

Abstract
Autophagy is a multistep process that involves the degradation and digestion of intracellular components by the lysosome. It has been proved that many core autophagy-related molecules participate in this event. However, new component proteins that regulate autophagy are still being discovered. At present, we report PHF23 (PHD finger protein 23) with a PHD-like zinc finger domain that can negatively regulate autophagy. Data from experiments indicated that the overexpression of PHF23 impaired autophagy, as characterized by decreased levels of LC3B-II and weakened degradation of endogenous and exogenous autophagic substrates. Conversely, knockdown of PHF23 resulted in opposite effects. Molecular mechanism studies suggested that PHF23 interacts with LRSAM1, which is an E3 ligase key for ubiquitin-dependent autophagy against invading bacteria. PHF23 promotes the ubiquitination and proteasome degradation of LRSAM1. We also show that the PHD finger of PHF23 is a functional domain needed for the interaction with LRSAM1. Altogether, our results indicate that PHF23 is a negative regulator associated in autophagy via the LRSAM1 signaling pathway. The physical and functional connection between the PHF23 and LRSAM1 needs further investigation.
Keywords: autophagy, PHD domain, PHF23, LRSAM1, ubiquitination
Abbreviations: Three-MA, 3-methyladenine; AML, acute myeloid leukemia; ATG, autophagy-related; BafA1, bafilomycin A1; CALCOCO2, calcium binding and coiled-coil domain 2; CQ, chloroquine; EBSS, Earle's balanced salt solution; FBS, fetal bovine serum; GFP, green fluorescent protein; GST, glutathione S-transferase; IP, immunoprecipitation; LRSAM1, leucine rich repeat and sterile α motif containing 1; MAP1LC3B/LC3B; microtubule-associated protein 1 light chain 3 β; PHD, plant homeodomain; PHF23, PHD finger protein 23; PIK3C3, phosphatidylinositol 3-kinase, catalytic subunit type 3; SQSTM1, sequestosome 1
Introduction
Autophagy is essential for maintaining homeostatic processes, such as organelle and protein turnover, but it is also crucial in responses to stress conditions such as nutrient deprivation, oxidative stress, pathogen infection, and hypoxia. Dysregulation of autophagy has been linked with many diseases, including cancer, chronic infection myopathies, heart disease, and neurodegeneration.1,2
The autophagic process is formed by the double-membrane vesicle, the autophagosome, which surrounds the damaged organelles and cytoplasmic components. The autophagosome finally fuses with the lysosome so that the contents are degraded by lysosomal enzymes.2 Generally, autophagy has a basal level in most cells, to regulate the long-lived protein turnover and damaged cellular structures. While in the condition of stress such as starvation, autophagy will be excited to a higher level.1 There are 2 highly conserved ubiquitin-like modification reactions in the process of autophagosome formation, i.e., the ATG12–ATG5 conjugation system and the MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 β) modification process. Both systems are crucial for expansion of the phagophore membrane and closure to form autophagosomes. The ubiquitin-like ATG12 is activated by the E1 enzyme ATG7, transferred to the E2 enzyme ATG10 and conjugated to ATG5 to form a high-molecular-weight complex with ATG16L1. The LC3B C terminus is removed by ATG4 to generate LC3B-I, then processed by ATG7, conveyed to the E2 enzyme ATG3 and conjugated to PE (phosphatidylethanolamine). The ATG12–ATG5-ATG16L1 complex apparently has an E3-like activity for formation of LC3B–PE (i.e., LC3B-II).3 LC3B-II locates to the phagophore and autophagosomal membranes and serves as an autophagic marker protein.4-8
It has been demonstrated that autophagy has certain selectivity to particular substrates. This selective autophagy requires receptor molecules essential for degradation of ubiquitinated substrates.9 These receptor molecules include SQSTM1/p62 (sequestosome 1), NBR1 (neighbor of BRCA1 gene 1), CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2), BNIP3L/Nix, etc. They generally contain an LC3B interacting region and ubiquitin-binding domains; consequently, these proteins promote the degradation of ubiquitinated substrates (such as protein aggregates, damaged mitochodria, or intracellular bacteria) by autophagy. SQSTM1 is the most studied protein among them, and its intracellular level has been proven to be regulated by autophagy. Therefore, the level of SQSTM1 is currently used to monitor autophagic flux in a negative correlation.5,8,10 Although some molecular machinery in autophagy regulation have been discovered in the past 10 years, many details regarding their cellular pathways remain uncovered.11,12
Recent findings have revealed that the E3 ubiquitin ligase LRSAM1 could recognize and ubiquitinate various bacteria and initiate the autophagic reaction.13,14 During this process, LRSAM1 interacts with CALCOCO2, transfers polyubiquitin chains to CALCOCO2, which in turn binds LC3B, thereby causing the reaction of autophagic cascade and the removal of bacteria. This means that LRSAM may play an important role in resistance to cellular bacteria by autophagy. Clinical reports indicate that mutations of LRSAM1 cause Charcot-Marie-Tooth disease.15-17 Loss of LRSAM1 increases the sensitivity of peripheral axon degeneration in a mouse model of Charcot-Marie-Tooth disease,18 indicating that dysregulation of LRSAM1 is implicated in neuronal development and homeostasis. A recent finding indicates that the level of LRSAM1 is significantly upregulated in patients with colorectal cancer, 19 implying that the aberrant expression of LRSAM1 may be involved in the cancer process. These data underline the functional multiplicity of LRSAM1 in human health.
We have previously established a screening platform for novel human autophagy-related genes, based on an automated fluorescence microscopy system, 20 and have identified several unreported genes linked to cell autophagy, including TM9SF1, EVA1A/TMEM166, TMEM74, EMC6,21 TMEM208,22 and PHF23. The NUP98-PHF23 fusion gene has been first identified in an AML (acute myeloid leukemia) patient, as a product of chromosome translocation t (11; 17) (p15; p13).23 This fusion protein contains the N-terminal half of NUP98 and the PHF23 C terminus.24 Overexpression of NUP98-PHF23 can weaken 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced differentiation in K562 cells with a PHD domain-dependent manner.24 Importantly, the PHD domain of PHF23 can specially recognize histone H3 lysine 4 trimethylation (H3K4me3) which is suggested as a pivotal component in the regulation of gene expression and epigenetic states.25 The limited data hint that this binding of PHF23 to H3K4me3 may result in the inactivation of differentiation-associated transcription factors or protein kinases, and concomitantly enforces the transcriptional activation of proliferation-related genes in haematopoietic stem cells, eventually leading to the transformation of haematopoietic cells and leukemogenesis during mammalian development. So far, the molecular mechanism of NUP98-PHF23−interrelated AML is not clear. In this study, we demonstrate that PHF23 is a novel negative regulator of autophagy, which interacts with LRSAM1 and facilitates its ubiquitylation and proteasome degradation. Our study may imply a potential link between leukemogenesis and autophagy regulation mediated by PHF23.
Results
Bioinformatic analysis and expression profile of human PHF23
The human PHF23 gene is located on chromosome 17p13.1 and contains 5 exons (Fig. 1A). The full-length cDNA and predicted amino acid sequences are shown in Fig. S1A. The PHF23 gene is highly evolutionarily conserved (Fig. S1B). The PHF23 protein consists of 403 amino acid residues and the relative molecular weight is 43.8 kDa. Using a homology search, we found that the amino acid sequence of PHF23 is well conserved across many species (Fig. S1C). Protein blast analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi) showed that PHF23 has a plant homeodomain (PHD)-like zinc finger domain at the C terminus, which is characterized by a canonical Cys4-His-Cys3 (or C4HC3) motif that coordinates 2 zinc ions (Fig. S1A, underlined). It is predicted to contain a nuclear localization signal (residues 177 to 228) 26 (Fig. S1A, gray highlighted).
Figure 1.

