

Abstract
How autophagic degradation is linked to endosomal trafficking routes is little known. Here we screened a collection of uncharacterized Drosophila mutants affecting membrane transport to identify new genes that also have a role in autophagy. We isolated a loss of function mutant in Snap29 (Synaptosomal-associated protein 29 kDa), the gene encoding the Drosophila homolog of the human protein SNAP29 and have characterized its function in vivo. Snap29 contains 2 soluble NSF attachment protein receptor (SNARE) domains and a asparagine-proline-phenylalanine (NPF motif) at its N terminus and rescue experiments indicate that both SNARE domains are required for function, whereas the NPF motif is in part dispensable. We find that Snap29 interacts with SNARE proteins, localizes to multiple trafficking organelles, and is required for protein trafficking and for proper Golgi apparatus morphology. Developing tissue lacking Snap29 displays distinctive epithelial architecture defects and accumulates large amounts of autophagosomes, highlighting a major role of Snap29 in autophagy and secretion. Mutants for autophagy genes do not display epithelial architecture or secretion defects, suggesting that the these alterations of the Snap29 mutant are unlikely to be caused by the impairment of autophagy. In contrast, we find evidence of elevated levels of hop-Stat92E (hopscotch-signal transducer and activator of transcription protein at 92E) ligand, receptor, and associated signaling, which might underlie the epithelial defects. In summary, our findings support a role of Snap29 at key steps of membrane trafficking, and predict that signaling defects may contribute to the pathogenesis of cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma (CEDNIK), a human congenital syndrome due to loss of Snap29.
Keywords: autophagy, dome, Notch, Snap29, SNARE, trafficking, usnp
Abbreviations: Atg, autophagy-related; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; CFP, cyan fluorescent protein; dome, domeless; EM, electron microscopy; ESCRT, endosomal sorting complex required for transport; E(spl)mβ-HLH, enhancer of split mβ, helix-loop-helix; FE, follicular epithelium; histone H3, His3; hop-Stat92E, hopscotch-signal transducer and activator of transcription protein at 92E; GFP, green fluorescent protein; MENE, mutant eye no eclosion; MVB, multivesicular body; N, Notch; NECD, N extracellular domain; NPF, asparagine-proline-phenylalanine; os, outstretched; ref(2)P, refractory to sigma P; Snap29, synaptosomal-associated protein 29 kDa; SNARE, soluble NSF attachment protein receptor; Socs36E, suppressor of cytokine signaling at 36E; Syb, Synaptobrevin; Syx, syntaxin; Vamp, vesicle-associated membrane protein; V-ATPase, vacuolar H+-ATPase; Vps25, vacuolar protein sorting 25; WT, wild type
Introduction
Organ development and homeostasis require concerted regulation of membrane trafficking routes, such as those governing protein secretion and endo-lysosomal degradation, and those controlling macroautophagy (autophagy hereafter), which regulates turnover of organelles and large cytoplasmic proteins.
Studies in model organisms have clearly shown that the endo-lysosomal degradation pathway is required for correct organ development, due to its ability to promote degradation of signaling receptors controlling tissue growth and polarity (for review see ref. 1). Such a major role of endocytosis on tissue architecture is underscored by the fact that Drosophila larval imaginal discs, a recognized model of epithelial organ development, when mutant for a number of the Endosomal Sorting Complexes Required for Transport (ESCRT) genes, display loss of polarity and overactivation of major signaling pathways, including N (Notch) and hop-Stat92E.2,3 In contrast, mutants in genes controlling autophagy often do not display loss of tissue architecture, or altered signaling phenotypes, indicating that impairment of endo-lysosomal or autophagic degradation have dramatically distinct consequences on tissue development.4,5 However, it is poorly understood which regulators of trafficking are required for formation and convergence of autophagosomes into the endosomal degradation route, and their relevance to organ development and homeostasis.
In autophagy, double-membrane organelles called autophagosomes are formed by a phagophore that sequesters portions of the cell cytoplasm. Autophagosomes then fuse with lysosomes, in which the autophagosome content is degraded.6 Studies have shown that 2 ubiquitin-like conjugation systems are required for autophagosome formation,7 and a number of organelles, such as the endoplasmic reticulum (ER), mitochondria, the Golgi apparatus, endosomes, and the plasma membrane have all been suggested to supply membranes and factors for autophagosome formation.8,9 Research in yeast indicates that, once formed, the autophagosome fuses with the vacuole, the yeast lysosome, in a manner dependent on the GTPase Ypt7/Rab7, on the homotypic fusion and protein sorting (HOPS) complex, and on SNARE-mediated membrane fusion.10,11 In metazoans, fusion events between autophagosomes and endosomal compartments are more complex, entailing the formation of amphisomes, which arise from fusion of autophagosomes with the multivesicular body (MVB), a late endosomal organelle.12,13 Consistent with this difference, in Drosophila and in mammalian cells ESCRT proteins, which regulate endosomal sorting and MVB formation,2,14 and the PtdIns3P 5-kinase fab1, which control endosome function,15 are required for amphisome and autolysosome formation.16 Also, differently from yeast, when formation of late endosomes is blocked in Drosophila and mammalian cells, autophagosomes accumulate in the cytoplasm, suggesting that amphisome formation helps clearance of autophagic cargoes.17,18
The nature of SNARE-mediated fusion events occurring during formation and clearance of autophagosomes via the endo-lysosomal system is partly obscure. SNARE-mediated fusion involves a stereotypic set of SNARE proteins forming a 4-helix bundle composed by distinct SNARE domains named Qa-, Qb-, Qc- or R-SNARE. Usually, a Qa-SNARE-containing protein (a syntaxin, or t-SNARE) and a R-SNARE -containing protein (a VAMP protein, or v-SNARE) are carried by opposing membranes, and each provide a SNARE domain to the fusion complex. These proteins are glued together by Qb- and Qc- containing proteins, providing the remaining 2 SNARE domains. The Qb- and Qc-SNAREs involved in fusion events can be contributed by members of the SNAP protein family, with SNAP25 and SNAP23 being the most extensively studied.19 However, metazoan genomes also contain SNAP29, which, unlike other SNAP family members, contains a N-terminal NPF (asparagine-proline-phenylalanine) motif that binds endocytic adaptors, such as EDH1, and lacks palmitoylation sites for membrane anchoring.20,21 Consistent with this, SNAP29 resides in the cytoplasm and associates with membranes transiently.21-23 In contrast to its paralogs, SNAP29 has been much less studied and its function is unclear. In tissue culture and in in vitro studies, SNAP29 has been suggested to interact with multiple Qa-SNAREs such as syntaxins, and to associate with a number of intracellular organelles to promote—as well as inhibit—membrane fusion.21-26 Using depletion approaches, it has been shown that SNAP29 and its homolog in C. elegans and zebrafish regulates trafficking between several organelles, and that it is required for integrity of various intracellular compartments.27-30 Finally, in Drosophila and human cells, the SNAREs STX17/syntaxin 17 (Syx17) and vesicle-associated membrane protein 7 (VAMP7/Vamp7) have been very recently reported to act with SNAP29/Snap29 in fusion of autophagosomes to lysosomes.5,31 Homozygous nonsense mutations leading to truncations of the human SNAP29 protein cause CEDNIK syndrome, a rare inherited congenital condition affecting skin and nervous system development and homeostasis, and resulting in short life span.32,33 Despite the evidence above, how SNAP29 functions and how its loss results in acquisition of CEDNIK traits is currently unclear.
