

Abstract
Background
Advanced non-small cell lung cancer (NSCLC) is an aggressive tumor that is treated with a combination of chemotherapy and radiation if the patient is not a candidate for surgery. Predictive biomarkers for response to radiotherapy are lacking in this patient population, making it a non-tailored therapy regimen with unknown outcome. Twenty to 30 % of NSCLC harbor an activating mutation in KRAS that may confer radioresistance. We hypothesized that mutant KRAS can regulate glutamine metabolism genes in NSCLC and maintain tumor redox balance through transamination reactions that generate cytosolic NADPH via malic enzyme 1 (ME1), which may contribute to radioresistance.
Findings
A doxycycline-inducible mouse model of KRASG12D driven NSCLC and patient data was analyzed from multiple publicly accessible databases including TCGA, CCLE, NCBI GEO and Project Achilles. ME1 expression was found to be mutant KRAS associated in both a NSCLC mouse model and human NSCLC cancer cell lines. Perturbing glutamine metabolism sensitized mutant KRAS, but not wild-type KRAS NSCLC cell lines to radiation treatment. NSCLC survival analysis revealed that patients with elevated ME1 and GOT1 expression had significantly worse outcomes after radiotherapy, but this was not seen after chemotherapy alone.
Conclusions
KRAS driven glutamine metabolism genes, specifically ME1 and GOT1 reactions, may be a predictive marker and potential therapeutic target for radiotherapy in NSCLC.
Electronic supplementary material
The online version of this article (doi:10.1186/s13014-015-0457-x) contains supplementary material, which is available to authorized users.
Keywords: Radioresistance, KRAS, Glutamine, NSCLC, ME1, GOT1, ROS
Background and findings
Patients with locally advanced NSCLC that are not candidates for surgery are treated with a combination of chemotherapy and radiation therapy [1]. Clinical trials assessing the efficacy of radiation therapy in this patient population have shown mixed results [2–4]. Furthermore, 20–30 % of all NSCLC harbor an activating mutation in KRAS [5]. Interestingly, several studies have demonstrated that the presence of mutant KRAS may act as a marker for radioresistance in NSCLC, yet the exact mechanism is not well understood [6–12]. Recent literature has demonstrated that mutant KRAS reprograms glutamine metabolism flux in pancreatic cancers through cytosolic aspartate aminotransferase (GOT1) and malic enzyme 1 (ME1) [13–18]. By synthesizing significant intracellular pools of NADPH via ME1, KRAS-reprogrammed pancreatic cancers rely on glutamine for redox balance in the face of reactive oxygen species (ROS) production from rapid proliferation and microenvironment stressors (Fig. 1a) [16]. In this context, NADPH is an essential co-factor to blunt ROS formation through the maintenance of intracellular reduced glutathione and thioredoxin [19]. However, to date, there are no studies evaluating whether mutant KRAS similarly reprograms glutamine metabolism genes in NSCLC for redox balance and whether this may be a potential mechanism to attenuate ionizing radiation (IR)-induced ROS and DNA damage. Therefore, we characterized glutamine metabolism genes in mutant vs wild-type KRAS NSCLC both in vitro and in vivo, demonstrated the necessity of ME1 in mutant, but not wild-type, KRAS cell lines, and demonstrated that ME1 gene expression is a predictive marker in the treatment response to radiation therapy in a cohort NSCLC patients.
Fig. 1.