PHF23 genetic information and expression profile. (A) Schematic of the gene and mRNA structure of PHF23. The PHF23 gene is located on chromosome 17, has 5 exons and encodes a protein with 403 amino acid residues. (B) Schematic representation of PHF23 and LRSAM1 constructs used in this study. (C) PHF23 mRNA expression was analyzed by RT-PCR in human normal tissues and cell lines (D). GAPDH expression was amplified as an internal control. (E) Protein expression of PHF23 in mammalian cell lines was detected by a rabbit anti-PHF23 antibody using western blot. ACTB was used as the loading control.
Expression profile analysis indicated that PHF23 mRNA was widely expressed human tissues and various cell lines detected by RT-PCR analysis (Fig. 1C and D). For subsequent experiments, we produced a rabbit anti-PHF23 polyclonal antibody using PHF23 peptides (Fig. S1A, boxed sequences). This rabbit anti-human PHF23 antibody was used to survey the expression and localization of the PHF23 proteins. Consistent with the results of RT-PCR, the PHF23 protein was also presented in many cell lines detected by western blot (Fig. 1E). The ubiquitous expression pattern of PHF23 protein was also measured by immunohistochemistry and tissue microarray (http://www.proteinatlas.org/ENSG00000040633). Immunofluorescence analysis demonstrated that the PHF23 protein was mainly present in the nucleus and part in the cytoplasm which is similar to a previous report.24
PHF23 overexpression impairs EBSS-induced autophagosome formation
PHF23 has originally been identified as an autophagy-related gene using an approach of functional genomics.20 To follow up, we designed a series of experiments to further explore the potential relationship between PHF23 and autophagy. It was noted that PHF23 overexpression failed to affect the occurrence of GFP-LC3B puncta (Fig. 2A and B, left panel) in U2OS cells under nutrient-rich conditions. We next performed experiments in starved U2OS cells treated with EBSS containing neither amino acids nor FBS. The results revealed that PHF23 overexpression reduced the punctate distribution of GFP-LC3B compared to that produced by the control vector (Fig. 2A and B, middle panel). Since GFP-LC3B puncta correlate with the number of autophagosomes, 5 the decrease of GFP-LC3B dots may result from either impaired autophagosome formation or autophagosome clearance by the lysosomal compartment. In order to analyze the autophagic status in PHF23-overexpressing cells under starvation, we cultured U2OS cells in the presence or absence of bafilomycin A1 (BafA1). BafA1 is a known inhibitor of the late phase of autophagy. It prevents the fusion between autophagosomes and lysosomes by inhibiting the vacuolar H+ ATPase (V-ATPase). Although BafA1 treatment resulted in an increase of GFP-LC3B dots in PHF23- and vector-transfected cells caused by EBSS, the punctate GFP-LC3B distribution in the former cells was less than that of the latter cells (Fig. 2A and B, right panel). The distribution of endogenous LC3B dots was similar to that of GFP-LC3B in PHF23-overexpressing U2OS cells (Fig. S2), indicating that overexpression of PHF23 impairs EBSS-induced synthesis of autophagosomes.
Figure 2.

PHF23 overexpression impairs EBSS-induced autophagosome formation. (A) Representative confocal microscopy images of GFP-LC3B distribution obtained from U2OS cells cotransfected with the indicated plasmids and treated with 10 nM bafilomycin A1 (BafA1) and/or EBSS for the last 2 h. (B) Quantification of GFP-LC3B puncta per cell treated as in (A). Data are means ± SD of at least 100 cells scored (*P < 0.05, **P < 0.01). (C) U2OS cells were transfected as indicated for 24 h, treated with 10 nM of BafA1 and/or EBSS or rapamycin (RAPA, 2 μM) for the last 2 h. The levels of LC3B-II were detected by protein gel blot. (D) Quantification of LC3B-II levels relative to ACTB in cells treated as in (C). Average value in vector-transfected cells without BafA1 treatment was normalized as 1. Data are means ± SD of results from 3 experiments (*P < 0.05, **P < 0.01). (E) U2OS cells were cotransfected with polyQ80-luciferase (or control polyQ19-luciferase), vector (or PHF23) as indicated for 30 h, and treated with or without EBSS for the last 2 h. Luciferase activities were monitored, and polyQ80-luciferase/polyQ19-luciferase ratios were calculated (*P < 0.05)..
We next detected the steady-state level of endogenous LC3B-II protein using protein gel blot. In comparison of vector-transfected U2OS cells, PHF23 alone did not affect the LC3B-II level under nutrient-rich conditions (Fig. 2C and D, lane 2 vs. lane 1). After treatment with BafA1, the LC3B-II signals detected in PHF23-overexpressing U2OS cells were weaker than that in the vector group with or without EBSS (Fig. 2C and D, lane 4 vs. lane 3, lane 8 vs. lane 7). To support this finding, we treated cells with rapamycin (RAPA, which induces autophagy by inhibiting the activity of MTOR) and monitored the accumulation of LC3B-II in the presence or absence of BafA1. Compared with the vector group, PHF23 overexpression attenuated the accumulation of LC3B-II (Fig. 2C and D, lane 10 vs. lane 9, lane 12 vs. lane 11). The results hinted that PHF23 overexpression attenuates the autophagosome formation in the state of stress.
We further examined the functional significance of autophagy inhibition by PHF23 overexpression using the accumulation of exogenously expressed polyQ80 aggregates as a surrogate marker for protein degradation in U2OS cells.27 As shown in Fig. 2E, compared with the vector group, the accumulation of exogenous polyQ80 aggregates was upregulated in PHF23-overexpressing cells after EBSS treatment, while PHF23 alone had no obvious effect in normal condition, indicating that PHF23 overexpression seemed to impair the autophagic substrate clearance under amino acid deprivation.
Knockdown of PHF23 increases autophagic flux
In order to discuss the physiological effect of PHF23 on the regulation of autophagy, we conducted a series of experiments in PHF23-depleted cells. Using RT-PCR and western blot assays, we identified 2 effective siRNAs against PHF23 (siPHF23-1 and siPHF23-2) (Fig. 3A). Unless otherwise noted, siPHF23-2 hereafter will be designated simply as siPHF23 and the control siRNA as siControl. Data from repeated experiments showed that PHF23 knockdown significantly increased the occurrence of endogenous LC3B puncta when compared with siControl-transfected cells (Fig. 3B and C, left). Likewise, chloroquine (CQ, an inhibitor of the autophagic flux through raising lysosomal pH) treatment caused a further increase of LC3B dots in PHF23-silenced cells (Fig. 3B and C, middle). A similar finding was observed in GFP-LC3B-transfected U2OS cells (Fig. S3 A and B). In line with these results, we next measured the LC3B conversion by protein gel blot. We observed that depletion of PHF23 obviously elevated the levels of endogenous LC3B-II with or without CQ treatment (Fig. 3D and E, lanes 2 vs. lane 1, lanes 4 vs. lane 3). We also obtained similar results in PHF23-depleted HeLa cells (Fig.S4 A and B, lanes 2 vs. lane 1, lanes 4 vs. lane 3). These data indicated that knockdown of PHF23 promotes autophagosome synthesis.
Figure 3 (see previous page).