In this study, we used Drosophila imaginal discs to identify novel regulators of membrane trafficking that might have a role in autophagy, and to assess the importance of identified genes for epithelial organ development. With this strategy, we identified the first Drosophila null mutant in Snap29 (also referred to as CG11173/usnp). Snap29 mutant imaginal discs present impairment of a late step of autophagy. In addition, we found that Snap29 exerts an inhibitory role in membrane fusion at the apical membrane. In fact, Snap29 mutant tissue secretes autophagosomes in the apical lumen and presents excess of receptors on the plasma membrane. These defects correlate with disruption of the epithelial organization of imaginal discs and with a dramatic alteration in developmental signaling. Taken together, our data highlight a novel point of contact between trafficking and autophagy routes that is critical for organ development and might advance our understanding of the CEDNIK pathogenesis.
Results
MENE (2R) B6-21, a novel regulator of protein trafficking and autophagy
To identify novel trafficking genes that may control autophagy, we screened a collection of Drosophila MENE (mutant eye no eclosion) mutants. These mutants have been selected for their ability to cause larval lethality when made homozygous in eye-antennal imaginal discs, and most of the mapped ones disrupt membrane trafficking genes.34,35 To monitor formation and clearance of autophagosomes in WT discs (Fig. 1A), and in discs containing predominantly homozygous mutant cells (referred to as “mutant discs” henceforth; see Materials and Methods) we stained with an antibody that recognizes Drosophila refractory to sigma P (ref(2)P), a SQSTM1/p62 ortholog shown to be a selective autophagy target (Fig. 1B, marked in red).36 To monitor both secretion and endocytic degradation routes, we also stained with an antibody that recognizes N, a transmembrane receptor actively trafficked and enriched at the apical plasma membrane and in late endosomes (Fig. 1B, marked in green).2,37-39 As previously reported in WT discs, N localizes to the apical plasma membrane and in endosomal puncta, while little signal of ref(2)p is detected, consistent with low levels of constitutive autophagy (Fig. 1C). Discs mutant for genes regulating autophagy, but not protein secretion or endocytic trafficking, such as autophagy-related gene 13 (Atg13), which is required for initiation of autophagy,40 display normal morphology, and accumulate high levels of ref(2)P with no alteration of N localization, when compared to WT discs (Fig. 1D). In contrast, discs mutant for Vacuolar protein sorting 25 (Vps25), display accumulation of both N and ref(2)P as well as major organ morphology alterations (Fig. 1E), consistent with the reported role as tumor suppressor.2,16 Discs mutant for fab1, also accumulate N and ref(2)P but display no organ morphology alterations (Fig. 1F), as previously shown.15,16 Among MENE mutants, MENE (2R)-E B6-21 shows accumulation of both N and ref(2)P, as well as epithelial morphology alterations (Fig. 1G). Similar defects are observed in mutant wing discs (Fig. S1A and B). Analysis of N and ref(2)P at the subcellular level indicated key differences among MENE (2R)-E B6-21, Vps25, and fab1 mutant discs (Fig. 1H to L). Compared to WT or Atg13 mutant discs (Fig. 1H and I), in discs mutant for both Vps25 and fab1, N and ref(2)P form adjoining intracellular accumulations (Fig. 1J and K). This phenotype is consistent with previously reported functions of these genes in endosomal sorting and formation of amphisomes (Vps25), and in lysosomal maturation and fusion of amphisomes to lysosomes (fab1).16 In contrast, in MENE (2R)-E B6-21 mutant discs, N appears to accumulate apically, while ref(2)P accumulates in the apical portion below the cell cortex (Fig. 1L). These data indicate that the MENE (2R)-E B6-21 mutation affects a gene controlling protein trafficking and autophagy, at a step likely to be distinct to those regulated by Vps25 or fab1.
Figure 1.

An assay to identify novel trafficking genes involved in autophagy. (A and B) Schematic view of subapical cross-section through a third-instar larval eye-antennal imaginal discs (A) and of an epithelial cell contained in it (B). B depicts the secretion and endocytic degradation routes followed by N, and the autophagic degradation routes highlighted by ref(2)P. AM, amphisome; Ant, anterior; AP, autophagosome; EE, early endosome; GA, Golgi apparatus; LY, lysosome; NU, Nucleus; PG, phagophore; PM, plasma membrane; Post, posterior. (C to G) WT and mutant discs of the indicated genotype immunostained to detect N, ref(2)p and nuclei. A′ to E″ show the N and ref(2)P channels, respectively. Compared to WT, discs mutant for the Atg13 display strong accumulation of ref(2)P, no N accumulation and normal organ morphology. In Vps25 and fab1 mutant discs N and ref(2)P accumulate, however only in Vps25 mutant discs disc morphology is aberrant. MENE (2R)-E B6 mutant discs display epithelial architecture defects, and strong ref(2)P and N accumulation. (H to L) High magnification of a cross-section of the anterior portion of mutant discs, as in C to G. Enlargements of the boxed area and its single channels are shown below each panel. Note the distinct patterns of accumulation of N, or ref(2)P, in the different mutants.
MENE (2R)-E B6-21 is a mutant in the gene encoding Drosophila Snap29
To identify the gene mutated in MENE (2R)-E B6-21, we mapped the mutation by complementation, recombination and direct sequencing (see Materials and Methods for details) to Snap29 (Fig. 2A). Drosophila Snap29 protein shares all the features of its human homolog, with which it shares 33% identity, including the presence of an acidic NPF motif at its N terminus, of 2 SNARE domains separated by a linker region, and the absence of cysteine residues for membrane anchoring in the linker region (Fig. S2). The identified mutation introduces a premature stop codon predicted to truncate the protein between the 2 SNARE domains (Fig. 2B). Discs containing mutant cells express almost normal levels of mRNA (Fig. 2C). However, with an anti-Snap29 antibody that we generated we observed that discs containing mutant cells express a Snap29 form of predicted size for the truncated protein (Snap29B6; Fig. 2D).
