvinylbipy are weakly solvatochromic. Therefore, comparing to 34 and 35 that contain single D-π-A type fluorophores, the solvent effect on the emission of 36 is reduced. The two emission bands of 36 and its dizinc(II) complex, as characterized in acetonitrile [109], are 100 nm apart and are not expected to be solvent sensitive. Therefore, in principle, the two segments of emission changes could be monitored using blue and green emission filter sets.
5.3. ICT/spiro–rhod/FRET
In addition to fluorescence enhancement (φ1/φ0) followed by emission band shift (λ2 – λ1) as the two segments of changes of a heteroditopic ligand, zinc(II)-dependent sequential emission band shifts (Δλ1 = λ1 – λ0; Δλ2 = λ2 – λ1) over a significant portion of the visible spectrum would also be a useful outcome. Compound 37 (Fig. 14) includes an AVB moiety and a spirolactam precursor of rhodamine [110]. The emission color of the alkoxyphenylvinylbipy fluorophore is blue, which turns to green when zinc(II) binds. After saturating the bipy site, zinc(II) would then occupy the tridentate pocket consisting the carbonyl, pyridyl nitrogen, and triazolyl N3, which leads to the cleavage of the spiro C–N bond and the formation of the fluorescent rhodamine (hence “spiro–rhod” in the section title) [111–114]. FRET from the zinc(II)-bound AVB to rhodamine switches the emission from green to orange.
Fig. 14.

(a) Fluorescence changes of compound 37 upon sequential binding of zinc(II) in acetonitrile; (b) the change of color of a solution of 37 as the concentration of zinc(II) perchlorate increases from left to right; (c) corresponding changes in the color of emission.
5.4. ICT1/ICT2
Compound 38 consists of an iminocoumarin moiety and a bipy substituent [115]. The excitation of 38 may lead to the formation of two different CT excited states depending on the applied excitation energy. The high-energy excited state primarily involves the iminocoumarin moiety (COUM state in Fig. 15), in which the imino nitrogen acts as an electron acceptor while the methoxy is the electron donor. At a lower energy the charge transfer from the methoxy to the bipy moiety could instead become dominant (BIPY state). This hypothesis is based on the calculations on the frontier molecular orbital arrangements of 38, which is consistent with the experimental observations [115]. Either the COUM or the BIPY transitions might be favored in two populations of ground state conformers that differ in the dihedral angle between the coumarin and bipy moieties. After the fluorescence properties of compound 38 was reported [115], we acquired the single crystal structure of the analogous compound 39 (Fig. 16a), which has a dihedral angle (C1–C2–C3–C4) of 45−, close to the 40− of the most stable conformer predicted from the conformational analysis [115].
Fig. 15.
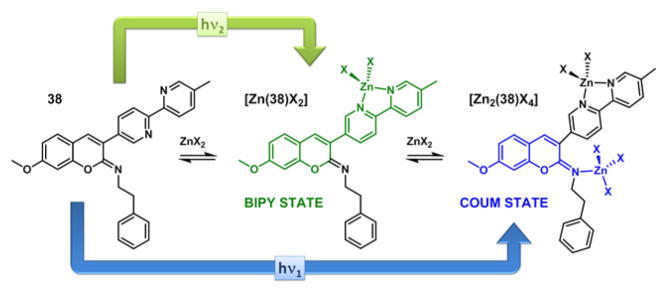
The zinc(II)-dependent dual-emitting iminocoumarin 38. X = monodentate anion.
Fig. 16.

X-ray single crystal structures of compound 39 and complex [Zn2(40)2]4+. Carbon and hydrogen: black; oxygen: red; nitrogen: blue; zinc: brown. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Both bipy and imino nitrogen in 38 are able to bind zinc(II), as supported in a crystal structure of the zinc(II) complex of 40 (Figure 16b), the N-(2-picolyl) analog of 38 [116]. Without the N-(2-picolyl) group in 40, it is assumed that the bidentate bipy has a higher affinity than the monodentate imino group, as in 38. Increasing the concentration of zinc(II) perchlorate in the acetonitrile solution of 38 first elicits an emission band in green color, followed by a hypsochromic shift to blue as zinc(II) concentration continues to rise (Fig. 15) [115]. It was concluded that the bipy moiety initially captures a zinc(II) ion to stabilize the lower energy BIPY excited state, which affords the green emission. Further addition of zinc(II) results in the imino/zinc(II) coordination, which stabilizes the higher energy, blue-emitting COUM state. This work opens the door to investigating fluorescent compounds that may exhibit dual emission from two orthogonal charge-transfer states, between which either the internal conversion is slow (a violation of Kasha’s Rule), or the ground state conformations differ.
We have reported four different types of fluorescent heteroditopic ligands thus far (34, 36–38); the emission wavelengths and quantum yields of their zinc(II)-free and bound forms are listed in Table 1. These compounds undergo sequential zinc(II) coordination, which alters the relaxation pathways of their fluorophores in different manners. Unraveling the metal-coordination modulated photophysical processes in these exquisitely designed molecules is intellectually satisfying. On the application side, the goal is to offer an indicator that is able to cover the zinc(II) concentration from nanomolar to millimolar range via fluorescence correlation using two emission bandpass filters on a fluorescence microscope. The structural framework embodied by compounds 36 and 37 are, in our opinion, mostly likely to achieve this objective. Both compounds are two-fluorophore systems, which provide large enough spectral separations to be analyzed by two bandpass filters. The challenge in molecular design is the selection of right pairs of zinc(II) ligands and fluorophores to maximize the dynamic ranges of the zinc(II)-sensitive emission in two channels within the desired (i.e., biologically relevant) concentration ranges.
Table 1.