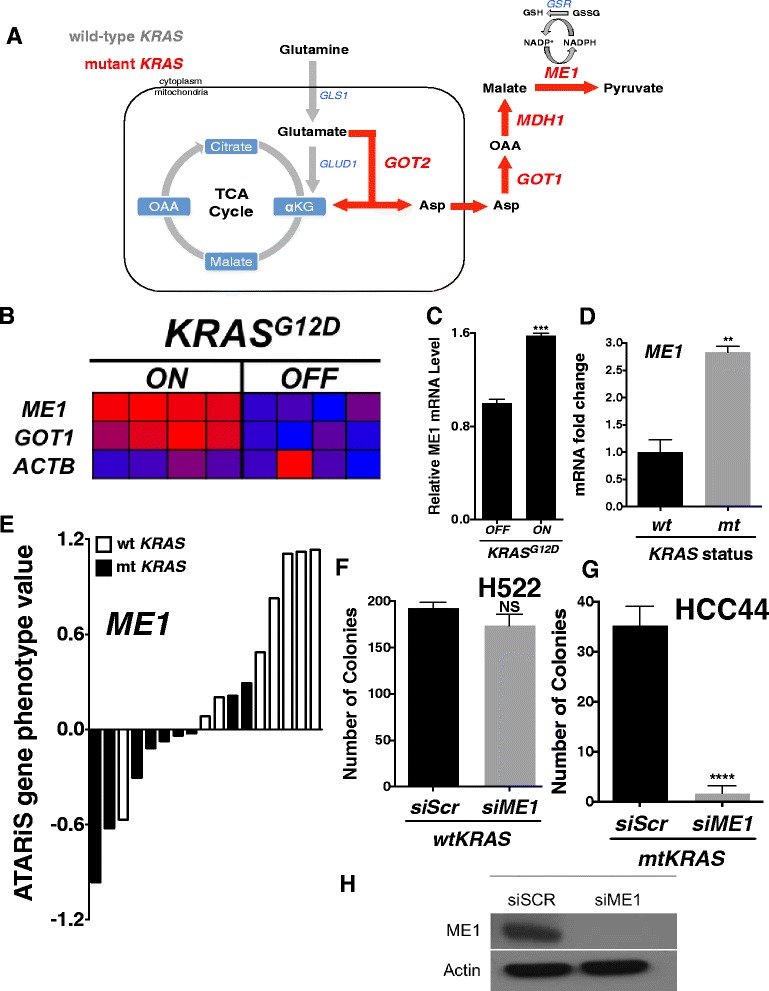
Mutant KRAS is associated with ME1 and GOT1 expression in NSCLC. a Model of mutant KRAS-reprogrammed glutamine utilization (red). GLS1 = glutaminase 1; GLUD1 = glutamate dehydrogenase 1; GOT2 = mitochondrial aspartate aminotransferase; ASP = aspartate; GOT1 = cytosolic aspartate aminotransferase; OAA = oxaloacetate; MDH1 = malate dehydrogenase 1; ME1 = malic enzyme 1; GSR = glutathione disulfide reductase. b When fed doxycycline, the mice develop lung tumors that are dependent on constitutive KRAS G12D expression [20]. Within 48 h of doxycycline withdrawal, KRAS G12D expression was extinguished and whole-genome gene expression analyses of lung tumors were performed. Consistent with mutant KRAS-driven reprogramming of glutamine metabolism, ME1 and GOT1 levels were up-regulated when KRAS G12D was induced vs 48 h extinction with doxycycline withdrawl. c KRAS G12D induction upregulated ME1 mRNA in mouse doxycycline inducible KRAS G12D embryonic fibroblasts derived from the transgenic mice. d mRNA expression of ME1 in mutant KRAS vs wild-type KRAS NSCLC cell lines. Mutant KRAS lines: A549, CALU6, NCIH1155, NCIH1373, NCIH1385, NCIH1573, NCIH2030, NCIH2122, NCIH2347, NCIH460 and NCIH647. Wild-type KRAS lines: CALU3, HCC2108, HCC2279, HCC2935, HCC4006, NCIH322, NCIH520, NCIH522, NCIH596, NCIH661 and NCIH838. e NSCLC cell line dependencies on ME1 based on ATARiS gene phenotype value assessed from Project Achilles. Black bars = mutant KRAS cell. White bars = wild-type KRAS cell. Mutant KRAS lines: A549, CALU1, CORL23, HCC44, NCIH1650, NCIH1792, NCIH2122, NCIH23 and NCIH441. Wild-type KRAS lines: HCC2814, HCC827, NCIH1299, NCIH1437, NCIH1975, NCIH661, NCIH838 and HCC827GR5. f-g Seven day clonogenic survival assay of H522 and HCC44 with RNAi knockdown of ME1. h ME1 western blot in H522; band at 64 kDa. All results were compared using Student’s t-tests as indicated. *p < 0.05; **p < 0.01; ***p < .001
Mutant KRAS is associated with ME1 and GOT1 expression in NSCLC
Gene set enrichment analysis (GSEA) of wild-type vs mutant KRAS NSCLC cell lines from the Cancer Cell Line Encyclopedia (CCLE) revealed that genes involved in glutamine dependent redox balance (ME1 and GOT1) were significantly upregulated in mutant KRAS cell lines with normalized enrichment scores (NES) >1.48 (Table 1, Additional file 1: Figure S1A).