Figure 2.

MENE(2R)-E B6 is a null mutant of Drosophila Snap29. (A) Schematic view of the Snap29 locus. Df(2R)egl2 (black) complements the B6-21 mutation, while Df(2R)3-659 and Df(2R)106 (red) fail to complement it, indicating that B6-21 maps to the genetic interval 60A3-A5 on the right arm of the Drosophila chromosome 2. The coding sequence of Snap29 is shown in orange, while the domains of Snap29 are indicated in yellow and blue. A black triangle marks the approximate position of the B6 mutation. (B) Sequencing of the B6 allele in heterozygosity with the parental chromosome on which the mutation was induced. A C-to-T change creates a premature stop codon that truncates the protein right after the first SNARE domain. (C) Expression of Snap29 mRNA is only 25% reduced in mutant eye-antennal and wing discs, relative to WT. (D) Analysis of Snap29 expression by protein gel blot in WT disc extracts and in extracts of discs containing Snap29B6 mutant cells indicates that Snap29B6, a truncated form of Snap29, is present in mutant cells. (E) Ubiquitous expression of CFP-Snap29 under tubulin-Gal4 (tub>) rescues lethality of homozygous Snap29 flies. Rescued flies (right) are indistinguishable from heterozygous animals (left). (F) Adult eyes of flies with the indicated genetic background. Eye-specific ectopic expression of CFP-Snap29, or of a Snap29 form with a mutated NPF motif (Snap29AAA) rescue defects of Snap29B6 mutant eye discs and yield adults with normal (CFP-Snap29), or reduced eyes (Snap29AAA). Mutant cells expressing the rescue construct give rise to orange photoreceptors, while the WT cells give rise to dark red ones. (G) Western blot of extracts from WT discs, or discs containing Snap29B6 mutant cells or discs containing Snap29B6 mutant cells and expressing CFP-Snap29, reveal expression of a truncated form of Snap29 (Snap29B6) and of CFP-Snap29 in rescued discs. The asterisks indicate unspecific signals.
To confirm that the identified mutation in Snap29 is responsible for the disc phenotype, we performed rescue experiments by ectopic expression of Snap29 in Snap29B6 mutant discs. Both Snap29B6 homozygous and Snap29B6 hemizygous animals are lethal shortly after larval hatching. In contrast, expression of a full-length CFP-tagged form of Snap29 (CFP-Snap29) under the tubulin promoter fully rescues homozygous Snap29B6 flies to adulthood (Fig. 2E). Taken together, these data indicate that impairment of Snap29 function is the cause of the phenotypes observed in Snap29B6 mutant tissue, and that Snap29B6 is likely to be a strong loss of function Snap29 allele.
To determine which domain of Snap29 is important for function, we performed rescue experiments by ectopic expression of full-length and mutated Snap29 forms in Snap29B6 mutant eye discs. Mutant forms of Snap29 do not appear to possess dominant negative activity when overexpressed at comparable levels in otherwise WT discs (Fig. S1C to G). This is the case also of Snap29SNARE2Δ, a mutant form of Snap29 that lacks the second SNARE domain and resembles the Snap29B6 protein, suggesting that Snap29B6 is most likely nonfunctional (Fig. 2D, S1C and D, S2). As expected, specific expression of CFP-Snap29 under the eyeless promoter rescues developmental defects and lethality of larvae bearing Snap29B6 homozygous mutant eye-antennal discs, and leads to eclosion of adults with eye structures indistinguishable to that of WT animals (Fig. 2F and G). In contrast, Snap29 forms lacking either the first or the second SNARE domain (Fig. S1C and D) are unable to rescue Snap29B6 mutant discs. Surprisingly, a Snap29 form with a mutated NPF motif (Snap29AAA; Fig. S1C and D) can rescue pupal lethality, yielding escaper adults with various degrees of eye defect (Fig. 2F). These data suggest that the presence of both SNARE domains is crucial for function, while the NPF motif is in part dispensable, at least upon overexpression.
Snap29 mutant tissue displays altered autophagy and Golgi apparatus organization
To understand the function of Drosophila Snap29 in epithelial tissue of the imaginal disc, we analyzed the ultrastructure of Snap29B6 mutant discs by electron microscopy (EM; Fig. 3). Compared to WT, Snap29B6 mutant tissue displays a striking accumulation of double membrane vesicular organelles (Fig. 3A and B). Accumulated double-membrane organelles present diameters ranging from 0.5 μm to 5 μm with most of them averaging 0.7 μm and contain a wide array of intact cellular structures, such as mitochondria, ER, or vesicles (Fig. 3B, arrows). By immuno-EM, we find that these organelles are positive for ref(2)p and for Atg8a, a master regulator of autophagy,41 indicating that most of these are autophagosomes (Fig. 3D, arrows).
Figure 3.

(See previous page). Snap29 mutant tissue possesses defective Golgi morphology and accumulates autophagosomes in the cytoplasm and apical lumen. (A and B) Electron micrograph of a section of eye disc tissue of the indicated genotype. A portion of the apical part of 3 to 5 epithelial cells above the level of the basal nuclei is shown. While double-membrane organelles are very rarely observed in WT cells, mutant cells are packed with them. In B, examples of double-membrane organelle containing other organelles are indicated with white arrows, while examples of organelles with a single membrane containing with vesicles or organelles in the apical lumen are indicated by white arrowheads. (C) Example of a thick 200-nm section of mutant tissue used for tomography reconstructions. Gold particles have been added for image registration. White boxes highlight a intracellular giant body containing autophagosomes, lysosomes and mitochondria (bottom box), and a similar apical extracellular structure (top box). Corresponding higher magnification images are shown in L and M and relative tomograms are presented as supplementary data. (D) ref(2)P and Atg8a label autophagosomes in Snap29B6 mutant cells by immuno-EM, as indicated by white arrows. A large double-membrane structure marked with Atg8a is also shown in the rightmost panel. (E) Example of amphisome (blue) in a WT cell close to a multilamellar organelle that could be a lysosome (purple). (F to I) Examples of autophagic organelles accumulated in mutant cells: Autophagosomes completely enclosed by a double membrane (F, light blue). Autophagosomes clustered together enclosed by folded membranes in part connected (G, light blue). The cytoplasmic space between them is shaded in red. Autophagosomes (H, light blue) close to a cluster of multilamellar organelles that could be lysosomes (H, purple). A rarely occurring amphisome (I, blue) in between 2 autophagosomes (I, light blue). (J to J″) Three representative planes of a tomographic reconstruction showing intracellular autophagosomes (one highlighted in light blue), exocytosed autophagosomes (2 highlighted in green), and one autophagosome in the process of being exocytosed (green) the lumen connecting to the apical extracellular space is highlighted in red. (K and L) Examples of large structures inside (K) and outside (L) mutant cells. Autophagosomes internal to these structures are highlighted in light blue and electrondense lysosomal material is shaded in purple. Tomograms of panels E to L are presented as supplementary data. (M and N) Examples of Golgi apparatus organization. Note the absence of stack organization in the Golgi apparatus of a mutant cell. Labels are as follows: AJ, adherens junctions; ER, endoplasmic reticulum; GA, Golgi apparatus; LU, apical lumen; MI, mitocondrium; NU, nucleus.