Emission wavelength maxima and quantum yields of heteroditopic compounds 34, 36, 37, and 38, monotopic compounds 41 and 42, and their zinc(II) complexes in acetonitrile.
| Ligand | λ0 (nm) | λ1 (nm) | λ2 (nm) | φ0 | φ1 | φ2 | Ref. # |
|---|---|---|---|---|---|---|---|
| 34 | 395 | 395 | 455 | 0.03 | 0.53 | 0.63 | [103] |
| 36 | 424 | 410/428a | 514 | 0.02 | 0.15 | 0.13 | [109] |
| 37 | 431 | 528 | 587 | N.D.b | 0.04 | 0.11 | [110] |
| 38 | 432 | 523 | 454 | 0.02 | 0.27 | 0.40 | [115] |
| 41 | 504 | 504 | – | 0.32 | 0.33 | – | [125] |
| 42 | 633 | 633 | – | 0.28 | 0.29 | – | [126] |
Two discernable vibronic bands of monozinc complex.
N.D.: not determined.
6. Intramolecular FRET strategy
Zinc(II)-sensitive fluorophores are being reported in growing numbers, and have been covered by numerous recent (since 2009) review articles [102,106,117–124]. A relatively small fraction of these compounds have been applied in fluorescence microscopy to illuminate zinc(II) ions in cellular or tissue specimens. It points to the fact that in addition to high brightness and zinc(II) sensitivity, other benchmarks have to be met in order for a zinc(II)-responding fluorescent ligand to be optimally useful in fluorescence microscopic experiments.
A fluorescent indicator needs to be photostable to endure the video time-lapse experiments for acquiring the dynamic information of an analyte during a biological event. A narrow emission band is preferred to allow multicolor imaging capacity. If an indicator could be used in the ratiometric mode that reduces the readout dependence on instrumental parameters and indicator loading concentration, that would also be a plus. The intramolecular FRET strategy described in this section may improve the performance of a zinc(II) indicator on these three fronts.
6.1. green, lysosome-targeting FRET-based zinc(II) indicator
In compound 41 (Fig. 17), the FRET donor AVB component (blue) is sensitive to zinc(II) coordination [125]. However, it suffers from low photostability, due in part to its propensity for photo-isomerization. Also, the CT emission band of the bipy-containing donor alone is inhomogeneously broadened (FWHM = 3870 cm−1) as a result of strong solvation, which could make it problematic in multicolor imaging. The intramolecular FRET from the donor to the acceptor BODIPY in 41 transfers the zinc(II) sensitivity of the AVB donor to the BODIPY fluorophore, in addition to reducing the bandwidth of the donor to that of BODIPY (FWHM = 1082 cm−1). Moreover, the extent of trans-cis photoisomerization is significantly reduced, which can be attributed to a fast FRET that competes with donor isomerization in the excited state (Fig. 17b) [125]. With a morpholino group that targets lysosomes in mammalian cells, the emission of 41 is shown to correlate with the zinc(II) increase in lysosomes using confocal fluorescence microscopy (Fig. 18, a and b). Using two-color structured illumination microscopy (SIM) [127,128], it is shown that 41 localizes in the interior of lysosomes of live HeLa cells, rather than anchoring on the lysosomal membranes, which are labeled by a red fluorescent protein fused with lysosome associated membrane protein (Fig. 19).
Fig. 17.
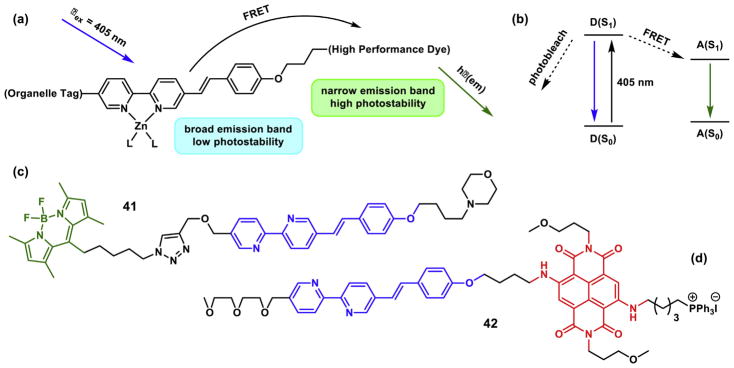
(a) The design of an intramolecularly FRETting zinc(II) indicator. The zinc(II)-bound AVB is the FRET donor. (b) Jablonski diagram to show how FRET (from D to A) protects a donor fluorophore from photobleaching. (c) Green FRET dye 41 [125]; and (d) red dye 42 [126]. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 18.

The overlay of differential interference contrast (DIC) and fluorescence images of HeLa cells that contain (a) compound 41 with no supplemental ZnCl2, (b) 41 in the presence of 50 μM of ZnCl2, (c) compound 42 with no supplemental ZnCl2, and (d) 42 in the presence of 50 μM of ZnCl2. The excitation source is a 405-nm diode laser. Emission window: 500–530 nm for panels (a) and (b); 580–680 nm for panels (c) and (d). Scale bar: 10 μm. Panels (a) and (b) are reproduced from Ref. [125] with permission from John Wiley and Sons; panels (c) and (d) are reproduced from Ref. [126] with permission from The Royal Society of Chemistry.
Fig. 19.
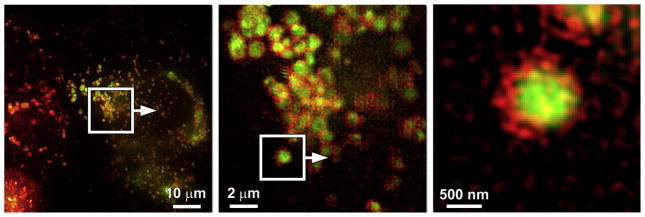
Localization of compound 41 (green) compared to that of a lysosome-associated membrane protein fused to the fluorescent protein FusionRed (red). Green: 495–550 nm; red: 570–620 nm. Reproduced from Ref. [125] with permission from John Wiley and Sons. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The rigorous characterization of the intramolecular FRET in 41 (using a methoxy analog) is carried out using femtosecond time-resolved transient absorption spectroscopy [66]. The decay time constant of FRET is 41 ps in acetonitrile. This value increases as the medium becomes more polar, which is consistent with the Förster-type resonance energy transfer that is based on excited state dipole/dipole interactions and is consequently attenuated by solvent dipoles. The solvent-relaxation of the donor CT excited state occurs on the same time scale as FRET, but can be distinguished by the opposite solvent polarity dependency [66].