Knockdown of PHF23 increases autophagic flux. (A) U2OS cells were transfected with siControl, siPHF23-1 or siPHF23-2 for 48 h. PHF23 mRNA or protein expression was detected by RT-PCR and western blot, respectively. (B) Representative confocal microscopy images of endogenous LC3B distribution in U2OS cells transfected with siControl or siPHF23 for 48 h and treated with CQ (25 μM) for the last 4 h and/or 10 mM 3-MA for the last 6 h. (C) Quantification of endogenous LC3B dots in siControl or siPHF23 cells treated with reagents as indicated in (B). Data are means ± SD of at least 100 cells scored (**P < 0.01, ***P < 0.001). (D) Western blot analysis of endogenous LC3B-II levels in U2OS cells treated as in (B). (E) Quantification of amounts of LC3B-II relative to ACTB in cells treated as in (D). Average value in siControl-transfected cells without CQ treatment was normalized as 1. Data are means ± SD of results from 3 experiments (**P < 0.01). (F) HeLa cells stably expressing GFP-LC3B were cotransfected with siControl, siPHF23-1, or siPHF23-2 for 48 h. Levels of SQSTM1 and free GFP were analyzed by protein gel blot. (G) Quantification of amounts of SQSTM1 protein or free GFP relative to ACTB in cells treated as in (F). The average value in siControl-transfected cells was normalized as 1. Data are means ± SD of results from 3 experiments (*P < 0.05). (H) U2OS cells were cotransfected with polyQ80-luciferase (or polyQ19-luciferase) and the indicated siRNAs for 48 h. PolyQ80–luciferase/polyQ19-luciferase ratios were analyzed using the Dual Luciferase Reporter System (*P < 0.05, **P < 0.01). (I) Representative confocal microscopy images of mTagRFP-mWasabi-LC3B distribution in U2OS cells cotransfected with mTagRFP-mWasabi-LC3B and siControl or siPHF23 and cultured for 48 h. (J) Representative confocal microscopy images of GFP-LC3B distribution in U2OS cells cotransfected with different combinations for 48 h, and treated with CQ (25 μM) for the last 4 h. (K) Quantification of GFP-LC3B dots in U2OS cells treated as in (J). Data are means ± SD of at least 100 cells scored (*P < 0.05, **P < 0.01). (L) Western blot analysis of endogenous LC3B-II levels in U2OS cells treated as in (J). (M) Quantification of endogenous LC3B-II levels relative to ACTB in cells treated as in (L). The average value in siControl-transfected cells was normalized as 1. Data are means ± SD of results from 3 experiments (*P < 0.05, **P < 0.01).
To explore which step is involved in the elevated autophagy in PHF23-silenced cells, we treated the U2OS cells with 3-methyladenine (3-MA), as it is a widely used inhibitor of autophagy by interfering with the PIK3C3 (phosphatidylinositol 3-kinase, catalytic subunit type 3) that is critical during the initial phase of phagophore formation. As shown in Fig. 3B and C (right panel), treatment with 3-MA could reduce the endogenous LC3B dots in both PHF23-silenced and control cells. In the same circumstance, 3-MA depressed the accumulation of LC3B-II detected by western blot (Fig. 3D and E, lanes 6 vs. lane 4). Similarly, 3-MA also attenuated the LC3B-II signal enhanced by CQ in PHF23-silenced HeLa cells (Fig.S4 A and B, lanes 6 vs. lane 4). These data imply that PHF23-mediated autophagy regulation in different cells may at least partly involve the activity of the PIK3C3 complex.
Accumulating data showed that the effect of GFP-LC3B fusion protein is similar to endogenous LC3B protein in autophagy. Since GFP protein is relatively resistant to lysosomal hydrolysis compared with LC3B, the levels of free GFP detected by protein gel blot have been used as a measure of functional autophagic flux.5 In the HeLa cells stably expressing GFP-LC3B, we observed that the free GFP band was stronger in PHF23-silenced cells than that in control cells (Fig. 3F and G, upper panel). This treatment also resulted in a reduction of the endogenous autophagy substrate SQSTM127-28 (Fig. 3F and G, middle panel). Likewise, knockdown of PHF23 diminished the accumulation of polyQ80 aggregates (Fig. 3H). Furthermore, we employed an mTagRFP-mWasabi-LC3B reporter to evaluate the maturation of the autophagosome.29 U2OS cells were cotransfected with mTagRFP-mWasabi-LC3B and siControl or siPHF23, then observed and documented by confocal microscopy. As shown in Fig. 4I, knockdown of PHF23 enhanced the transition of mTagRFP-mWasabi-LC3B-positive autophagosomes to mTagRFP-positive, mWasabi-negative autolysosomes (Fig. 3I, lower panel). This result suggested that autophagosome–lysosome fusion was promoted in PHF23 knockdown cells. Taken together, these data suggested that PHF23 is a negative regulator for autophagic degradation of cellular components.
Figure 4.

PHF23 associates with LRSAM1 via the PHD domain. (A) HEK293 cells were cotransfected with GFP-PHF23 and LRSAM1-MYC-FLAG for 24 h. Total cell extracts were subjected to IP using either an anti-GFP or a control IgG. LRSAM1 was detected in the immunoprecipitated proteins by western blot. (B) HEK293 cells were treated as in (A), except that cell lysates were immunoprecipitated with an anti-FLAG antibody. PHF23 was detected in the immunoprecipitated proteins by protein gel blot. (C) HEK293 cells were cotransfected with plasmids expressing PHF23PHDΔ and LRSAM1-MYC-FLAG for 24 h. Total cell extracts were subjected to IP using either an anti-FLAG or a control IgG, as indicated. PHF23PHDΔ was not detected in the immunoprecipitated proteins, by western blot. (D and E) GST-PHF23 or GST-PHF23PHDΔ fusion protein and the GST protein immobilized on Glutathione-Sepharose beads were incubated with LRSAM1-FLAG-transfected HEK293 cell lysates at 4°C for 4 h. LRSAM1 and GST were detected in the washed beads, by protein gel blot.
Next, the specific effects of PHF23 knockdown were assessed in an approach of recovery experiment. In this context, U2OS cells were transfected with siControl or siPHF23 alone, or together with the PHF23 expression plasmid and then treated with CQ. As shown in Fig. 3J and K, the GFP-LC3B dots in PHF23-silenced cells plus the PHF23 plasmid was reduced compared with siPHF23 alone. Consistent with these observations, the levels of endogenous LC3B-II was downregulated, detected by western blot (Fig. 3L and M, lane 3 vs. lane 2) in PHF23-silenced cells transfected with the PHF23 plasmid. Taken together, our results confirmed that PHF23 could indeed negatively regulate cellular autophagy processes.
PHF23 is associated with LRSAM1 via the PHD domain
PHF23 was implicated as a novel binding partner of LRSAM1 through a large-scale yeast 2-hybrid screen.30 However, no experimental evidence has been reported in mammalian cells or further functional investigation conducted to support this finding. To confirm the correlation of PHF23 and LRSAM1, we carried out a coimmunoprecipitation assay. LRSAM1-FLAG and GFP-PHF23 plasmids were cotransfected into HEK293 cells. 24 h later, cells were lysed and the cell lysates were subjected to immunoprecipitation (IP) with an anti-GFP antibody. Immunoblotting showed that GFP-PHF23 coprecipitated with LRSAM1-FLAG (Fig. 4A). Furthermore, GFP-PHF23 was detected in the reciprocal LRSAM1-FLAG immunoprecipitates (Fig. 4B), demonstrating that the 2 proteins were present in a complex in HEK293 cells.
Bioinformatic analysis suggested that PHF23 has a PHD-like zinc finger domain at the C terminus, which is required for the functionality of PHD finger-containing proteins.31,32 We next wanted to know whether the interaction between PHF23 and LRSAM1 is mediated by the PHD finger. To confirm this speculation, LRSAM1-FLAG and PHF23PHDΔ plasmids were cotransfected into HEK293 cells, and the cell extracts were submitted to IP with an anti-FLAG antibody. Immunoblotting assay uncovered that the loss of the PHD domain of PHF23 abolished the interaction between PHF23 and LRSAM1 (Fig. 4C). We further followed an affinity-isolation approach. As shown in Fig. 4D and E, the GST-PHF23 fusion protein could precipitate LRSAM1-FLAG from cell lysates. Nevertheless, GST-PHF23PHDΔ was inactive. These results showed that PHF23 binds to LRSAM1 in a PHD domain-dependent manner.