In WT eye disc cells autophagic structures are very rare and tomographic analysis reveals that the few found are late-stage amphisomes or autolysosomes, as judged by their single and double-limiting membrane and by their partly degraded luminal content (Fig. 3E; Movie S1). In contrast, the vast majority of the autophagosomes accumulated in Snap29B6 mutant cells are completely delimited by double membrane filled with a continuous protein-poor space. In addition, the accumulated autophagosomes in Snap29B6 mutant cells contain cytosolic structures in their lumen that are perfectly preserved, indicating that no degradation occurs in these organelles (Fig. 3F; Movie S2). The outer and inner limiting membranes of the accumulated autophagosomes is often convoluted and folded on a limited part of the organelle surface, and accumulated autophagosomes were often found close to each other (Fig. 3G; Movie S3), or close to multilamellar organelles that could be lysosomes (Fig. 3H; Movie S4). Rarely, amphisomes were found in Snap29B6 mutant cells, suggesting that fusion of autophagosomes with MVBs can still occur to a certain degree. In this case, signs of content degradation, such as broken membranes, are apparent (Fig. 3I; Movie S5).
In addition to intracellular accumulation of autophagosomes, Snap29B6 mutant cells present a prominent apical extracellular accumulation of large vesicles, possessing a single-delimiting membrane and displaying approximately the size of the autophagosomes accumulated intracellularly (Fig. 3B, arrowhead). Importantly, tomographic analysis reveals that these also can contain intact undigested cellular structures, and are often found juxtaposed to the apical membrane, or can be found at various stages of fusion with the apical plasma membrane (Fig. 3J, Movie S6), indicating that they might represent exocytosed autophagic structures.
Occasionally, we observed the presence of very large (>2 μm in diameter) cytoplasmic bodies surrounded by a double membrane containing smaller autophagosomes and other organelles (Fig. 3C and D, K; Movie S7). Similar giant structures could be also found outside of the cells apically (Fig. 3C, L; Movie S8). While these aberrant organelles could occur by multiple rounds of autophagic sequestration, the presence of apoptotic cells in Snap29B6 mutant discs (Fig. S3A and B) suggests that they could be apoptotic bodies, possibly engulfed by epithelial cells.
Finally, compared to WT cells, Snap29B6 mutant cells show a prominent disorganization of the cisternae of the Golgi apparatus (Fig. 3M and N). Tomographic reconstructions show that while in WT cells the Golgi cisternae are typically stacked (Movie S9), in Snap29B6 mutant cells the cisternae are highly perforated and their number is decreased (Movie S10). Also, while in WT cells the length of individual cisternae is uniform, in Snap29B6 mutant cells their size is highly variable (Movie S10). Taken together, these data indicate that Drosophila Snap29 is required to prevent accumulation of autophagosomes and to maintain a correct Golgi apparatus morphology.
Snap29 controls fusion of autophagosomes to degradative organelles
In agreement with our data on mutant discs, in imaginal discs containing clones of Snap29B6 mutant cells (referred to as “mosaic discs” henceforth; Materials and Methods), mutant cells also display accumulation of ref(2)P, when compared to surrounding GFP-positive WT cells (Fig. 4A and B, D). Similar ref(2)P accumulation is seen in cells mutant for Vps25 (Fig. 4C and D). As expected from failure of autophagy to clear ubiquitinated protein aggregates marked by ref(2)P, Snap29B6 mutant discs display moderate accumulation of ubiquitin, when compared to WT or Vps25 mutant discs (Fig. 4B and C, E). Accumulation of autophagosomes and ref(2)P in mutant cells could be due to a block in the autophagy flux or to induction of autophagy, or both. To distinguish between these possibilities, we tested S6k phosphorylation and expression of Atg genes, as it has been shown that, in conditions of starvation-induced autophagy, levels of phospho-S6k are low and expression of certain Atg genes is high.42,43 Compared to WT discs, mutant discs possess high levels of phospho-S6k and low Atg8a and Atg18b expression, suggesting that accumulation of autophagosomes in mutant cells is not due to induction of autophagy (Fig. 4F and G). To test whether a block in autophagosome fusion to degradative organelles is the cause of autophagosome accumulation, we analyzed the fat body, a tissue used as model to study autophagy. Fat body cells in which autophagy has been induced by starvation, when depleted of Snap29 show accumulation of ref(2)P and decreased levels of punctate structures positive for mCherry-Atg8a, a marker of autophagosomes and autolysosomes, when compared to WT cells (Fig. S3C and D). Consistent with our EM findings, these data suggest that Snap29 is required for fusion of autophagosomes with degradative endocytic organelles.
Figure 4.

Snap29 mutant cells fail to complete autophagy. (A to C) Clones of Snap29B6 (A and B) or Vps25A3 (C) mutant cells in mosaic eye-antennal discs accumulate high levels of ref(2)p and ubiquitin, compared to surrounding WT cells. (B and C) show a high magnification image of an anterior portion of an eye discs. Single ref(2)P and Ubiquitin channels are shown. (D to F) Immunoblots of protein extracts from eye-antennal discs of the indicated genotypes to detect ref(2)P (D), ubiquitin (E) and pS6k (F). Compared to protein extracts of WT discs, discs mutant for Snap29 and for the autophagy and trafficking regulator Vps25 accumulate ref(2)P, ubiquitin and pS6k. Loading controls are shown below each blot. (G) Relative expression of Atg8a or Atg18b by Q-PCR analysis of mRNA extracts from WT and mutant discs. Mutant discs do not show induction of expression of Atg genes. (H and I) A single medial confocal cross-section of the Drosophila FE of a stage 9 egg chamber. FE cells overexpressing GFP-LAMP1 are stained for Snap29 and Atg8a. H′ to H"’ show respectively the LAMP1 and Snap29, the LAMP1 and Atg8a, and the Snap29 and Atg8a merged channels. A high magnification of a typical cluster formed by GFP-LAMP1, Snap29 and Atg8a-positive vesicles is shown in (I). The 3 proteins are in close proximity.