6.2. red, mitochondria-targeting FRET-based zinc(II) indicator
Another example of the FRET strategy is compound 42 (Fig. 17d), in which the diamino-substituted naphthalenediimide (NDI) is the acceptor fluorophore [126]. Therefore, this compound exhibits zinc(II)-dependent red fluorescence. The triphenylphosphonium (TPP) moiety directs the compound to mitochondria of mammalian cells so that the organellar-specific zinc(II) signals could be recorded (Fig. 18, c and d). One may envision that the co-administration of 41 and 42, which have identical zinc(II)-responding fluoroionophores but FRET acceptors of different colors and localization tags for different subcellular targets, may reveal the correlated changes of zinc(II) ions in different organelles.
7. Summary and outlook
When bipy and a fluorophore are non-conjugatively connected, the bipy/zinc(II) complex often acts as an electron acceptor from an excited fluorophore, by which the emission is quenched. If bipy is conjugated with another π system, e.g., an arylvinyl moiety as described in this work, the coordination with zinc(II) often elicits a bathochromic shift of emission because in the zinc(II) complex the excited state polarizing toward the bipy end is being stabilized. The restriction of single bond rotation upon zinc(II) coordination might have contributed to the uncompromised fluorescence quantum yields of the zinc(II) complexes of these fluorophores. The sensitivity of AVB ligands to zinc(II) has been utilized to develop fluorescent indicators for this ion. However, AVBs are prone to photoisomerization, and the emission of AVBs is broad due to the strong solvent–solute interactions that a large excited state dipole enjoys. The zinc(II) sensitivity of AVBs can be preserved, their emission width be reduced, and their photostability be increased, by employing an intramolecular FRET strategy. Our group has also incorporated AVBs in heteroditopic ligand structures to afford fluorescent compounds that are responsive to zinc(II) concentration changes over broad ranges. The lessons learned from the zinc (II)-sensitive AVBs and the efforts to transform them into indicators useful in fluorescence imaging of intracellular zinc(II) ions could conceivably be applied in indicator designs targeting other analytes.
Supplementary Material
Acknowledgments
This research has been supported by the National Institutes of Health (GM081382) and the National Science Foundation (CHE0809201, CHE1213574). We are grateful to Prof. Ken Knap-penberger (FSU Chemistry and Biochemistry) on the collaboration in photophysical characterizations of the fluorescent ligands and complexes, in particular the application of femtosecond time-resolved transient absorption spectroscopy. We thank Prof. Cathy Levenson (FSU College of Medicine) for offering guidance on the biology of zinc. We are particularly indebted to Mike Davidson and his coworkers Chris Murphy, John Allen, and Michelle Baird at the National High Magnetic Field Laboratory for providing expertise on the fluorescence microscopy front of this work over the past seven years.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, athttp://dx.doi.org/10.1016/j.jphotochem.2015.05.008.
References
- 1.Constable EC. Homoleptic complexes of 2,2′-bipyridine. Adv Inorg Chem. 1989;34:1–63. [Google Scholar]
- 2.Kaes C, Katz A, Hosseini MW. Bipyridine: the most widely used ligand. A review of molecules comprising at least two 2,2′-bipyridine units. Chem Rev. 2000;100:3553–3590. doi: 10.1021/cr990376z. [DOI] [PubMed] [Google Scholar]
- 3.Kassierer EF, Kertes AS. Synergic extraction of some lanthanides and transition metals by two bidentate chelating agents. J Inorg Nucl Chem. 1972;34:3221–3231. [Google Scholar]
- 4.Kavallieratos K, Rosenberg JM, Bryan JC. Pb(II) coordination and synergistic ion-exchange extraction by combinations of sulfonamide chelates and 2,2′-bipyridine. Inorg Chem. 2005;44:2573–2575. doi: 10.1021/ic050047r. [DOI] [PubMed] [Google Scholar]
- 5.Wang JS, Matyjaszewski K. Controlled living radical polymerization. Atom transfer radical polymerization in the presence of transition-metal complexes. J Am Chem Soc. 1995;117:5614–5615. [Google Scholar]
- 6.Schneider C, Sreekanth AR, Mai E. Scandium–bipyridine-catalyzed enantioselective addition of alcohols and amines to meso-epoxides. Angew Chem Int Ed. 2004;43:5691–5694. doi: 10.1002/anie.200460786. [DOI] [PubMed] [Google Scholar]
- 7.Nicewicz DA, MacMillan DWC. Merging photoredox catalysis with organocatalysis: the direct asymmetric alkylation of aldehydes. Science. 2008;322:77–80. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bourrez M, Molton F, Chardon-Noblat S, Deronzier A. [Mn(bipyridyl) (CO)3Br]: An abundant metal carbonyl complex as efficient electrocatalyst for CO2 reduction. Angew Chem Int Ed. 2011;50:9903–9906. doi: 10.1002/anie.201103616. [DOI] [PubMed] [Google Scholar]
- 9.Hayashi Y, Santoro S, Azuma Y, Himo F, Ohshima T, Mashima K. Enzyme-like catalysis via ternary complex mechanism: alkoxy-bridged dinuclear cobalt complex mediates chemoselective O-esterification over N-amidation. J Am Chem Soc. 2013;135:6192–6199. doi: 10.1021/ja400367h. [DOI] [PubMed] [Google Scholar]
- 10.Cagle FW, Smith GF. 2,2′-Bipyridine ferrous complex ion as indicator in determination of iron. Anal Chem. 1947;19:384–385. [Google Scholar]
- 11.Stookey LL. Ferrozine—a new spectrophotometric reagent for iron. Anal Chem. 1970;42:779–781. [Google Scholar]