PHF23PHDΔ increases cell autophagy
As mentioned above, PHF23-mediated autophagy regulation may occur through the LRSAM1 pathway, and the PHD domain of PHF23 may be essential for its functionality. To verify this hypothesis, U2OS cells were transfected with PCDB-vector and PHF23PHDΔ in the presence or absence of CQ. Overexpression of PHF23PHDΔ resulted in more endogenous LC3B puncta than that with the vector control (Fig. 5A and B, left panel). Likewise, CQ treatment further increased the LC3B puncta in PHF23PHDΔ-overexpressing cells (Fig. 5A and B, right panel), indicating that PHF23PHDΔ promoted autophagesome formation. Data obtained from protein gel blot revealed that the levels of LC3B-II was increased in PHF23PHDΔ-overexpressing cells with or without CQ, compared with vector control (Fig. 5C and D, lane 3 vs lane 1, lane 6 vs. lane 4) or PHF23-transfected cells (Fig. 5C and D, lane 3 vs. lane 2, lane 6 vs. lane 5). Consistent with these phenomena, overexpression of PHF23PHDΔ induced a reduction of the SQSTM1 protein (Fig. 5E and F). These results indicated that the PHD domain of PHF23 is required for the PHF23 activity, and PHF23PHDΔ may function as an antagonist of PHF23 to promote autophagy.
Figure 5.
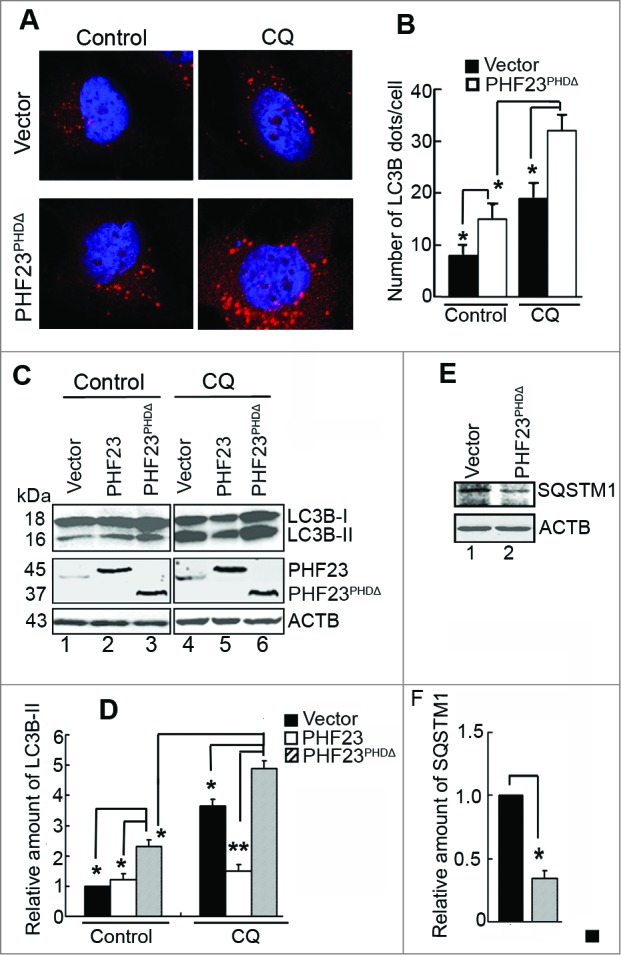
PHF23PHDΔ increases cell autophagy. (A) Representative confocal microscopy images of endogenous LC3B distribution in U2OS cells transfected for 24 h with the indicated plasmids and then treated with or without CQ (25 μM) for the last 4 h. (B) Quantification of endogenous LC3B dots in U2OS cells treated as in (A). Data are means ± SD of at least 100 cells scored (*P < 0.05). (C) Western blot analysis of endogenous LC3B-II levels in U2OS cells transfected with the indicated plasmids for 24 h and then treated with or without CQ (25 μM) for the last 4 h. (D) Quantification of amounts of LC3B-II relative to ACTB in cells treated as in (C). The average value in the control vector-transfected cells without CQ treatment was normalized as 1. Data are means ± SD of results from 3 experiments (*P < 0.05, **P < 0.01). (E) Western blot analysis of endogenous SQSTM1 levels in U2OS cells transfected with vector or PHF23PHDΔ for 24 h. (F) Quantification of SQSTM1 levels relative to ACTB in cells treated as in (E). The average value in control vector-transfected cells was normalized as 1. Data are means ± SD of results from 3 experiments (*P < 0.05).
LRSAM1 regulates autophagy
Previous studies have indicated that the LRSAM1 is an E3 ligase which is essential for ubiquitin-dependent anti-Salmonella autophagy.13 However, whether LRSAM1 mediates conventional autophagy in mammalian cell lines has not been reported in detail. Here, we wanted to explore the autophagy level in LRSAM1-overexpressing and LRSAM1-silenced U2OS cells. Compared to the vector control, overexpression of LRSAM1 caused an increase of GFP-LC3B dots with or without CQ (Fig. S5A and B), suggesting that LRSAM1 enhances the formation of autophagesomes. Consistent with the results of fluorescence microscopy, the level of endogenous LC3B-II protein was upregulated in LRSAM1-transfected cells (Fig. 6A and B, lane 2 vs. lane 1). CQ treatment further enhanced accumulation of LC3B-II in LRSAM1-transfected cells (Fig. 6A and B, lane 4 vs. lane 3). Similarly, LRSAM1 weakly enhanced EBSS-induced LC3B conversion (Fig. 6A and B, lane 6 vs. lane 5). The above-mentioned data indicated that LRSAM1 enhanced the formation of autophagosomes.
Figure 6.

LRSAM1 regulates autophagy. (A) Western blot analysis of endogenous LC3B-II levels in U2OS cells transfected with vector or LRSAM1 for 24 h. CQ (25 μM) was added for the last 4 h and with or without EBSS for the last 2 h. (B) Quantification of LC3B-II levels relative to ACTB in cells treated as in (A). The average value in control vector-transfected cells (lane 1) was normalized as 1. Data are means ± SD of results from 3 experiments (*P < 0.05). (C) U2OS cells were transfected with the indicated siRNAs for 48 h, and then LRSAM1 mRNA was analyzed by RT-PCR. GAPDH was amplified as an internal control. (D) Western blot analysis of endogenous LC3B-II levels in U2OS cells transfected for 48 h with siControl or siLRSAM and treated with CQ (25 μM) for the last 4 h and/or EBSS for the last 2 h. (E) Quantification of amounts of LC3B-II relative to ACTB in cells treated as in (D). The average value in siControl-transfected cells (lane 1) was normalized as 1. Data are means ± SD of results from 3 experiments (*P < 0.05). (F) HeLa cells stably expressing GFP-LC3B were cotransfected for 48 h with siControl, siLRSAM1-1 or siLRSAM1-2. Levels of SQSTM1 and free GFP were analyzed by western blot. (G) Quantification of amounts of SQSTM1 protein or free GFP relative to ACTB in cells treated as in (F). The average value in siControl-transfected cells was normalized as 1 (lane 1). Data are means ± SD of results from 3 experiments (*P < 0.05).
Next, experiments were carried out in LRSAM1-depressed U2OS cells. Two candidate siRNAs against LRSAM1 (siLRSAM1-1 and siLRSAM1-2)14 were validated by RT-PCR (Fig. 6C). Hereafter, siLRSAM1-1 will be referred to simply as siLRSAM1. Data from western blot showed that the levels of endogenous LC3B-II diminished in LRSAM1-depletd cells with or without CQ treatment (Fig. 6D and E, lane 2 vs. lane 1, lane 6 vs. lane 5), suggesting that the decreased expression of LRSAM1 attenuated autophagosome synthesis. Additionally, depletion of LRSAM1 also reduced EBSS-induced LC3B-II accumulation in the presence or absence of CQ (Fig. 6D and E, lane 4 vs. lane 3, lane 8 vs. lane 7). These phenomena were accompanied by increased SQSTM1 protein accumulation (Fig. 6F and G, lane 2 vs. lane 1, upper panel) and a decrease of free GFP fragments (Fig. 6F and G, lane 2 vs. lane 1, middle panel). These results suggested that the autophagic flux was damaged in LRSAM1-silenced cells and promoted in LRSAM1-overexpressing U2OS cells.