Table 1.
GSEA results for mutant vs. wild-type KRAS NSCLC cell lines
| Name | NES | Genes | NOM p-val |
|---|---|---|---|
| NITROGEN_METABOLISM | 1.59 | GLS | .001 |
| GLUTAMATE_METABOLISM | 1.59 | GOT1, GOT2, GLS | .001 |
| CARBON_FIXATION | 1.48 | ME1, ME3, GOT1, GOT2, MDH1 | .037 |
Next, we utilized gene expression data (GSE40606) of a tetracycline operator-regulated Tet-op-KRASG12D; p53−/− transgenic mouse model of NSCLC to examine mRNA expression in the KRAS induced (“ON”) and extinguished (“OFF”) states (Fig. 1b). When fed doxycycline, the mice develop lung tumors that are dependent on constitutive KRASG12D expression [20]. Within 48 h of doxycycline withdrawal, KRASG12D expression was extinguished and whole-genome gene expression analyses of lung tumors were performed. Consistent with our cell line results, ME1 and GOT1 levels were significantly upregulated when KRASG12D (n = 4 mice) was induced vs 48 h extinction with doxycycline withdrawal (n = 4 mice) (Fig. 1b). We found that KRASG12D induction similarly upregulated ME1 and GOT1 mRNA in mouse doxycycline inducible KRASG12D embryonic fibroblasts derived from the transgenic mice (Fig. 1c, Additional file 1: Figure S1B).
Next, we measured mRNA levels of ME1 and GOT1 in 11 mutant and 11 wild-type KRAS NSCLC cell lines and found both genes to be significantly upregulated in the mutant cell lines (Fig. 1d, Additional file 1: Figure S1C). Next, to determine if mutant KRAS NSCLC cell lines relied on ME1 for survival, we analyzed 17 NSCLC cell lines from the Project Achilles database, an openly accessible platform of large-scale functional RNAi screens of cancer cell lines to identify genes that affect cell survival [21]. We found that 7 out of 9 mutant KRAS cell lines relied on ME1 for viability, while ME1 was dispensable in all but one of the wild-type cell lines (Fig. 1e). To verify these results, we knocked down ME1 (Fig. 1h) in H522, a wild-type KRAS line, and in HCC44, a mutant KRAS line. Using clonogenic survival assays, we found that ME1 loss rendered HCC44, but not H522, unable to form visible colonies (Fig. 1f, g). Taken together, our analyses indicate that mutant KRAS is associated with ME1 gene expression in NSCLC and that ME1 is an essential viability gene in mutant, but not wild-type, KRAS cell lines. In support of this observation, ME1 is a known NRF2 transcriptional target, which itself is positively regulated by mutant KRAS signaling via the MAPK pathway [22, 23].
Targeting glutamine metabolism sensitizes mutant KRAS NSCLC cell lines to radiation treatment
Mutant KRAS HCC44 and wild-type KRAS H522 cells were grown in Gln-free or Gln-containing (2 mM) media for 16 h, then exposed to ionizing radiation and allowed to form colonies for 7 days. Short-term Gln deprivation did not significantly alter clonogenic survival on its own, but did sensitize HCC44 and not H522 cells to radiation, at normally sub-lethal doses (Fig. 2a, b). Using this short term glutamine deprivation protocol, we next screened the mutant KRAS NSCLC cell lines H2009, H1573 and A549; and the wild-type KRAS NSCLC cell lines H661, H322 and H596 (Fig. 2c). Interestingly, we found that upon glutamine deprivation, mutant, but not wild-type, KRAS lines were sensitized to radiation (Fig. 2c). To pharmacologically mimic these results, we pre-treated HCC44 and H522 with the glutaminase 1 (GLS1) inhibitor, CB-839 [24], for 48 h at 1 μM followed by radiation treatment. Consistent with our glutamine deprivation results, HCC44, but not H522, was sensitized to radiation treatment (Fig. 2d).
Fig. 2.