- 12.Kalyanasundaram K. Photophysics, photochemistry and solar energy conversion with tris(bipyridyl) ruthenium(II) and its analogues. Coord Chem Rev. 1982;46:159–244. [Google Scholar]
- 13.O’Regan B, Grätzel M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature. 1991;353:737–740. [Google Scholar]
- 14.Nazeeruddin MK, Kay A, Rodicio I, Humphry-Baker R, Muller E, Liska P, Vlachopoulos N, Grätzel M. Conversion of light to electricity by cis-X2Bis(2,2′-bipyridyl-4,4′-dicarboxylate) ruthenium(II) charge-transfer sensitizers (X = Cl−, Br−, I−, CN−, and SCN−) on nanocrystalline TiO2 electrodes. J Am Chem Soc. 1993;115:6382–6390. [Google Scholar]
- 15.Palmer RA, Piper TS. 2,2′-Bipyridine complexes. I. Polarized crystal spectra of tris(2,2′-bipyridine) copper(II), -nickel(II), -cobalt(II), -iron(II), and -ruthenium(II) Inorg Chem. 1966;5:864–878. [Google Scholar]
- 16.Crosby GA. Spectroscopic investigations of excited states of transition-metal complexes. Acc Chem Res. 1975;8:231–238. [Google Scholar]
- 17.Braterman PS, Song JI, Peacock RD. Electronic absorption spectra of the iron(II) complexes of 2,2′-bipyridine, 2,2′-bipyrimidine, 1,10-phenanthroline, and 2,2′:6′,2″-terpyridine and their reduction products. Inorg Chem. 1992;31:555–559. [Google Scholar]
- 18.Rensmo H, Lunell S, Siegbahn H. Absorption and electrochemical properties of ruthenium(II) dyes, studied by semiempirical quantum chemical calculations. J Photochem Photobiol A: Chem. 1998;114:117–124. [Google Scholar]
- 19.Rebek J, Jr, Trend JE. On binding to transition states and ground states: remote catalysis. J Am Chem Soc. 1978;100:4315–4316. [Google Scholar]
- 20.Rebek J, Jr, Trend JE, Wattley RV, Chakravorti S. Allosteric effects in organic chemistry. site-specific binding. J Am Chem Soc. 1979;101:4333–4337. [Google Scholar]
- 21.Bolm C, Zehnder M, Bur D. Optically active bipyridines in asymmetric catalysis. Angew Chem Int Ed Engl. 1990;29:205–207. [Google Scholar]
- 22.Kelly TR, Bowyer MC, Bhaskar KV, Bebbington D, Garcia A, Lang F, Kim MH, Jette MP. A molecular brake. J Am Chem Soc. 1994;116:3657–3658. [Google Scholar]
- 23.Damrauer NH, Cerullo G, Yeh A, Boussie TR, Shank CV, McCusker JK. Femtosecond dynamics of excited-state evolution in [Ru(bpy)3]2+ Science. 1997;275:54–57. doi: 10.1126/science.275.5296.54. [DOI] [PubMed] [Google Scholar]
- 24.Kalyanasundaram K, Grätzel M. Applications of functionalized transition metal complexes in photonic and optoelectronic devices. Coord Chem Rev. 1998;77:347–414. [Google Scholar]
- 25.Matyjaszewski K, Jo SM, Paik HJ, Shipp DA. An investigation into the CuX/ 2,2′-bipyridine (X) Br or Cl) mediated atom transfer radical polymerization of acrylonitrile. Macromolecules. 1999;32:6431–6438. [Google Scholar]
- 26.Newkome GR, Patri AK, Holder E, Schubert US. Synthesis of 2,2′-bipyridines: versatile building blocks for sexy architectures and functional nanomaterials. Eur J Org Chem. 2004:235–254. [Google Scholar]
- 27.Yeh AT, Shank CV, McCusker JK. Ultrafast electron localization dynamics following photo-induced charge transfer. Science. 2000;289:935–938. doi: 10.1126/science.289.5481.935. [DOI] [PubMed] [Google Scholar]
- 28.Boller TM, Murphy JM, Hapke M, Ishiyama T, Miyaura N, Hartwig JF. Mechanism of the mild functionalization of arenes by diboron reagents catalyzed by iridium complexes. Intermediacy and chemistry of bipyridine-ligated iridium trisboryl complexes. J Am Chem Soc. 2005;127:14263–14278. doi: 10.1021/ja053433g. [DOI] [PubMed] [Google Scholar]
- 29.Balzani V, Bergamini G, Marchioni F, Balzani PC, Bergamini G, Marchioni F, Ceroni P. Ru(II)-bipyridine complexes in supramolecular systems, devices and machines. Coord Chem Rev. 2006;250:1254–1266. [Google Scholar]
- 30.Castellano FN, Pomestchenko IE, Shikhova E, Hua F, Muro ML, Rajapakse N. Photophysics in bipyridyl and terpyridyl platinum(II) acetylides. Coord Chem Rev. 2006;250:1819–1828. [Google Scholar]
- 31.Hapke M, Brandt L, Lützen A. Versatile tools in the construction of substituted 2,2′-bipyridines-cross-coupling reactions with tin, zinc and boron compounds. Chem Soc Rev. 2008;37:2782–2797. doi: 10.1039/b810973g. [DOI] [PubMed] [Google Scholar]
- 32.Tian LL, Wang C, Dawn S, Smith MD, Krause JA, Shimizu LS. Macrocycles with switchable exo/endo metal binding sites. J Am Chem Soc. 2009;131:17620–17629. doi: 10.1021/ja906474z. [DOI] [PubMed] [Google Scholar]
- 33.Lancaster KM, Gerken JB, Durrell AC, Palmer JH, Gray HB. Electronic structures, photophysical properties, and electrochemistry of ruthenium(II) (bpy)2 pyridylimidazole complexes. Coord Chem Rev. 2010;254:1803–1811. [Google Scholar]
- 34.Happ B, Winter A, Hager MD, Schubert US. Photogenerated avenues in macromolecules containing Re(I), Ru(II), Os(II), and Ir(III) metal complexes of pyridine-based ligands. Chem Soc Rev. 2012;41:2222–2255. doi: 10.1039/c1cs15154a. [DOI] [PubMed] [Google Scholar]
- 35.Morimoto T, Nishiura C, Tanaka M, Rohacova J, Nakagawa Y, Funada Y, Koike K, Yamamoto Y, Shishido S, Kojima T, Saeki T, Ozeki T, Ishitani O. Ring-shaped Re(I) multinuclear complexes with unique photofunctional properties. J Am Chem Soc. 2013;135:13266–13269. doi: 10.1021/ja406144h. [DOI] [PubMed] [Google Scholar]
- 36.Li L, Zeng M, Herzon SB. Broad-spectrum catalysts for the ambient temperature anti-markovnikov hydration of alkynes. Angew Chem Int Ed. 2014;53:7892–7895. doi: 10.1002/anie.201404320. [DOI] [PubMed] [Google Scholar]