PHF23 mediates autophagy by promoting the ubiquitination and degradation of LRSAM1
We next sought to determine the functional connections between PHF23 and LRSAM1 in the regulation of autophagy. Data obtained from protein gel blot indicated that LRSAM1 overexpression augmented the accumulation of endogenous LC3B-II compared with the vector control in the presence or absence of CQ (Fig. 7A and B, lane 2 vs. lane 1, lane 6 vs. lane 5). PHF23 overexpression attenuated the LC3B conversion induced by LRSAM1 (Fig. 7A and B, lane 3 vs. lane 2), while PHF23 knockdown increased the accumulation of LC3B-II caused by LRSAM1 (Fig. 7A and B, lane 4 vs. lane 2). Similar effects were also observed in CQ-treated U2OS cells (Fig. 7A and B, lane 7 vs. lane 6, lane 8 vs. lane 6). These results suggested that LRSAM1-mediated autophagy signaling is inactivated by PHF23 overexpression and promoted by PHF23 silencing.
Figure 7.

PHF23 mediates autophagy by promoting ubiquitination and degradation of LRSAM1. (A) Western blot analysis of endogenous LC3B-II levels in U2OS cells transfected with the indicated plasmids for 24 h. CQ (25 μM) was added for the last 4 h. (B) Quantification of amounts of LC3B-II relative to ACTB in cells treated as in (A). The average value in control vector-transfected cells was normalized as 1. Data are means ± SD of results from 3 experiments (*P < 0.05). (C) Western blot analysis of endogenous LC3B-II levels in U2OS cells transfected for 48 h with different siRNAs and then treated with CQ (25 μM) for the last 4 h. (D) Quantification of LC3B-II levels relative to ACTB in cells treated as in (C). The average value in siControl-transfected cells (lane 1) was normalized as 1. Data are means ± SD of results from 3 experiments (*P < 0.05, **P < 0.01). (E) HeLa cells stably expressing GFP-LC3B were cotransfected with different siRNAs for 48 h. Levels of free GFP were analyzed by protein gel blot. (F) Quantification of free GFP levels relative to ACTB in cells treated as in (E). The average value in siControl-transfected cells was normalized as 1. Data are means ± SD of results from 3 experiments (*P < 0.05, **P < 0.01). (G) U2OS cells were cotransfected with polyQ80-luciferase (or polyQ19-luciferase) and the indicated siRNAs for 48 h. PolyQ80 /polyQ19 ratios were analyzed using the Dual Luciferase Reporter System (*P < 0.05, **P < 0.01). (H) HEK293 cells were transiently transfected with different combinations of mammalian expression vectors for FLAG-ubiquitin, PHF23, PHF23PHDΔ, and LRSAM1. After 24 h, cells were treated with 10 μM MG132 for 6 h. The cells lysates were then immunoprecipitated with an anti-LRSAM1 antibody and probed with an anti-ubiquitin antibody. (I) HEK293 cells were transiently transfected with different combinations of mammalian expression vectors for PHF23, PHF23PHDΔ, and LRSAM1 for 24 h, cells were treated with 10 μM MG132 for 6 h. The cells lysates were then probed with anti- LRSAM1 and PHF23 antibodies. ACTB was used as the loading control.
We next detected the effects of LRSAM1 on PHF23-mediated autophagy. Cells were transfected using siLRSAM1, siPHF23, or both in the presence or absence of CQ. The results as shown in Fig. 7C and D, knockdown of PHF23 increased the accumulation of LC3B-II compared to that of siControl group with or without of CQ treatment (Fig. 7C and D, lane 2 vs. lane 1, lane 6 vs. lane 5). By contrast, LRSAM1 knockdown reduced the LC3B conversion compared to that of the siControl group (Fig. 7C and D, lane 7 vs. lane 5). Furthermore, knockdown of LRSAM1 significantly decreased LC3B-II accumulation in PHF23-silenced U2OS cells with or without of CQ (Fig. 7C and D, lane 4 vs. lane 2, lane 8 vs. lane 6).
The effect of LRSAM1 on siPHF23-mediated autophagy was further determined by GFP-LC3B cleavage in HeLa cells stable expressing the GFP-LC3B fusion protein. As shown in Fig. 7E and F, PHF23-silenced HeLa cells displayed more free GFP than siControl-treated cells. However, cotransfection of siPHF23 and siLRSAM1 markedly reduced the accumulation of free GFP compared to siPHF23 alone (Fig. 7E and F, lane 4 vs. lane 2). Likewise, knockdown of LRSAM1 diminished the degradation of polyQ80 aggregates in PHF23-silenced U2OS cells (Fig. 7G). These data suggested that cellular autophagic flux was impaired by the inhibition of LRSAM1 expression. Taken together, our results implied that the interactions between PHF23 and LRSAM1 are mutually antagonistic in the regulation of cellular autophagy.
LRSAM1 has been reported to be a ubiquitin ligase that promotes anti-Salmonella autophagy in a ubiquitin-dependent manner.13 The degradation of LRSAM1 is regulated by the ubiquitin-proteasome system as well. Therefore, we wondered whether PHF23 could influence the ubiquitination of LRSAM1 through the PHD domain. To prove this hypothesis, U2OS cells were transfected with different combinations of mammalian expression vectors for Flag-ubiquitin, PHF23, PHF23PHDΔ and LRSAM1 for 24 h, followed by treatment with the proteasome inhibitor MG132. Subsequently, the cell lysates were immunoprecipitated by an anti-LRSAM1 antibody, and the samples were probed in a western blot with an anti-ubiquitin antibody to detect ubiquitinated LRSAM1. As shown in Fig. 7H, the level of ubiquitinated LRSAM1 was increased in cells overexpressing PHF23 compared with LRSAM1 alone, whereas PHF23PHDΔ failed to influence this effect. At the same time, we observed that overexpression of PHF23 could decrease the levels of total LRSAM1 protein (Fig. 7H, middle panel), suggesting that PHF23 may regulate the stability of LRSAM1. As LRSAM1 is degraded through the proteasome, the effect of PHF23 on proteasome-mediated proteolysis of LRSAM1 was further tested with or without MG132 treatment. As shown in Fig. 7I, the reduction of LRSAM1 levels by PHF23 overexpression was significantly reversed by MG132 treatment (Fig. 7I, lanes 4 vs. lane 2), while PHF23PHDΔ did not influence this effect. In addition, knockdown of PHF23 slightly increased the endogenous LRSAM1 level (Fig. S6A). But LRSAM1 overexpression had no obvious effect on the level of PHF23 (Fig. S6B). Consistent with PHF23-mediated ubiquitylation causing LRSAM1 degradation, these data suggested that LRSAM1 is destabilized in the presence of PHF23. Collectively, these results indicated that the PHD domain of PHF23, which interacts with LRSAM1 is required for its biological activities, and the PHF23-mediated negative regulation of autophagy may result from promoting ubiquitination and degradation of LRSAM1 through a yet unknown pathway.
Discussion
In order to identify human genes involved in autophagy regulation, we established a cell-based functional screening platform based on an automated fluorescence microscopy system in which we can analyze the images of GFP-LC3B dots in cotransfected cells. We have previously identified a number of autophagy-related human genes and some of them have been characterized in our laboratory.20-22 In this study, we characterized a human protein termed PHF23 with a PHD-like zinc finger domain (http://www.ncbi.nlm.nih.gov/protein/NP_077273.2). We successfully determined that PHF23 functions as a negative regulator of the autophagic process.