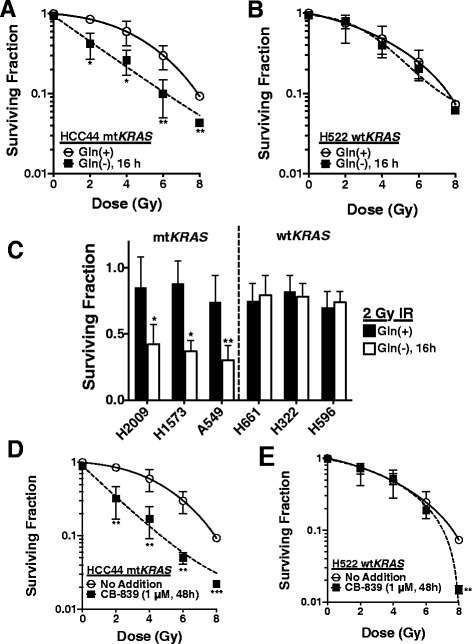
Targeting glutamine metabolism sensitizes mutant KRAS NSCLC cell lines to radiation treatment. a, b Seven day clonogenic survival of HCC44 or H522 after radiation treatment after growth in either complete media or Gln deprived media for 16 h. c Clonogenic survival screen of mutant KRAS (H2009, H1573 and A549) or wild-type KRAS (H661, H322 and H596) NSCLC cell lines grown in either complete media or Gln deprived media for 16 h followed by treatment with 2 Gy of ionizing radiation. d Clonogenic survival of HCC44 and H522 pre-treated with 1 μM CB-839 for 48 h followed by treatment with various doses of ionizing radiation. All results were compared using Student’s t-tests as indicated. *p < 0.05; **p < 0.01; ***p < .001
GOT1 and ME1 expression predicts response to radiation therapy in NSCLC patients
To expand our in vivo and in vitro findings into a clinical context, we analyzed mutant KRAS status, tumor mRNA expression and RECIST outcomes data from the TCGA in lung adenocarcinoma (LUAD) NSCLC patients who were treated with IR (patient characteristics Additional file 2: Table S1 and Additional file 3: Table S2, https://tcga-data.nci.nih.gov/tcga/tcgaCancerDetails.jsp?diseaseType=LUAD&diseaseName=Lungadenocarcinoma) [25]. Of the 14 LUAD NSCLC patients who had a complete response (CR) to IR treatment, ~93 % (13/14) of the patient’s tumors were wild-type KRAS, while only ~7 % (1/14) of the tumors were mutant KRAS, suggesting that wild-type KRAS tumors may be more radiosensitive compared to mutant KRAS tumors, consistent with previous reports (Fig. 3a) [6–12]. ME1 and GOT1 expression levels were significantly elevated in those patients who had progressive disease (PD) when treated with IR vs patients who demonstrated a CR after radiation therapy (Fig. 3b, c). Furthermore, we assessed overall survival outcomes in IR treated NSCLC patients (n = 73) grouped into high or low GOT1 and ME1 expressers. Interestingly, we found that patients with high expression of GOT1 or ME1 had significantly worse prognosis over a 140 month time period when compared to low GOT1 or ME1 expressers (Fig. 3d, e). Lastly, we did not observe a significant median survival difference between high and low GOT1/ME1 expressers in NSCLC patients who received chemotherapy, but not IR (Additional file 4: Figure S2A, B). Taken together, this suggests that ME1 and GOT1 are predictors to radiation, but not chemotherapeutic, response in NSCLC.
Fig. 3.