- 37.Zhang W, Alonso-Mori R, Bergmann U, Bressler C, Chollet M, Galler A, Gawelda W, Hadt RG, Hartsock RW, Kroll T, Kjær KS, Kubicek K, Lemke HT, Liang HW, Meyer DA, Nielsen MM, Purser C, Robinson JS, Solomon EI, Sun Z, Sokaras D, Driel TBV, Vankó G, Weng TC, Zhu D, Gaffney KJ. Tracking excited-state charge and spin dynamics in iron coordination complexes. Nature. 2014;509:345–348. doi: 10.1038/nature13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu J, Yan Y, Helbig BJ, Huang Z, Lian T, Schmehl RH. The influence of ligand localized excited states on the photophysics of second row and third row transition metal terpyridyl complexes: recent examples and a case study. Coord Chem Rev. 2015;282–283:100–109. [Google Scholar]
- 39.Knoll JD, Turro C. Control and utilization of ruthenium and rhodium metal complex excited states for photoactivated cancer therapy. Coord Chem Rev. 2015;282–283:110–126. doi: 10.1016/j.ccr.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Silva AP, Dixon IM, Gunaratne HQN, Gunnlaugsson T, Maxwell PRS, Rice TE. Integration of logic functions and sequential operation of gates at the molecular-scale. J Am Chem Soc. 1999;121:1393–1394. [Google Scholar]
- 41.Sohna JES, Jaumier P, Fages F. Zinc(II)-driven fluorescence quenching of a pyrene-labelled bis-2,2′-bipyridine ligand. J Chem Res. 1999:134–135. [Google Scholar]
- 42.Rodriguez AL, Peron G, Duprat C, Vailier M, Fouquet E, Fages F. The use of a monoorganotin derivative of pyrene in the palladium(0)-catalyzed synthesis of a new metal-cation complexing molecule displaying excited state charge transfer properties. Tetrahedron Lett. 1998;39:1179–1182. [Google Scholar]
- 43.Ueno T, Urano Y, Setsukinai KI, Takakusa H, Kojima H, Kikuchi K, Ohkubo K, Fukuzumi S, Nagano T. Rational principles for modulating fluorescence poperties of fluorescein. J Am Chem Soc. 2004;126:14079–14085. doi: 10.1021/ja048241k. [DOI] [PubMed] [Google Scholar]
- 44.Ajayaghosh A, Carol P, Sreejith S. A ratiometric fluorescence probe for selective visual sensing of Zn2+ J Am Chem Soc. 2005;127:14962–14963. doi: 10.1021/ja054149s. [DOI] [PubMed] [Google Scholar]
- 45.Leroy S, Soujanya T, Fages F. Zinc(II)-operated intramolecular charge transfer fluorescence emission in pyrene-2,2′-bipyride conjugated molecular rods. Tetrahedron Lett. 2001;42:1665–1667. [Google Scholar]
- 46.Chen LX, Jäger WJH, Gosztola DJ, Niemczyk MP, Wasielewski MR. Ionochromic effects and structures of metalated poly(p-phenylenevinylene) polymers incorporating 2,2′-nipyridines. J Phys Chem B. 2000;104:1950–1960. [Google Scholar]
- 47.Wang X-y, Guerzo AD, Schmehl RH. Preferential solvation of an ILCT excited state in bis(terpyridine–phenylene–vinylene) Zn(II) complexes. Chem Commun. 2002:2344–2345. doi: 10.1039/b205042k. [DOI] [PubMed] [Google Scholar]
- 48.Abbotto A, Bellotto L, Angelis FD, Manfredi N, Marinzi C. Heteroaromatic donor–acceptor–π-conjugated 2,2′-bipyridines. Eur J Org Chem. 2008:5047–5054. [Google Scholar]
- 49.Divya KP, Sreejith S, Balakrishna B, Jayamurthy P, Anees P, Ajayaghosh A. A Zn2+-specific fluorescent molecular probe for the selective detection of endogenous cyanide in biorelevant samples. Chem Commun. 2010;46:6069–6071. doi: 10.1039/c0cc01159b. [DOI] [PubMed] [Google Scholar]
- 50.Sreejith S, Divya KP, Jayamurthy P, Mathew J, Anupama VN, Philips DS, Anees P, Ajayaghosh A. Heteroaromatic donors in donor–acceptor–donor based fluorophores facilitate zinc ion sensing and cell imaging. Photochem Photobiol Sci. 2012;11:1715–1723. doi: 10.1039/c2pp25110h. [DOI] [PubMed] [Google Scholar]
- 51.Sarma M, Chatterjee T, Ghanta S, Das SK. D–π–A–A–π–D prototype 2,2′-bipyridine dyads exhibiting large structure and environment-sensitive fluorescence: synthesis, photophysics, and computation. J Org Chem. 2012;77:432–444. doi: 10.1021/jo202015m. [DOI] [PubMed] [Google Scholar]
- 52.Divya KP, Sreejith S, Ashokkumar P, Yuzhan K, Peng Q, Maji SK, Tong Y, Yu H, Zhao Y, Ramamurthy P, Ajayaghosh A. A ratiometric fluorescent molecular probe with enhanced two-photon response upon Zn2+ binding for in vitro and in vivo bioimaging. Chem Sci. 2014;5:3469–3474. [Google Scholar]
- 53.Younes AH, Zhang L, Clark RJ, Zhu L. Fluorescence of 5-arylvinyl-5′-methyl-2,2′-bipyridyl ligands and their zinc complexes. J Org Chem. 2009;74:8761–8772. doi: 10.1021/jo901889y. [DOI] [PubMed] [Google Scholar]
- 54.Lakowicz JR. Principles of Fluorescence Spectroscopy. Springer; 2006. Solvent and environmental effects; pp. 205–235. 2015. [Google Scholar]
- 55.Suppan P, Ghoneim N. Solvatochromism. The Royal Society of Chemistry; 1997. [Google Scholar]
- 56.Valeur B. Principles and Applications. WILEY-VCH; 2002. Effect of polarity on fluorescence emission. Polarity probes, Molecular Fluorescence; pp. 200–225. 2015. [Google Scholar]
- 57.Reichardt C, Welton T. Solvents and Solvent Effects in Organic Chemistry. Wiley-VCH; 2010. Solvent effects on the absorption spectra of organic compounds. 2015. [Google Scholar]
- 58.Painelli A, Terenziani F. Optical spectra of push–pull chromophores in solution: a simple model. J Phys Chem A. 2000;104:11041–11048. [Google Scholar]
- 59.Kosower EM. Intramolecular donor–acceptor systems. 9. Photophysics of (phenylamino) naphthalenesulfonates: a paradigm for excited-state intramolecular charge transfer. Acc Chem Res. 1982;15:259–266. [Google Scholar]