If Snap29 regulates fusion of autophagosomes with lysosomes, it should be found associated to these organelles. To investigate localization of Snap29 in epithelial cells, we first assessed specificity of the anti-Snap29 for immunolocalization. In discs (Fig. S4A), in Schneider-2 (S2) cells (Fig. S4B and C) and in the follicular epithelium (FE) enwrapping the germline in the adult ovary (Fig. S4E), anti-Snap29 marks cytoplasmic puncta, and such signal is reduced in clones of Snap29B6 mutant cells (Fig. S4A) or upon Snap29 depletion (Fig. S4C and D), indicating that the antibody is specifically recognizing endogenous Snap29. In FE of animals expressing GFP-LAMP1, a marker of late endosomes, Snap29 puncta only rarely overlap with GFP-LAMP1 puncta (Fig. 4H). However, they are sometimes found in close proximity of clusters of Atg8a puncta surrounding GFP-LAMP1 positive structures (Fig. 4I), indicating that Snap29 could transiently associate to autophagosomes to mediate lysosomal fusion.
Snap29 is a negative regulator of autophagosome secretion
Since we found presence of organelles with the morphology of autophagosomes in the apical lumen of Snap29B6 mutant discs (Fig. 3B and C, J, L), we hypothesized that Snap29 could negatively regulate secretion at the plasma membrane. Consistent with this, we observed that secreted organelles in mutant cells are ref(2)P-and Atg8a-positive (Fig. 5A to E). Secretion of autophagosomes could be an indirect consequence of intracellular accumulation of autophagosomes. To assess whether this is the case, we tested whether secretion of autophagosomes is also present in discs mutant for Syx17 and Vamp7, which have been proposed to regulate autophagosome fusion to lysosomes together with Snap29.5,31 Surprisingly, both by immunofluorescence and EM, we find that, while Syx17 and Vamp7 mutant tissues display accumulation of autophagosomes intracellularly, they are devoid of secreted autophagosomes (Fig. 5F–I). In addition, in contrast to Snap29B6 mutant discs (Fig. 1G, L), Syx17 and Vamp7 mutant discs possess normal morphology and N localization (Fig. 5J and K). Taken together, these data indicate that autophagosome secretion is unlikely a consequence of impaired fusion of autophagosomes with lysosomes and suggest that Snap29 might negatively regulate fusion at the plasma membrane independent of Syx17 and Vamp7.
Figure 5.

Autophagosomes are secreted in Snap29 mutant discs. (A to D) Cross sections of WT and Snap29B6 mutant wing discs (A and B), or eye discs (C and D) stained as indicated. ref(2)P and Atg8a accumulate inside mutant cells and above the apical lumen of Snap29B6 mutant discs. White arrowheads highlight the apical plasma membrane of the tissue above which is the lumen between 2 epithelial folds. (E) ImmunoEM of a Snap29B6 mutant disc reveals Atg8a labeling of a large vesicle in the extracellular lumen. The white arrowheads indicate the apical plasma membrane. (F to I) Sections of eye discs mutant for Syx17 or Vamp7. ref(2)P and autophagosomes accumulate within mutant cells, but they are not present in the extracellular lumen. (J and K) Cross-sections of Syx17 and Vamp7 mutant eye discs stained as indicated. High magnifications (right panels and insets) show that unperturbed N localization and no colocalization of N with accumulated ref(2)P. Labels are as in Figure 3.
Snap29 interacts with trafficking regulators and localizes to trafficking compartments
Spurred by the requirement for Snap29 in possibly at least 2 distinct fusion processes, we assessed which SNAREs can be found in complex with Snap29. Thus, we immuno-precipitated endogenous Snap29 from S2 cell extracts and subjected the recovered complexes to mass spectrometry (Fig. 6A). In 4 experiments using 2 independently-raised polyclonal anti-Snap29 (this study and5), we repeatedly identified a set of SNARE proteins that consists of the plasma membrane Qa-SNAREs Syx1A and Syx4, of the endosomal Qa-SNARE Syx7, and of the R-SNAREs Sec22, Synaptobrevin (Syb) and Vamp7. We have also found the SNARE-specific factors Nsf2, αSnap and γSnap1 and a limited set of other proteins (Fig. 6B; Table S1). These data are consistent with large-scale protein complex identification approaches,44 and suggest that Snap29 associates with multiple SNARE complexes. Consistent with this, Snap29 depletion in the posterior half of developing wing imaginal discs leads to a reduction of adult wing tissue that is enhanced by depletion of Syx17 (Fig. S3E to G) or Vamp7 (Fig. S3H and I), or Sec22 (Fig. S3J and K). In addition, we find that, when overexpressed in the FE, the functional CFP-Snap29 localizes to the plasma membrane, partially to the Golgi apparatus and to the early endosome (Fig. 6C and D). Finally, endogenous Snap29 partially localizes with the marker of the endosomal recycling compartment Rab11 (Fig. 6E). Together, this evidence suggests that Drosophila Snap29 can associate to proteins and compartments important for secretion and recycling and degradation in endosomes.
Figure 6.

Snap29 interacts with SNARE proteins and localizes to multiple trafficking organelles. (A) Anti-Snap29 immunoblotting of Schneider-2 (S2) cell extracts immunoprecipitated with protein G only (IP G) and corresponding supernatant fraction (SN G), or with anti-yeast Mad2 as unrelated control (IP C) and corresponding supernatant fraction (SN C), or with rabbit anti-Snap29 (IP Snap29) and corresponding supernatant fraction (SN Snap29). Snap29 is efficiently immunoprecipitated only using anti-Snap29. (B) Membrane fusion proteins coimmunoprecipitated with Snap29 in at least 2 out of 4 experiments using 2 independently-raised anti-Snap29. The proportion of immunoprecipitations containing each protein is indicated. (C to E) Colocalization of CFP-Snap29 or Snap29 with markers of the indicated trafficking compartments in FE cells. Single medial confocal cross-sections are shown, with single channel below each panel. The insets show higher magnification of colocalized proteins. CFP-Snap29 localizes to the plasma membrane upon overexpression and partially colocalizes with markers of the Golgi apparatus (GM130; C), early endosomes (Syx7; D), while endogenous Snap29 partially colocalizes with recycling endosomes (GFP-Rab11; E).