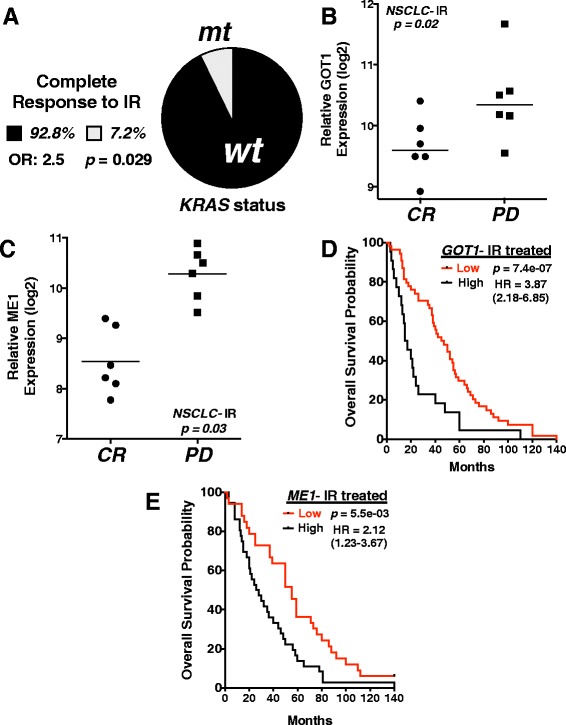
GOT1 and ME1 expression predicts response to radiation therapy in NSCLC patients. a Percent of complete responders to ionizing radiation (IR) in NSCLC patients separated based on KRAS status. Total number of complete responders in TCGA database = 14; wild-type KRAS = 13, mutant KRAS responders = 1. OR = odds ratio. Results compared using Fisher’s exact test. b, c ME1 and GOT1 log2 mRNA expression levels with calculated mean from TCGA NSCLC patients prior to radiation treatment with associated patient outcome after radiation treatment, CR = complete response, disappearance of all target lesions; PD = progressive disease, >20 % increase in the sum of the longest diameter of target lesions. Multiple probes integrated for each gene. d, e Kaplan-Meier overall survival curves in IR-treated NSCLC patients from KMPLOT database separated into high and low GOT1 and ME1 expression. Total number of NSCLC patients analyzed = 73; number of patients with high expression: ME1 = 40, GOT1 = 45; number of patients with low expression: ME1 = 33, GOT1 = 28. All results were compared using Student’s t-tests or a Cox regression analysis unless otherwise stated. *p < 0.05; **p < 0.01; ***p < .001
Conclusions
This multi-database translational study is the first to identify mutant KRAS associated glutamine metabolism genes, GOT1 and ME1, as potential radioresistance biomarkers in NSCLC. Our study revealed that elevated expression of GOT1 or ME1 is a highly predictive biomarker in radiation treatment, but not chemotherapeutic, outcomes. Additionally, ~93 % of patients with a complete response to IR treatment harbored wild-type KRAS in their tumors. To explain these observations, we hypothesize that KRAS-reprogrammed glutamine flux through GOT1 and ME1 is critical to maintain cytosolic NADPH levels for redox balance and lipid synthesis in NSCLC. In the face of ROS stress, as observed with IR treatment, NADPH is preferentially used to maintain reduced glutathione and thioredoxin 1 to protect cells from ROS damage [19]. In this context, KRAS may reprogram NSCLC glutamine metabolism similar to that observed in pancreatic cancer to maintain redox balance, thus providing an oncogene driven mechanism of radioresistance. While there are currently no known specific inhibitors of ME1 or GOT1, targeting upstream glutamine utilization via glutaminase 1 (GLS1, Fig. 1a) inhibition (with BPTES or CB-839) may blunt downstream utilization of glutamine/glutamate through GOT1 and ME1, thus depleting tumor, but not normal tissue, NADPH/GSH production, leading to tumor-specific radiosensitivity while sparing normal tissue [24].
Materials and methods
Databases
GSEA of mutant vs wild-type KRAS NSCLC cell lines was completed using the Broad Institute’s publically available Cancer Cell Line Encyclopedia (CCLE) (http://www.broadinstitute.org/ccle) [26]. Transgenic mouse data was obtained through GEO Series accession number GSE40606 at Transgenic mouse data was obtained through GEO Series accession number GSE40606. We obtained NSCLC expression, mutation, treatment and outcomes patient data from The Cancer Genome Atlas (TCGA) using the lung adenocarcinoma (LUAD) dataset (https://tcga-data.nci.nih.gov/tcga/tcgaCancerDetails.jsp?diseaseType=LUAD&diseaseName=Lung adenocarcinoma) [25]. Level 2, tumor somatic mutation data was obtained for KRAS for each patient in the analysis (Fig. 3a). Level 2, normalized gene expression data was obtained for GOT1 and ME1 for each patient in the analysis (Fig. 3b, c). Patient characteristics are shown in Additional file 2: Table S1 and Additional file 3: Table S2. Cell line gene dependency data was obtained from Broad Institute’s Project Achilles (http://www.broadinstitute.org/achilles) [21].
Kaplan-Meier statistics
Survival analysis in radiation treated NSCLC patients (n = 73) was conducted using the Kaplan-Meier Plotter webtool (kmplot.com) [27]. Briefly, kmplot segregates each gene into percentile of expression between the lower and upper quartiles and the best performing threshold is used as the final cutoff in a univariate Cox regression analysis. Kaplan-Meier survival plot and the hazard ratio with 95 % confidence intervals and logrank P value is calculated with the Bioconductor package in R.