- 60.Kovalenko SA, Ernsting NP, Ruthmann J. Femtosecond hole-burning spectroscopy of the dye DCM in solution: the transition from the locally excited to a charge-transfer state. Chem Phys Lett. 1996;258:445–454. [Google Scholar]
- 61.Grabowski ZR, Rotkiewicz K, Rettig W. Structural changes accompanying intramolecular electron transfer: focus on twisted intramolecular charge-transfer states and structures. Chem Rev. 2003;103:3899–4031. doi: 10.1021/cr940745l. [DOI] [PubMed] [Google Scholar]
- 62.Sissa C, Calabrese V, Cavazzini M, Grisanti L, Terenziani F, Quici S, Painelli A. Tuning the nature of the fluorescent state: a substituted polycondensed dye as a case study. Chem Eur J. 2013;19:924–935. doi: 10.1002/chem.201202154. [DOI] [PubMed] [Google Scholar]
- 63.Olsen S. Locally-excited (LE) versus charge-transfer (CT) excited state competition in a series of para-substituted neutral green fluorescent protein (GFP) chromophore models. J Phys Chem B. 2015;119:2566–2575. doi: 10.1021/jp508723d. [DOI] [PubMed] [Google Scholar]
- 64.Turro NJ. Modern Molecular Photochemistry. University Science Books; Sausalito, CA: 1991. [Google Scholar]
- 65.This shift can be considered as the constant term in the Lippert–Mataga equation.
- 66.Sreenath K, Yi C, Knappenberger JKL, Zhu L. Distinguishing Förster resonance energy transfer and solvent-mediated charge-transfer relaxation dynamics in a zinc(II) indicator: a femtosecond time-resolved transient absorption spectroscopic study. Phys Chem Chem Phys. 2014;16:5088–5092. doi: 10.1039/c3cp55382e. [DOI] [PubMed] [Google Scholar]
- 67.Mataga N, Kaifu Y, Koizumi M. Solvent effects upon fluorescence spectra and the dipolemoments of excited molecules. Bull Chem Soc Jpn. 1956;29:465–470. [Google Scholar]
- 68.Koutek B. Dipole moments in excited state. Statistical investigation of methods employing solvatochromism. Collect Czech Chem Commun. 1978;43:2368–2386. [Google Scholar]
- 69.Ravi M, Samanta A, Radhakrishnan TP. Excited state dipole moments from an efficient analysis of solvatochromic stokes shift data. J Phys Chem. 1994;98:9133–9136. [Google Scholar]
- 70.Painelli A, Terenziani F. A non-perturbative approach to solvatochromic shifts of push–pull chromophores. Chem Phys Lett. 1999;312:211–220. [Google Scholar]
- 71.Rettig W. Charge separation in excited states of decoupled systems—TICT compounds and implications regarding the development of new laser dyes and the primary process of vision and photosynthesis. Angew Chem Int Ed Engl. 1986;25:971–988. [Google Scholar]
- 72.Singh TS, Mitra S, Chandra AK, Tamai N, Kar S. A combined experimental and theoretical study on photoinduced intramolecular charge transfer in trans-ethyl p-(dimethylamino) cinamate. J Photochem Photobiol A: Chem. 2008;197:295–305. [Google Scholar]
- 73.Pistolis G, Malliaris A. Effect of substituents on the spectroscopic properties of all-trans-1,6-diphenyl-1,3,5-hexatriene. Chem Phys. 1998;226:83–99. [Google Scholar]
- 74.Lobo BC, Abelt CJ. Does PRODAN Possess a planar or twisted charge-transfer excited state? Photophysical properties of two prodan derivatives. J Phys Chem A. 2003;107:10938–10943. [Google Scholar]
- 75.Katritzky AR, Zhu DW, Schanze KS. Intramolecular charge transfer properties of dicyanovinyl-substituted aromatics. J Phys Chem. 1991;95:5137–5142. [Google Scholar]
- 76.Diwu Z, Lu Y, Zhang C, Klaubert DH, Haugland RP. Fluorescent molecular probes II. The synthesis, spectral properties and use of fluorescent solvatochromic dapoxyl dyes. Photochem Photobiol. 1997;66:424–431. [Google Scholar]
- 77.Merritt LL, Schroeder E. The crystal structure of 2,2′-bipyridine. Acta Cryst. 1956;9:801–804. [Google Scholar]
- 78.Göller A, Grummt UW. Torsional barriers in biphenyl, 2,2′-bipyridine and 2-phenylpyridine. Chem Phys Lett. 2000;321:399–405. [Google Scholar]
- 79.Younes AH, Zhang L, Clark RJ, Davidson MW, Zhu L. Electronic structural dependence of the photophysical properties of fluorescent heteroditopic ligands –implications in designing molecular fluorescent indicators. Org Biomol Chem. 2010;8:5431–5441. doi: 10.1039/c0ob00482k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Joshi HS, Jamshidi R, Tor Y. Conjugated 1,10-phenanthrolines as tunable fluorophores. Angew Chem Int Ed. 1999;38:2722–2725. doi: 10.1002/(sici)1521-3773(19990917)38:18<2721::aid-anie2721>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 81.Mataga N, Murata Y. Electron donor–acceptor interactions in the fluorescent state of tetracyanobenzene-aromatic hydrocarbon complexes. J Am Chem Soc. 1969;91:3144–3152. [Google Scholar]
- 82.Englman R, Jortner J. The energy gap law for radiationless transitions in large molecules. Mol Phys. 1970;18:145–164. [Google Scholar]
- 83.Freed KF, Jortner J. Multiphonon processes in the nonradiative decay of large molecules. J Chem Phys. 1970;52:6272–6291. [Google Scholar]
- 84.Yuan AH, Younes JR, Allen MW. Enhancing the Photostability of Arylvinylbipyridyl Compounds as Fluorescent Indicators for Intracellular Zinc(II) Ions. J Org Chem. 2015 doi: 10.1021/acs.joc.5b00503. Revision under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.The bipy/zinc(II) complex has been reported to have low cytotoxicity. See: Shi Y, Toms BB, Dixit N, Kumari N, Mishra L, Goodisman J, Dabrowiak JC. Cytotoxicity of Cu(II) and Zn(II) 2,2′-Bipyridyl Complexes: Dependence of IC50 on Recovery Time. Chem Res Toxicol. 2010;23:1417–1426. doi: 10.1021/tx100126f.