Snap29 regulates N secretion and endocytic degradation
To further identify at which steps of membrane trafficking Snap29 might function, we investigated localization and trafficking of N in mosaic eye-antennal imaginal discs containing Snap29B6 mutant cells. As already observed in Snap29B6 mutant discs (Fig. 1L) in Snap29B6 mutant cells we found that N accumulates, compared to WT cells (GFP-positive; Fig. 7A to D). The site of N accumulation is most likely the plasma membrane. In fact, excess N in mutant cells does not colocalize with markers of the Golgi apparatus or with the early endosomal marker Syx7 (Fig. 7C and D). To assess whether excess N is exposed on the surface of Snap29B6 mutant cells, we labeled nonpermeabilized mosaic discs with an anti-N antibody that recognizes the N extracellular domain (anti-NECD). Compared to WT cells, localization of N at the plasma membrane in Snap29B6 mutant cells is increased and visible in punctate subdomain of the cell surface (Fig. 7E), indicating that either delivery of N to the apical plasma membrane is increased in mutant cells, or that N internalization from the surface is decreased. To distinguish between these 2 possibilities, we cultured mosaic discs for 15 or 210 min after surface labeling with anti-NECD. This makes it possible to follow internalization in early endosomes (15′) and lysosomal degradation (210′) of N.2 In both WT and mutant cells, N is able to access Syx7-positive early endosomes 15 min after labeling (Fig. 7F). In mutant cells a pool of N is still present in Syx7-negative puncta, representing either late endosomes or lysosomes, 210 min after pulse (Fig. 7G and H). As expected for both upregulation of N secretion and decrease of N degradation, protein extracts from Snap29B6 mutant discs contain more N than WT discs and about the same amount of N compared to discs mutant for Vps25, which is required for endosomal degradation of N (Fig. 7I).2 Overall, these data suggest that Snap29 is required to prevent excess N secretion to the plasma membrane, as well as to promote N degradation in a postendosomal compartment.
Figure 7.

Snap29 mutant cells display altered N trafficking. (A and B) Clones of Snap29B6 mutant cells in mosaic eye-antennal discs accumulate high levels of N, compared to surrounding WT cells. A′ and B″ are single channels. (C and D) Clones of Snap29B6 mutant cells in the anterior portion of a mosaic eye discs stained as indicated. N and GM130 do not colocalize in both WT and mutant cells, while a fraction of N colocalizes with Syx7 in both WT and mutant cells, excluding accumulation in these compartments. (C′ and D″) are single channels. (E) Labeling of nonpermeabilized Snap29B6 mosaic eye-antennal discs with an anti-N NECD. Compared to WT cells marked by expression of GFP, clones of mutant cells in the eye disc display higher N surface levels (Surf. N). E′ shows the single confocal channel for anti-N. (F and G) Z-sections of eye disc epithelia subjected to 15′ (F) and 210′ (G) internalization of anti-NECD and staining as indicated. Similar to WT cells, N is present on the apical plasma membrane of mutant cells (F, arrow) and is able to access early endosomes (F, arrowhead). N is efficiently internalized over time in both WT and mutant cells (G, arrow); however, in mutant cells it fails to be degraded and accumulates in a Syx7-negative compartment (G, arrowhead). (F′-G′) show the single confocal channel for anti-N. (H) 210′ min internalization of anti-NECD. Compared to WT cells, mutant cells display intracellular N accumulations. The actin-rich cell cortex is marked with phalloidin. (G′) shows the single confocal channel for anti-N. (I) Immunoblotting of protein extracts with an antibody recognizing the intracellular domain of N. Full-length N is approximately 300 kDa while the intracellular domain (iN) is 120 kDa. Both forms accumulated in Snap29 and Vps25 mutant discs, compared to WT. The same membrane blotted with anti-β-tubulin provides a loading control.
Snap29 controls disc development in part by downregulating hop-Stat92E signaling
Our data indicate that Snap29 controls key steps of protein trafficking and autophagy in epithelial tissue, and that loss of Snap29 compromises epithelial organ development. To determine how Snap29 regulates disc development, we first tested whether N signaling is altered in Snap29B6 mutant tissue. In fact, activation of N signaling is known to promote proliferation and differentiation of disc tissue.45 By monitoring expression of the N reporter Enhancer of split mβ, helix-loop-helix (E(spl)mβ-HLH)-lacZ, which is transcribed in response to N activation,46 we found that Snap29B6 eye discs present decreased N signaling activity, compared to WT discs (Fig. 8A to C). We then extended our analysis to a panel of signaling pathways important for development. We found that mutant discs express high levels of outstretched (os), a ligand known to activate hop-Stat92E signaling (Fig. 8C). Consistent with this, we observed high expression of the hop-Stat92E signaling reporter 10XSTAT-GFP47 in Snap29B6 mutant discs, revealing that Snap29 is required to downregulate hop-Stat92E signaling (Fig. 8D and E).
Figure 8.

Elevated hop-Stat92E signaling contributes to altered development of Snap29 mutant eye discs. (A and B) WT and Snap29 mutant eye discs expressing E(Spl)mβ-HLH-lacZ, a N signaling reporter, stained as indicated. Snap29 mutant discs display a reduction of N signaling compared to WT. A′ and B′ show single anti-β-Gal channels. (C) Expression of the indicated signaling target genes in eye disc extracts. The mRNA of a N target is decreased in Snap29 mutant eye-antennal disc extract, while the mRNA of the hop-Stat92E ligand os is greatly elevated, compared to WT. (D and E) The expression of the hop-Stat92E reporter gene 10XSTAT-GFP is increased in mutant discs compared to WT discs. D′-E′ are the single GFP channels. (F and G) WT and Snap29 mutant eye-antennal discs stained with anti-phospho-HistoneH3 (pHis3) to detect dividing cells. Compared to WT tissue in which proliferation is mostly limited to the differentiating photoreceptors along the morphogenetic furrow (arrowheads), mutant eye discs display areas containing several pH3-positive cells, suggesting that mutant cells overproliferate. The average number of pH3-positive cells in samples of the indicated genotype is indicated. P value (Wilcoxon-Mann Whitney Test) is 0.0625. F′-G′ are the single pHis3 channels. (H and I) Clones of Snap29 mutant cells in a mosaic eye discs stained as indicated. Compared to WT (GFP-positive) cells, mutant cells accumulate dome, mostly at the cell cortex. H′ and I′ shows single dome channels. (J and K) Adult eyes of flies of the indicated genotypes. Eye specific ectopic expression of Socs36E, a hop-Stat92E signaling inhibitor does not impair eye development (J). Socs36E expression in Snap29 mutant eye discs rescues in part eye development and yields adults with reduced eyes (K). In (K), mutant cells expressing the Socs36E give rise to orange photoreceptors, while the few WT cell to the dark red ones.