Our investigations indicated that overexpression of PHF23 impaired EBSS- or rapamycin-induced cellular autophagy, while knockdown of its expression enhanced autophagy. This notion was evidenced by increased steady-state levels of LC3B-II and degradation of substrate proteins in PHF23-silenced cells. It is well known that the increased LC3B-II levels in autophagy can have 2 possible causes: either increased autophagosome formation or a block in autophagosome maturation or degradation. Here, we have performed a series of assays, including analysis of LC3B-II synthesis (in the presence of BafA1 or CQ), studies of autophagic flux by means of the GFP-RFP-LC3B reporter. The presence of free GFP fragments together with assays of both endogenous (SQSTM1) and exogenous (polyQ80) autophagic substrate accumulation or degradation, demonstrate that PHF23 depletion enhances autophagosome formation and increases autophagic flux. These effects may involve the activity of the PIK3C3 complex, as PHF23 regulated-autophagy was found to be sensitive to the PIK3C3 inhibitor 3-MA. However, the overexpression of PHF23 alone had no evident effects either on LC3B-II synthesis or on the clearance of autophagic substrates under nutrient-rich conditions, suggesting that the levels of this component are normally saturating for these processes. Interestingly, we discovered that PHF23PHDΔ (lacking the PHD finger domain) is an antagonist of PHF23, which functions a positive effect in the regulation of autophagy.
The exploration of molecular mechanisms in the current study indicated that PHF23 could bind the E3 ligase LRSAM1, and the PHD finger domain of PHF23 is functionally required for its interaction with LRSAM1. A recent report has found that LRSAM1 recognizes and ubiquitinates unknown molecules of bacterial membranes. 13 Meanwhile, LRSAM1 binds the autophagy adaptor CALCOCO2, which in turn recognizes polyubiquitinated bacteria and binds LC3B proteins, thereby triggering the autophagic cascade and bacterial removal.33 Cells lacking LRSAM1 fail to inhibit bacterial proliferation, as do cells depleted of CALCOCO2. Additionally, the ubiquitin signal provided by LRSAM1 also may be required for the adaptor SQSTM1, which acts as a bridging molecule between ubiquitylated proteins and components of the autophagy machinery.34-36 Whether LRSAM1 is capable of ubiquitinating other target molecules and mediating nonbacterial autophagy is unclear. In our experimental system, we found that LRSAM1 knockdown attenuated starvation-induced autophagy in U2OS cells, indicating that LRSAM1 may be involved in conventional autophagy.
The biological significance of the interaction between PHF23 and LRSAM1 is preliminarily discussed in our laboratory. Data from our experiments indicated that overexpression of PHF23 impaired autophagosome formation induced by LRSAM1, while PHF23 silencing enhanced this effect, suggesting that PHF23 negatively regulates the function of LRSAM1 on autophagy. Conversely, depletion of LRSAM1 attenuated PHF23 knockdown-induced autophagy, implying that PHF23-regulated cell autophagy at least in part depends on LRSAM1 signaling, and interactions between PHF23 and LRSAM1 are mutually antagonistic in the regulation of cellular autophagy.
LRSAM1 itself was found to be auto-ubiquitinated.13 In the present study, we found that PHF23 overexpression increased ubiquitination and proteasome degradation of LRSAM1 in a PHD dependent manner, illustrating that the function and stability of LRSAM1 may be regulated by PHF23 through a still-uncharacterized pathway. Therefore, it was deduced that PHF23 may exert antagonistic effects on LRSAM1 in normal physiological conditions. In view of the functional multiplicity of LRSAM1 on anti-infection,13 Charcot-Marie-Tooth disease,15-17 and colorectal cancer,19 whether PHF23 is involved in these diseases deserves further study.
Acute myeloid leukemia is often linked with chromosomal translocations in which produce specific fusion genes that play important roles for leukemogenesis.37 NUP98 (nucleoporin 98kDa) is a common fusion partner which has been identified in human leukemia. NUP98-PHF23 fusion is a novel product of chromosomal translocation in AML.23 This potent oncoprotein caused an impairment of haematopoietic differentiation and induced AML in a murine model.25 In this process, a PHD domain that recognizes H3K4me3 was crucial for leukemogenesis,25 suggesting a potential functional connection between PHF23 and leukemia. As we all know, autophagy has both pro- and anti-tumor effects in tumor biology. Autophagic activity is finely adjusted and continuously switched on and off during the different phases of tumorigenesis.38 Linked to the relationship between PHF23 and autophagy as we observed, the role of PHF23 in leukemia or other tumors is worthy of further investigation. Similarly, the physical and functional connection between the PHF23 and LRSAM1 in different conditions will be also elucidated in the further experiments.
Materials and Methods
Antibodies and reagents
Polyclonal antibodies against PHF23 were prepared by immunizing rabbits with chemically synthesized PHF23 peptides (Fig. S1A, boxed sequences) and purified by peptide affinity chromatography via CNBr-activated SepharoseTM 4 Fast Flow (GE Healthcare, 17-0981-01) according to the manufacturer's instructions. Other primary antibodies used in this study were: anti-LC3B (Sigma Aldrich, L7543), anti-FLAG (Tianjin Sungene, km8002L), anti-myc (Tianjin Sungene, km8003L), anti-GFP (Tianjin Sungene, km8009L), anti-β-actin (ACTB, Tianjin Sungene, KM9001) and IRDye 800-conjugated anti-GFP (Rockland, 600-432-215) antibodies. Secondary antibodies included DyLight 800/DyLight 680-conjugated IgG against mouse (Rockland, 610-145-002/610-144-002) or rabbit (Rockland, 611-145-002/611-144-002) and FITC/RBITC/APC-conjugated IgG against mouse (Bioss Inc., bs-0296G-FITC/bs-0296G-RBITC/bs-0296G-APC) or rabbit (Bioss Inc., bs-0295G-FITC/bs-0295G-RBITC/bs-0295G-APC). Other reagents used in this study were: cDNA libraries of human normal adult tissues (Clontech, 636742, 636743, 636748), Dual-Luciferase ®Reporter (DLR™) Assay System (Promega, E1910), bafilomycin A1 (Sigma Aldrich, B1793), chloroquine (C6628-25G), 3-methyladenine (Sigma Aldrich, M9281) and RIPA Lysis Buffer (Beyotime, P0013D).
Plasmid construction
The PHF23 cDNA was amplified from a human kidney cDNA library (Clontech, 637204) by PCR with the forward primer P1 (5′- CGGAATTCATGCTGGAAGCCATGGCGGAGC-3′) and reverse primer P2 (5′- CCCAAGCTTTCAGGGCTCTCCAGATTTGGG-3′). The insert was released by EcoRI and HindⅢ then subcloned into the EcoRI site of pcDNA.3.1/myc-His(-)B (Invitrogen, V85520) to construct the pcDB-PHF23 plasmid, abbreviated PHF23 in this study. Based on this plasmid, we constructed the following plasmids in succession: PHF23-MYC, PHF23PHDΔ-MYC, GFP-PHF23, GST-PHF23, and GST- PHF23PHDΔ (Fig. 1B). We also constructed the following LRSAM1-expression plasmids: LRSAM1, LRSAM1-FLAG, and LRSAM1-GFP (Fig. 1B). All constructs were confirmed by DNA sequencing.
The mTagRFP-mWasabi-LC3B plasmid was kindly provided by Jian Lin (Peking University, Beijing, China). The polyQ80-luciferase and polyQ19-luciferase plasmids were kindly provided by Conrad C. Weihl (Washington University School of Medicine, USA). The GFP-LC3B plasmid was kindly provided by Zhenyu Yue (Mount Sinai School of Medicine, New York, USA). The following are sequences of double-stranded siRNAs against PHF23 and LRSAM1, which were designed and chemically synthesized by Genechem Corporation (Shanghai, China): siPHF23-1: 5′-GUGCUACCUUGCUUGAGAATT-3′, siPHF23-2: 5′-GACCGAA AGAACCGAAAGUTT-3′; siLRSAM1-1: 5′-GCAGAUGACAUUCUCGACAUCUCUATT-3′, siLRSAM1-2: 5′-CCGGCUCAUCCAGAUGGCCUACGAATT-3′. The Control siRNA (siControl) was confirmed to have no matches with the complete human genome by a BLAST search in NCBI (www.ncbi.nlm.nih.gov).