- 86.Eom GH, Park HM, Hyun MY, Jang SP, Kim C, Lee JH, Lee SJ, Kim SJ, Kim Y. Anion effects on the crystal structures of ZnII complexes containing 2,2′-bipyridine: their photoluminescence and catalytic activities. Polyhedron. 2011;30:1555–1564. [Google Scholar]
- 87.Kozhevnikov DN, Shabunina OV, Kopchuk DS, Slepukhin PA, Kozhevnikov VN. 5-Aryl-2,2′-bipyridines as tunable fluorophores. Tetrahedron Lett. 2006;47:7025–7029. [Google Scholar]
- 88.James PV, Yoosaf K, Kumar J, Thomas KG, Listorti A, Accorsi G, Armaroli N. Tunable photophysical properties of phenyleneethynylene based bipyridine ligands. Photochem Photobiol Sci. 2009;8:1432–1440. doi: 10.1039/b9pp00002j. [DOI] [PubMed] [Google Scholar]
- 89.Dennis AE, Smith RC. Turn-on fluorescent sensor for the selective detection of zinc ion by a sterically-encumbered bipyridyl-based receptor. Chem Commun. 2007:4641–4643. doi: 10.1039/b710740d. [DOI] [PubMed] [Google Scholar]
- 90.Sun Y, Wang S. Extending π-conjugation of triarylborons with a 2,2′-bpy core: impact of donor–acceptor geometry on luminescence, anion sensing, and metal ion binding. Inorg Chem. 2010;49:4394–4404. doi: 10.1021/ic1004159. [DOI] [PubMed] [Google Scholar]
- 91.Peng X, Xu Y, Sun S, Wu Y, Fan J. A ratiometric fluorescent sensor for phosphates: Zn2+-enhanced ICT and ligand competition. Org Biomol Chem. 2007;5:226–228. doi: 10.1039/b614786k. [DOI] [PubMed] [Google Scholar]
- 92.Goze C, Ulrich G, Charbonniere L, Cesario M, Prange T, Ziessel R. Cation sensors based on terpyridine-functionalized boradiazaindacene. Chem Eur J. 2003;9:3748–3755. doi: 10.1002/chem.200305084. [DOI] [PubMed] [Google Scholar]
- 93.Goodall W, Williams JAG. A new, highly fluorescent terpyridine which responds to zinc ions with a large red-shift in emission. Chem Commun. 2001:2514–2515. [PubMed] [Google Scholar]
- 94.Bergamini G, Boselli L, Ceroni P, Manca P, Sanna G, Pilo M. Terthiophene appended with terpyridine units as receptors for protons and Zn2+ ions: photoinduced energy and electron transfer orocesses. Eur J Inorg Chem. 2011:4590–4595. [Google Scholar]
- 95.Compound 25 lacks a vinyl or alkynyl type π bridge.