hop-Stat92E signaling is known to control proliferation during disc development, and ectopic signaling has been shown to contribute to aberrant architecture of mutant organs.48,49 Thus, we next assessed whether mutant discs contain more proliferating cells than controls. Proliferation in third instar eye discs from WT animals mostly occurs at the morphogenetic furrow (Fig 1A), consisting in a few rows of cells forming photoreceptors, as shown by immunolocalization of phospho-Histone H3 (pHis3)-positive cells (Fig. 8F, arrowheads). In Snap29B6 mutant eye discs, pH3-positive cells are not statistically more abundant. However, parts of the mutant tissue with altered epithelial architecture display a noticeably high number of pHis3-positive cells, suggesting that some Snap29B6 mutant cells might overproliferate (Fig. 8G).
hop-Stat92E signaling in Drosophila is transduced by dome (domeless), the only hop-Stat92E receptor encoded by the fly genome.50 Because the level of hop-Stat92E signaling is controlled by endocytic trafficking of dome, a transmembrane protein,51 we next determined whether levels of dome are altered in Snap29B6 mutant cells. In mosaic eye discs, we could detect significant accumulations of dome on the surface of Snap29B6 mutant cells, compared to surrounding WT cells (GFP-Positive; Fig. 8H and I). These data indicate that mutant discs possess higher levels of a hop-Stat92E ligand, of the hop-Stat92E receptor, and of hop-Stat92E signaling activity that could affect tissue proliferation.
Finally, to test whether elevated hop-Stat92E signaling is responsible for the phenotype of Snap29B6 mutant discs, we expressed in mutant-disc tissue Socs36E (suppressor of cytokine signaling at 36E), a negative regulator of the hop-Stat92E pathway thought to control endocytic degradation of dome.52 Surprisingly, eye disc-specific overexpression of Socs36E is sufficient to rescue the lethality of animals bearing Snap29B6 mutant eye discs. Animals rescued by increased Socs36E expression present very reduced eyes, bearing a few photoreceptors originating from mutant cells (Fig. 8J and K), indicating that reduction of hop-Stat92E signaling rescues in part the phenotype of Snap29B6 mutant discs. These data indicate that elevated hop-Stat92E signaling contributes to the developmental defects observed in Snap29B6 mutant tissue.
Discussion
Our analysis of the first Drosophila loss of function mutant in Snap29 indicates that it acts at key steps of autophagy, secretion, and endolysosomal trafficking, as summarized in the model presented in Fig. 9. The implications of our findings on our understanding of Snap29 function and its impact on the biology of developing epithelial organs is discussed in detail below.
Figure 9.
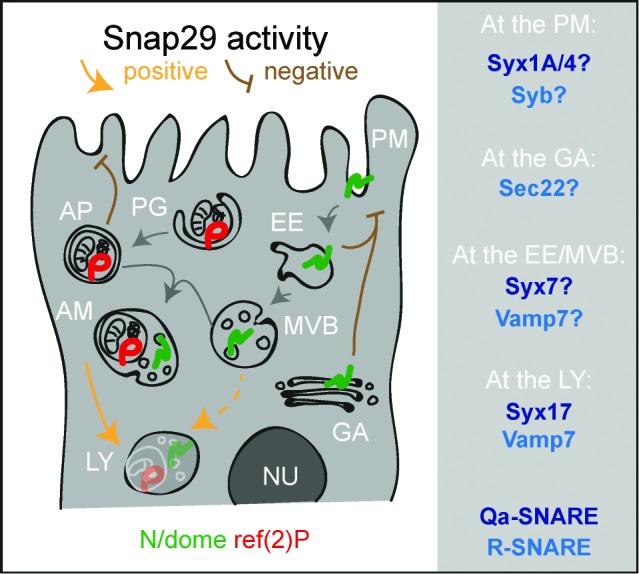
A model for Snap29 function at distinct steps of trafficking in epithelial cells. Schematic model illustrating Snap29 activity on membrane trafficking routes and organelle morphology in epithelial cells. Labels as in Figure 1. Qa- and R-SNARE likely to interact with Snap29 at distinct steps of trafficking based on our study and the literature are listed on the right. AM, amphisome; AP, autophagosome. EE, early endosome; GA, Golgi apparatus; LY, lysosome; MVB, multivesicular body; NU, nucleus; PG, phagophore; PM, plasma membrane.
The role of Snap29 in membrane fusion during autophagy in Drosophila
The identity of SNARE proteins regulating the subsequent steps of fusion required for autophagosome formation and maturation into autolysosomes is a long-standing question, on which significant progress has been reported recently.31,53-55 The SNAREs STX12/STX13, Ykt6, Vamp7, and Sec22 have been recently proposed to be required for autophagosome formation in yeast, Drosophila and mammals.53,54,56 In yeast, the SNAREs Vam3, Vam7, and Vti1 have all been suggested to control fusion of autophagosomes with vacuoles. While Vam3 and Vam7 have no clear homologs in metazoan animals, the mammalian SNAREs VAMP7, VAMP8, and VTI1B are all suggested to be involved in autophagosomal fusion events (for review see ref. 57). The Qa-SNARE protein STX17 is required for membrane fusion at 2 distinct steps of autophagy: Early autophagosome formation and fusion of autophagosomes with lysosomes to form autolysosomes.31,55 An association of Syx17 with Snap29 and the R-SNARE protein Vamp7 to form a fusion complex specific for late step of autophagy has been also very recently reported in the Drosophila fat tissue.5 Additionally, a certain degree of accumulation of autophagosomes has been observed in C. elegans depleted of Snap-29.28 Our ultrastructural analysis showing clearly accumulation of almost exclusively fully formed autophagosomes with preserved luminal content strongly favors the model that Snap29 is required with Syx17 and Vamp7 for fusion of autophagosomes with lysosomes. Consistent with this evidence, we found accumulation of autophagosomes in Syx17 and Vamp7 mutant discs and we detected a genetic interaction between Snap29 and Syx17, and Snap29 and Vamp7.
An aspect that demands further investigation is whether Snap29 acts elsewhere in the endolysosomal system. We find contrasting evidence for this. On one end, we find partial colocalization of Snap29 with the endosomal Qa-SNARE Syx7, and repeatedly find Syx7 in our immunoprecipitations. In addition, in our uptake assays, in mutant cells the endocytic cargo N accumulates in an endosomal compartment. On the other end, such compartment is Syx7 negative. Since accumulation of N in a Syx7-positive endosomes has been reported to promote ectopic N activation,2 and we have found reduced N signaling in Snap29 mutant discs, the point of N accumulation could be a postsorting compartment, such as the late endosome/MVB, or the lysosome. Despite this, we do not observe MVB accumulation in Snap29 mutant discs. These data are in sharp contrast with the accumulation of MVBs, but not of autophagosomes, that is observed in epithelial tissue mutant for vacuolar H+-ATPase (V-ATPase) subunit genes.58 Interestingly, in addition to enabling lysosomal functioning, V-ATPase have been proposed to play a role in membrane fusion and in autophagy.59,60 However, in addition to lack of accumulation of MVBs, we also find very little sign of acid-induced degradation in the autophagosomes accumulated in Snap29 mutant cells. Thus, the comparison between the EM findings in Snap29 and V-ATPase mutants58 suggests that Snap29 functions upstream of V-ATPase in autophagy and argues against a role of V-ATPase in autophagosome formation or fusion to lysosomes.