Cell culture, transfections, and treatments
U2OS, HEK293 and HeLa cell lines were maintained in DMEM (Invitrogen, 12800-017) supplemented with 10% fetal bovine serum (FBS). Other cell lines used for RT-PCR were cultured routinely in our laboratory. Cells were transfected using MegaTran 1.0 Transfection Reagent (ORIGEN, TT200004) according to the manufacturer's instruction. All experiments were performed on logarithmically growing cells. Cell autophagy was induced by nutrient deprivation through incubation with EBSS (Sigma Aldrich, E7510), which contains neither amino acids nor FBS. Autophagy inhibition was achieved by treating cells with 10 nM of bafilomycin A1 or 25 μM of chloroquine, which can block the fusion of autophagosomes and lysosomes.
RT-PCR assay
Different cell lines were cultured to the exponential phase, and total RNA samples were extracted with the TRIzol reagent (Invitrogen, 15596-026). RT-PCR was performed using the ThermoScript RT-PCR System (Invitrogen, 11146-024). Primers used for amplifying PHF23 were 5′- TGGACGAGGACATCATGGTA -3′ and 5′- ACAGAGTGGGGAGGAAGGAT-3′.
Poly Q degradation assay
U2OS cells were cotransfected with the polyQ80-Firefly luciferase construct (or the control polyQ19-Firefly luciferase construct) together with the indicated plasmids using MegaTran 1.0 Transfection Reagent (ORIGEN, TT200004) according to the manufacturer's protocol. Each group was divided into 2 groups: polyQ80-luciferase and polyQ19-luciferase. After treatment at the desired time, cells were lysed using Passive Lysis Buffer from the DLR™ Assay System and centrifuged in a refrigerated centrifuge. The cleared lysates were transferred into a fresh tube and the firefly luciferase activity of polyQ80-luciferase or polyQ19-luciferase in each group was measured; polyQ19-luciferase was used as an internal control. This approach was designed to measure only firefly luciferase reporter activity in the treated cell lysates, not Renilla luciferase. Briefly, 20 μl of cell lysate prepared from polyQ80-luciferase or polyQ19-luciferase transfected cells were dispensed in triplicate into a 96-well assay plate (Corning Inc. COSTAR, 3925) containing 100 μl of Luciferase Assay Buffer II (containing luciferase assay substrate) from the DLR™ Assay System according to the manufacturer's protocol. Then the assay plate was mixed by pipetting 2 or 3 times, and the stabilized luminescent signal was measured by the Veritas Microplate Luminometer (Turner Biosystems, CA, USA). Data were expressed as the ratio of polyQ80-luciferase/polyQ19-luciferase luminescence signal values in each group as described previously.39 All samples were assayed in triplicate, and the results were shown with 3 independent experiments.
Immunofluorescence, fluorescence, and confocal microscopy
U2OS cells were cultured in confocal dishes and treated as indicated, fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 (Beyotime, ST795). The dishes were then incubated with FBS overnight and exposed to primary antibody for 1 h at 4°C. After being washed 3 times with phosphate-buffered saline (Solarbio, P1010), the dishes were immersed with TRITC-conjugated secondary antibody solution (Jackson ImmunoResearch, 111-025-003). Nuclei were stained with Hoechst 33342 (Sigma-Aldrich, 14533). Morphological alterations in the cells were observed and documented with an Olympus FluoView™ FV1000 Confocal Microscope (Olympus, Melville, NY, USA).
To observe the endogenous LC3B puncta, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, incubated with FBS overnight, exposed to LC3B antibody, stained with TRITC-conjugated secondary antibody, and then observed by confocal microscopy. The number of LC3B puncta per cell was assessed in 5 non-overlapping fields, and statistical data were obtained from 3 independent experiments. Cells transfected with GFP-LC3B plasmids were observed and assessed as endogenous LC3B.
Immunoprecipitation and protein gel blot
For the IP analysis, treated cells were collected and disrupted in RIPA Lysis Buffer containing protease inhibitors (Roche Diagnostics, 04693116001). Total cell extracts (1 mg per sample) were mixed with precleared protein G sepharoseTM Fast Flow (GE Healthcare, 17-0618-01) and appropriate antibodies, followed by incubation for 4 h at 4°C. The beads were collected by centrifugation, washed 5 times using washing buffer, resuspended in 2× SDS loading buffer and then analyzed by western blot as described previously.21 The protein bands were visualized using DyLight 800/DyLight 680-conjugated secondary antibodies, and the infrared fluorescence image was obtained using an Odyssey infrared imaging system (LI-CORBiosciences, Lincoln, NE, USA).
GST affinity isolation assay
Recombinant GST or GST-PHF23 or GFP-PHF23PHDΔ fusion proteins were expressed in Escherichia coli strain BL21 (DE3) and purified. Equal amounts of these proteins were mixed with whole cell lysates extracted from transfected cells and glutathione-SepharoseTM4B (GE Healthcare, 17-0756-01) for 4 h at 4°C. After 5 washes, the beads were resuspended in 2× SDS loading buffer and analyzed by protein gel blot.21
Statistical analysis
Data are presented as the mean ± SD. Differences between groups were analyzed using the Student t test for continuous variables. Statistical significance in this study was set at P < 0.05. All reported P values are 2-sided. All analyses were performed with GraphPad Prism 5.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by a grant from the National Key Basic Research Program of China (973, 2011CB910103) and the Doctoral Program of the Ministry of Education (20130001110015).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/ 10.1038/nature06639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463-77; PMID:15068787; http://dx.doi.org/ 10.1016/S1534-5807(04)00099-1 [DOI] [PubMed] [Google Scholar]
- 3. Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 2007; 282:37298-302; PMID:17986448; http://dx.doi.org/ 10.1074/jbc.C700195200 [DOI] [PubMed] [Google Scholar]
- 4. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/ 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. . Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID:17611390; http://dx.doi.org/ 10.4161/auto.4600 [DOI] [PubMed] [Google Scholar]
- 7. Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S, Klionsky DJ. In search of an “autophagomometer”. Autophagy 2009; 5:585-9; PMID:19411822; http://dx.doi.org/ 10.4161/auto.5.5.8823 [DOI] [PubMed] [Google Scholar]
- 8. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/ 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279-96; PMID:21189453; http://dx.doi.org/ 10.4161/auto.7.3.14487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Larsen KB, Lamark T, Øvervatn A, Harneshaug I, Johansen T, Bjørkøy G. A reporter cell system to monitor autophagy based on SQSTM1SQSTM1. Autophagy 2010; 6:784-93; PMID:20574168; http://dx.doi.org/ 10.4161/auto.6.6.12510 [DOI] [PubMed] [Google Scholar]