Ethical approval and consent
All human data is sourced through The Cancer Genome Atlas (http://cancergenome.nih.gov/), no patients were approached for this study. No consent and no ethical approval were required to utilize this database.
Survival assay
For clonogenic survival assays, cells were trypsinized and plated onto 6-well plates at 100, 500, or 1000 cells per well in 2 ml of complete media, Gln deprived media for 16 h or complete media containing 1 μM CB-839 for 48 h. Cells were then exposed to IR (at various doses as indicated), allowed to grow for 7 days, washed with PBS and stained with crystal violet solution. Colonies with >50 normal appearing cells were counted and percent survival calculated and graphed with dose.
RNAi transfection
For siRNA transfection, cells were plated in 10 cm plates at 2 × 105 cells per plate and transfected with either control siRNA or siRNA against ME1 for 48 h followed by clonogenic survival assay.
Acknowledgements
The author thanks Dr. David A. Boothman for his salary support within the Simmons Comprehensive Cancer Center, which was funded by NIH/NCI grant 5P30CA142543.
Additional files
(A) Raw GSEA data of mutant vs wild-type KRAS NSCLC cell lines. Red = overexpressed across all cell lines; blue = under expressed across all cell lines. Absolute top row indicates specific cell lines used in analysis. Gray = mutant KRAS; yellow = wild-type KRAS. (B) KRAS G12D induction upregulated GOT1 mRNA in mouse doxycycline inducible KRAS G12D embryonic fibroblasts derived from the transgenic mice. (C) mRNA expression of GOT1 in mutant KRAS vs wild-type KRAS NSCLC cell lines. Same cell lines as in Fig. 1d.
TCGA lung adenocarcinoma patient description.
GOT1 and ME1 expression in TCGA lung adenocarcinoma patient tumors with treatment response after IR.
(A, B) Kaplan-Meier overall survival curves in chemotherapy treated NSCLC patients from TCGA database separated into high and low GOT1 and ME1 expression. Total number of chemotherapy treated NSCLC patients analyzed = 176; number of patients with high expression: ME1 = 69, GOT1 = 127; number of patients with low expression: ME1 = 107, GOT1 = 49. Logrank p-values not significant.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GC conceived and designed the study, performed the statistical analysis, carried out the: database analyses, experimental cell culture work, programming survival analysis script in R and drafted the manuscript. All authors read and approved the final manuscript.
References
- 1.Bi N, Wang L. Superiority of Concomitant Chemoradiation Over Sequential Chemoradiation in Inoperable, Locally Advanced Non-Small Cell Lung Cancer: Challenges in the Selection of Appropriate Chemotherapy. Semin Radiat Oncol. 2015;25(2):122–132. doi: 10.1016/j.semradonc.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Rowell NP, Williams CJ. Radical radiotherapy for stage I/II non-small cell lung cancer in patients not sufficiently fit for or declining surgery (medically inoperable) Cochrane Database Syst Rev. 2001;1:CD002935. doi: 10.1002/14651858.CD002935. [DOI] [PubMed] [Google Scholar]
- 3.Warram J. Preoperative irradiation of cancer of the lung: final report of a therapeutic trial. A collaborative study. Cancer. 1975;36(3):914–925. doi: 10.1002/1097-0142(197509)36:3<914::AID-CNCR2820360312>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 4.Dilling TJ. Radiation dose in non-small cell lung cancer: too much of a good thing? Int J Radiat Oncol Biol Phys. 2014;90(5):979–982. doi: 10.1016/j.ijrobp.2014.08.333. [DOI] [PubMed] [Google Scholar]
- 5.Westcott PM, Halliwill KD, To MD, Rashid M, Rust AG, Keane TM, Delrosario R, Jen KY, Gurley KE, Kemp CJ, et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature. 2015;517(7535):489–492. doi: 10.1038/nature13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mak RH, Hermann G, Lewis JH, Aerts HJ, Baldini EH, Chen AB, Colson YL, Hacker FH, Kozono D, Wee JO, et al. Outcomes by tumor histology and KRAS mutation status after lung stereotactic body radiation therapy for early-stage non-small-cell lung cancer. Clin Lung Cancer. 2015;16(1):24–32. doi: 10.1016/j.cllc.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernhard EJ, Stanbridge EJ, Gupta S, Gupta AK, Soto D, Bakanauskas VJ, Cerniglia GJ, Muschel RJ, McKenna WG. Direct evidence for the contribution of activated N-ras and K-ras oncogenes to increased intrinsic radiation resistance in human tumor cell lines. Cancer Res. 2000;60(23):6597–6600. [PubMed] [Google Scholar]