- 96.Loren JC, Siegel JS. Synthesis and fluorescence properties of manisyl-substituted terpyridine, bipyridine, and phenanthroline. Angew Chem Int Ed. 2000;40:754–757. [PubMed] [Google Scholar]
- 97.Zhu L, Zhang L, Younes AH. Mini review: fuorescent heteroditopic ligands of metal ions. Supramol Chem. 2009;21:268–283. [Google Scholar]
- 98.Outten CE, O’Halloran TV. Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science. 2001;292:2488–2492. doi: 10.1126/science.1060331. [DOI] [PubMed] [Google Scholar]
- 99.Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci. 2005;6:449–462. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- 100.Colvin RA, Holmes WR, Fontaine CP, Maret W. Cytosolic zinc buffering and muffling: their role in intracellular zinc homeostasis. Metallomics. 2010;2:306–317. doi: 10.1039/b926662c. [DOI] [PubMed] [Google Scholar]
- 101.de Silva AP, Gunaratne HQN, Gunnlaugsson T, Huxley AJM, McCoy CP, Rademacher JT, Rice TE. Signaling recognition events with fluorescent sensors and switches. Chem Rev. 1997;97:1515–1566. doi: 10.1021/cr960386p. [DOI] [PubMed] [Google Scholar]
- 102.Zhu L, Yuan Z, Sreenath K, Simmons JT. Zn(II)-coordination modulated ligand photophysical processes—the development of fluorescent indicators for imaging biological Zn(II) ions. RSC Adv. 2014;4:20398–20440. doi: 10.1039/C4RA00354C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang L, Clark RJ, Zhu L. A heteroditopic fluoroionophoric platform for constructing fluorescent probes with large dynamic ranges for zinc ions. Chem Eur J. 2008;14:2894–2903. doi: 10.1002/chem.200701419. [DOI] [PubMed] [Google Scholar]
- 104.Valeur B, Leray I. Design principles of fluorescent molecular sensors for cation recognition. Coord Chem Rev. 2000;205:3–40. [Google Scholar]
- 105.de Silva AP, Moody TS, Wright GD. Fluorescent PET (photoinduced electron transfer) sensors as potent analytical tools. Analyst. 2009;134:2385–2393. doi: 10.1039/b912527m. [DOI] [PubMed] [Google Scholar]
- 106.Xu Z, Yoon J, Spring DR. Fluorescent chemosensors for Zn2+ Chem Soc Rev. 2010;39:1996–2006. doi: 10.1039/b916287a. [DOI] [PubMed] [Google Scholar]
- 107.Kuang GC, Allen JR, Baird MA, Nguyen BT, Zhang L, Morgan TJJ, Levenson CW, Davidson MW, Zhu L. Balance between fluorescence enhancement and association affinity in fluorescent heteroditopic indicators for imaging zinc ion in living cells. Inorg Chem. 2011;50:10493–10504. doi: 10.1021/ic201728f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Murphy DB, Davidson MW. Fundamentals of Light Microscopy and Electronic Imaging. 2. Wiley-Blackwell; Hoboken, NJ: 2013. [Google Scholar]
- 109.Wandell RJ, Younes AH, Zhu L. Metal-coordination mediated sequential chelation-enhanced fluorescence (CHEF) and fluorescence resonance energy transfer (FRET) in a heteroditopic ligand system. New J Chem. 2010;34:2176–2182. [Google Scholar]
- 110.Sreenath K, Clark RJ, Zhu L. Tricolor emission of a fluorescent heteroditopic ligand over a concentration gradient of zinc(II) ions. J Org Chem. 2012;77:8268–8279. doi: 10.1021/jo3016659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim HN, Lee MH, Kim HJ, Kim SK, Yoon J. A new trend in rhodamine-based chemosensors: application of spirolactam ring-opening to sensing ions. Chem Soc Rev. 2008;37:1465–1472. doi: 10.1039/b802497a. [DOI] [PubMed] [Google Scholar]
- 112.Quang DT, Kim JS. FLuoro- and chromogenic chemodosimeters for heavy metal ion detection in solution and biospecimens. Chem Rev. 2010;110:6280–6301. doi: 10.1021/cr100154p. [DOI] [PubMed] [Google Scholar]
- 113.Jun ME, Roy B, Ahn KH. “Turn-on” fluorescent sensing with “reactive” probes. Chem Commun. 2011;47:7583–7601. doi: 10.1039/c1cc00014d. [DOI] [PubMed] [Google Scholar]
- 114.Chen X, Pradhan T, Wang F, Kim JS, Yoon J. Fluorescent chemosensors based on spiroring-opening of xanthenes and related derivatives. Chem Rev. 2012;112:1910–1956. doi: 10.1021/cr200201z. [DOI] [PubMed] [Google Scholar]
- 115.Younes AY, Zhu L. Tunable dual fluorescence of 3-(2,2′-bipyridyl)-substituted iminocoumarin. ChemPhysChem. 2012;13:3827–3835. doi: 10.1002/cphc.201200583. [DOI] [PubMed] [Google Scholar]
- 116.This structure was also acquired after the publication of ref. 115.
- 117.McRae R, Bagchi P, Sumalekshmy S, Fahrni CJ. In situ imaging of metals in cells and tissues. Chem Rev. 2009;109:4780–4827. doi: 10.1021/cr900223a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nolan EM, Lippard SJ. Small-molecule fluorescent sensors for investigating zinc metalloneurochemistry. Acc Chem Res. 2009;42:193–203. doi: 10.1021/ar8001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tomat E, Lippard SJ. Imaging mobile zinc in biology. Curr Opin Chem Biol. 2010;14:225–230. doi: 10.1016/j.cbpa.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pluth MD, Tomat E, Lippard SJ. Biochemistry of mobile zinc and nitric oxide revealed by fluorescent sensors. Annu Rev Biochem. 2011;80:333–355. doi: 10.1146/annurev-biochem-061009-091643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Huang Z, Lippard SJ. Illuminating mobile zinc with fluorescence: from cuvettes to live cells and tissues. Methods Enzymol. 2012;505:445–468. doi: 10.1016/B978-0-12-388448-0.00031-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Formica M, Fusi V, Giorgi L, Micheloni M. New fluorescent chemosensors for metal ions in solution. Coord Chem Rev. 2012;256:170–192. [Google Scholar]
- 123.Liu Z, He W, Guo Z. Metal coordination in photoluminescent sensing. Chem Soc Rev. 2013;42:1568–1600. doi: 10.1039/c2cs35363f. [DOI] [PubMed] [Google Scholar]
- 124.Carter KP, Young AM, Palmer AE. Fluorescent sensors for measuring metal ions in living systems. Chem Rev. 2014;114:4564–4601. doi: 10.1021/cr400546e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sreenath K, Yuan Z, Allen JR, Davidson MW, Zhu L. A fluorescent indicator for imaging lysosomal zinc(II) with FRET-enhanced photostability and a narrow band of emission. Chem Eur J. 2015;21:867–874. doi: 10.1002/chem.201403479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sreenath K, Allen JR, Davidson MW, Zhu L. A FRET-based indicator for imaging mitochondrial zinc ions. Chem Commun. 2011;47:11730–11732. doi: 10.1039/c1cc14580k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gustafsson MGL. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microscopy. 2000;198:82–87. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- 128.Gustafsson MGL, Shao L, Carlton PM, Wang CJR, Golubovskaya IN, Cande WZ, Agard DA, Sedat JW. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys J. 2008;94:4957–4970. doi: 10.1529/biophysj.107.120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.