- 11. Chen Y, Klionsky DJ. The regulation of autophagy – unanswered questions. J Cell Sci 2011; 124:161-70; PMID:21187343; http://dx.doi.org/ 10.1242/jcs.064576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Loos B, Engelbrecht AM, Lockshin RA, Klionsky DJ, Zakeri Z. The variability of autophagy and cell death susceptibility: unanswered questions. Autophagy 2013; 9:1270-85; PMID:23846383; http://dx.doi.org/ 10.4161/auto.25560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huett A, Heath RJ, Begun J, Sassi SO, Baxt LA, Vyas JM, Goldberg MB, Xavier RJ. The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular Salmonella Typhimurium. Cell Host Microbe 2012; 12:778-90; PMID:23245322; http://dx.doi.org/ 10.1016/j.chom.2012.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ng AC, Eisenberg JM, Heath RJ, Huett A, Robinson CM, Nau GJ, Xavier RJ. Human leucine-rich repeat proteins: a genome-wide bioinformatic categorization and functional analysis in innate immunity. Proc Natl Acad Sci USA 2011; 108(Suppl 1): 4631-8; PMID:20616063; http://dx.doi.org/ 10.1073/pnas.1000093107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weterman MA, Sorrentino V, Kasher PR, Jakobs ME, van Engelen BG, Fluiter K, de Wissel MB, Sizarov A, Nürnberg G, Nürnberg P, et al. . A frameshift mutation in LRSAM1 is responsible for a dominant hereditary polyneuropathy. Hum Mol Genet 2012; 21:358-70; PMID:22012984; http://dx.doi.org/ 10.1093/hmg/ddr471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nicolaou P, Cianchetti C, Minaidou A, Marrosu G, Zamba-Papanicolaou E, Middleton L, Christodoulou K. A novel LRSAM1 mutation is associated with autosomal dominant axonal Charcot-Marie-Tooth disease. Eur J Hum Genet 2013; 21(2):190-4; PMID:22781092; http://dx.doi.org/ 10.1038/ejhg.2012.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guernsey DL, Jiang H, Bedard K, Evans SC, Ferguson M, Matsuoka M, Macgillivray C, Nightingale M, Perry S, Rideout AL, et al. . Mutation in the gene encoding ubiquitin ligase LRSAM1 in patients with Charcot-Marie-Tooth disease. PLoS Genet 2010 Aug 26; 6(8). pii: e1001081; PMID:20865121; http://dx.doi.org/ 10.1371/journal.pgen.1001081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bogdanik LP, Sleigh JN, Tian C, Samuels ME, Bedard K, Seburn KL, Burgess RW. Loss of the E3 ubiquitin ligase LRSAM1 sensitizes peripheral axons to degeneration in a mouse model of Charcot-Marie-Tooth disease. Dis Model Mech 2013; 6:780-92; PMID:23519028; http://dx.doi.org/ 10.1242/dmm.010942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Piepoli A, Palmieri O, Maglietta R, Panza A, Cattaneo E, Latiano A, Laczko E, Gentile A, Carella M, Mazzoccoli G, et al. . The expression of leucine-rich repeat gene family members in colorectal cancer. Exp Biol Med (Maywood) 2012; 237:123-8; PMID:23045723; http://dx.doi.org/ 10.1258/ebm.2012.012042 [DOI] [PubMed] [Google Scholar]
- 20. He P, Peng Z, Luo Y, Wang L, Yu P, Deng W, An Y, Shi T, Ma D. High-throughput functional screening for autophagy-related genes and identification of TM9SF1 as an autophagosome-inducing gene. Autophagy 2009; 5:52-60; PMID:19029833; http://dx.doi.org/ 10.4161/auto.5.1.7247 [DOI] [PubMed] [Google Scholar]
- 21. Li Y, Zhao Y, Hu J, Xiao J, Qu L, Wang Z, Ma D, Chen Y. A novel ER-localized transmembrane protein, EMC6, interacts with RAB5A and regulates cell autophagy. Autophagy 2013; 9:150-63; PMID:23182941; http://dx.doi.org/ 10.4161/auto.22742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhao Y, Hu J, Miao G, Qu L, Wang Z, Li G, Lv P, Ma D, Chen Y. Transmembrane protein 208: a novel ER-localized protein that regulates autophagy and ER stress. PLoS One 2013; 8:e64228; PMID:23691174; http://dx.doi.org/ 10.1371/journal.pone.0064228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reader JC, Meekins JS, Gojo I, Ning Y. A novel NUP98-PHF23 fusion resulting from a cryptic translocation t(11;17)(p15;p13) in acute myeloid leukemia. Leukemia 2007; 21(4):842-4; PMID:17287853 [DOI] [PubMed] [Google Scholar]
- 24. Reader JC, Leng Q, Rassool FV, Ning Y. Regulation of differentiation by a PHD domain in the NUP98-PHF23 fusion protein. Leuk Res 2010; 34(8):1094-7; PMID:20219246; http://dx.doi.org/ 10.1016/j.leukres.2010.02.015 [DOI] [PubMed] [Google Scholar]
- 25. Wang GG, Song J, Wang Z, Dormann HL, Casadio F, Li H, Luo JL, Patel DJ, Allis CD. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 2009; 459(7248):847-51; PMID:19430464; http://dx.doi.org/ 10.1038/nature08036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nguyen Ba AN, Pogoutse A, Provart N, Moses AM. NLStradamus: a simple Hidden Markov Model for nuclear localization signal prediction. BMC Bioinformatics 2009 Jun 29; 10:202; PMID:19563654; http://dx.doi.org/ 10.1186/1471-2105-10-202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ravikumar B, Duden R, Rubinsztein DC. Aggregateprone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 2002; 11:1107-17; PMID:11978769; http://dx.doi.org/ 10.1093/hmg/11.9.1107 [DOI] [PubMed] [Google Scholar]
- 28. Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603-14; PMID:16286508; http://dx.doi.org/ 10.1083/jcb.200507002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou C, Zhong W, Zhou J, Sheng F, Fang Z, Wei Y, Chen Y, Deng X, Xia B, Lin J. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 2012; 8:1215-26; PMID:22647982; http://dx.doi.org/ 10.4161/auto.20284 [DOI] [PubMed] [Google Scholar]
- 30. Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, et al. . A human protein-protein interaction network: a resource for annotating the proteome. Cell 2005; 122:957-68; PMID:16169070; http://dx.doi.org/ 10.1016/j.cell.2005.08.029 [DOI] [PubMed] [Google Scholar]
- 31. Musselman CA, Kutateladze TG. PHD fingers: epigenetic effectors and potential drug targets. Mol Interv 2009; 9:314-23; PMID:20048137; http://dx.doi.org/ 10.1124/mi.9.6.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Musselman CA, Kutateladze TG. Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res 2011; 39:9061-71; PMID:21813457; http://dx.doi.org/ 10.1093/nar/gkr613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. von Muhlinen N, Thurston T, Ryzhakov G, Bloor S, Randow F. NDP52, a novel autophagy receptor for ubiquitin-decorated cytosolic bacteria. Autophagy 2010; 6:288-9; PMID:20104023; http://dx.doi.org/ 10.4161/auto.6.2.11118 [DOI] [PubMed] [Google Scholar]
- 34. Moscat J, Diaz-Meco MT. Feedback on fat: p62-mTORC1-autophagy connections. Cell 2011; 147:724-7; PMID:22078874; http://dx.doi.org/ 10.1016/j.cell.2011.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mostowy S, Sancho-Shimizu V, Hamon MA, Simeone R, Brosch R, Johansen T, Cossart P. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem 2011; 286:26987-95; PMID:21646350; http://dx.doi.org/ 10.1074/jbc.M111.223610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cemma M, Kim PK, Brumell JH. The ubiquitin-binding adaptor proteins p62SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy 2011; 7:341-5; PMID:21079414; http://dx.doi.org/ 10.4161/auto.7.3.14046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moore MA, Chung KY, Plasilova M, Schuringa JJ, Shieh JH, Zhou P, Morrone G. NUP98 dysregulation in myeloid leukemogenesis. Ann N Y Acad Sci 2007; 1106:114-42; PMID:17442773; http://dx.doi.org/ 10.1196/annals.1392.019 [DOI] [PubMed] [Google Scholar]
- 38. Kubisch J, Türei D, Földvári-Nagy L, Dunai ZA, Zsákai L, Varga M, Vellai T, Csermely P, Korcsmáros T. Complex regulation of autophagy in cancer - integrated approaches to discover the networks that hold a double-edged sword. Semin Cancer Biol 2013; 23:252-61; PMID:23810837; http://dx.doi.org/ 10.1016/j.semcancer.2013.06.009 [DOI] [PubMed] [Google Scholar]
- 39. Ju JS, Miller SE, Jackson E, Cadwell K, Piwnica-Worms D, Weihl CC. Quantitation of selective autophagic protein aggregate degradation in vitro and in vivo using luciferase reporters. Autophagy 2009; 5:511-9; PMID:19305149; http://dx.doi.org/ 10.4161/auto.5.4.7761 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.