- 8.Affolter A, Drigotas M, Fruth K, Schmidtmann I, Brochhausen C, Mann WJ, Brieger J. Increased radioresistance via G12S K-Ras by compensatory upregulation of MAPK and PI3K pathways in epithelial cancer. Head Neck. 2013;35(2):220–228. doi: 10.1002/hed.22954. [DOI] [PubMed] [Google Scholar]
- 9.Minjgee M, Toulany M, Kehlbach R, Giehl K, Rodemann HP. K-RAS(V12) induces autocrine production of EGFR ligands and mediates radioresistance through EGFR-dependent Akt signaling and activation of DNA-PKcs. Int J Radiat Oncol Biol Phys. 2011;81(5):1506–1514. doi: 10.1016/j.ijrobp.2011.05.057. [DOI] [PubMed] [Google Scholar]
- 10.Xu D, Allsop SA, Witherspoon SM, Snider JL, Yeh JJ, Fiordalisi JJ, White CD, Williams D, Cox AD, Baines AT. The oncogenic kinase Pim-1 is modulated by K-Ras signaling and mediates transformed growth and radioresistance in human pancreatic ductal adenocarcinoma cells. Carcinogenesis. 2011;32(4):488–495. doi: 10.1093/carcin/bgr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toulany M, Dittmann K, Kruger M, Baumann M, Rodemann HP. Radioresistance of K-Ras mutated human tumor cells is mediated through EGFR-dependent activation of PI3K-AKT pathway. Radiother Oncol: J of the European Soc for Therapeutic Radiology and Oncology. 2005;76(2):143–150. doi: 10.1016/j.radonc.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 12.Caron RW, Yacoub A, Zhu X, Mitchell C, Han SI, Sasazuki T, Shirasawa S, Hagan MP, Grant S, Dent P. H-RAS V12-induced radioresistance in HCT116 colon carcinoma cells is heregulin dependent. Mol Cancer Ther. 2005;4(2):243–255. [PubMed] [Google Scholar]
- 13.Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar-Sagi D, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015;75(3):544–553. doi: 10.1158/0008-5472.CAN-14-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brunelli L, Caiola E, Marabese M, Broggini M, Pastorelli R. Capturing the metabolomic diversity of KRAS mutants in non-small-cell lung cancer cells. Oncotarget. 2014;5(13):4722–4731. doi: 10.18632/oncotarget.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saqcena M, Mukhopadhyay S, Hosny C, Alhamed A, Chatterjee A, Foster DA. Blocking anaplerotic entry of glutamine into the TCA cycle sensitizes K-Ras mutant cancer cells to cytotoxic drugs. Oncogene. 2014;34(20):2672–80. doi: 10.1038/onc.2014.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kong B, Qia C, Erkan M, Kleeff J, Michalski CW. Overview on how oncogenic Kras promotes pancreatic carcinogenesis by inducing low intracellular ROS levels. Front Physiol. 2013;4:246. doi: 10.3389/fphys.2013.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyssiotis CA, Son J, Cantley LC, Kimmelman AC. Pancreatic cancers rely on a novel glutamine metabolism pathway to maintain redox balance. Cell Cycle. 2013;12(13):1987–1988. doi: 10.4161/cc.25307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakrabarti G, Gerber DE, Boothman DA. Expanding antitumor therapeutic windows by targeting cancer-specific nicotinamide adenine dinucleotide phosphate-biogenesis pathways. Clin Pharmacol: Advances and Applications. 2015;7:57–68. doi: 10.2147/CPAA.S79760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ, Whitsett JA, Koretsky A, Varmus HE. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15(24):3249–3262. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, Hinkle G, Boehm JS, Beroukhim R, Weir BA, et al. Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci U S A. 2008;105(51):20380–20385. doi: 10.1073/pnas.0810485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, Motohashi H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22(1):66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 23.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. 2014;13(4):890–901. doi: 10.1158/1535-7163.MCT-13-0870. [DOI] [PubMed] [Google Scholar]
- 25.Cancer Genome Atlas Research N Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013;8(12):e82241. doi: 10.1371/journal.pone.0082241. [DOI] [PMC free article] [PubMed] [Google Scholar]