Is Snap29 a positive regulator of membrane fusion?
We observed traits in Snap29 mutant cells that could be the result of excess or inappropriate membrane fusion events, rather than of reduced fusion. These are: the large amount of membranes forming the accumulated autophagosomes; the presence in these of folded, multilamellar membranes; the secretion of autophagosomes extracellularly. It is unlikely that these events are an indirect result from the need of mutant cells to get rid of autophagic cargoes. In fact, we find no autophagosome secretion or excess membrane around autophagosomes in Syx17 and Vamp7 mutant discs. Alternatively, excess autophagosome membrane and secretion could both arise from failure to inhibit excess vesicle fusion. Inhibitory SNAREs have been postulated to occur naturally to control Golgi stack fusion patterns,61 while bacteria encode inhibitory SNAREs containing 2 SNARE domains, that can act with STX7 and VAMP8 (the homologs of Drosophila Syx7 and Vamp7) to inhibit secretion of lysosomes in mammalian cells.62 Interestingly, negative regulation of fusion by SNAP29 at the plasma membrane has been observed in rat neurons.26 A direct role of Snap29 in inhibition of membrane fusion at the plasma membrane during secretion could account also for the elevated N and dome levels on the surface of mutant cells. Consistent with this possibility, we find that Snap29 interacts with Syx1A and Syx4, 2 plasma membrane syntaxins and can localize to the plasma membrane upon overexpression. Of note, unconventional secretion routes involving autophagy regulators have been recently described,63-65 suggesting a scenario in which the autophagy and secretion functions of Snap29 could be connected to a putative negative role in fusion. The nature of Snap29 function in fusion events, and its involvement in unconventional secretion routes are currently under investigation.
The function of Snap29 in Drosophila tissue architecture and CEDNIK pathogenesis
Despite the large body of evidence on SNAP29, the pathogenesis of CEDNIK is obscure. Our genetic analysis reveals that the Drosophila Snap29B6 mutant behaves as a strong loss of function and expresses a nonfunctional Snap29 protein, a similar situation to that reported for CEDNIK.32,33 Considering the absence of mouse mutants for Snap29, our findings in Drosophila could provide an initial framework to understand the pathogenesis of CEDNIK, which starts during fetal development and affects epithelial organs.32,33 In this regard, we observed that the in vivo effect of lack of Snap29 during development in Drosophila is also epithelial tissue disorganization. This phenotype is unlikely to be due to impaired autophagy. In fact, we find that genes specifically acting during autophagy, such as Atg13, Syx17, and Vamp7 are dispensable for eye disc development. In addition, Atg7 appears dispensable for skin barrier formation in mice and flies.66,67 This evidence predicts that impairment of autophagy does not cause the developmental alterations associated to CEDNIK at least in the skin, which have been fairly well characterized.29,32,33 It is well possible that impaired autophagy plays a role in the unexplored neuronal traits of CEDNIK, considering that autophagy is a major process preventing neurodegeneration (for review see ref. 68.)
Which of the nonautophagy defects associated to lack of Snap29 could then be relevant to skin pathogenesis in CEDNIK? Could it be the defect highlighted by N accumulation in late endosomal and lysosomal compartments in our uptake experiment? We do not favor this hypothesis. In fact, we do not detect ectopic N activation, which is a feature of mutants of ESCRT genes controlling endosomal sorting.2 Such difference suggests that in Snap29 mutant cells, the pool of N accumulating intracellularly has been subjected to MVB sorting and resides in the late endosomal and lysosomal lumen. Considering also that loss of genes that control post MVB sorting events generally does not perturb disc epithelium development,15,69,70 the defect highlighted by intracellular N accumulation in Snap29 mutant cells is per se unlikely to contribute to the developmental phenotypes of Snap29 mutant organs.
Excluding routes that converge on the lysosomes, a further possibility is that the epithelial defects are due to alteration of secretory trafficking. Increased N presence at the plasma membrane, coupled with decreased N activation, could be relevant, since loss of N signaling is known to lead to epithelial alterations in skin.71 Alternatively, excess hop-Stat92E signaling could be important. In this case, excess signaling could directly originate from increased levels of active dome on the surface of Snap29 mutant cells. This scenario is consistent with the fact that Drosophila mutants preventing cargo internalization, such as those disrupting clathrin, display increased level of cargoes at the plasma membrane and possess elevated hop-Stat92E signaling and reduced N signaling.51,72 Underscoring a possible problem at the plasma membrane, expression of Socs36E, a negative regulator of hop-Stat92E signaling reported to act also by enhancing endosomal degradation of dome,52 rescues part of the epithelial defects of Snap29 mutant discs. Alternatively, elevated hop-Stat92E signaling could be a secondary effect of epithelial architecture or trafficking alterations.3,48 Detailed analysis of secretion and of signaling activity in CEDNIK samples will reveal whether alteration of these processes play a role in the pathogenesis of the syndrome.
In summary, our study clarifies the function of Snap29 in membrane trafficking and its consequences for epithelial tissue development, which might prove relevant for human health.
Materials and Methods
Fly strains, mapping, and genetics
Flies were maintained on standard yeast/cornmeal/agar media. All experiments were performed at 25°C. Mosaic eye imaginal discs were generated using yw eyFLP; ubiGFP[w+] FRT42, while mutant eye discs were generated using yw, eyFLP; cl[w+], FRT42/CyO, TwiGal4, UAS-GFP. Similar lines with different FRTs, or with UbxFLP, were used to generate mutant tissue for genes located on different chromosome arms or to generate mutant wing disc tissue, as previously described.73,74 Other alleles used were w; FRT42D, Vps25A3,2 yv; UAS Snap29 RNAi (Bloomington Drosophila Stock Center [BDSC] # 25862), Atg13Δ81,40 FRT42D, fab121,15 Syx17LL06330, Vamp7G7738, Df(2R)BSC132 and Df(3L)Exel8098 (all from5), UAS-Syx17 RNAi (Vienna Drosophila Research Center - [VDRC] #GD36596), UAS Vamp7 RNAi (VDRC #GD13317), UAS Sec22 RNAi (VDRC #GD9888), UAS Socs36E (a gift from M. Zeidler).
To identify the locus affected by B6 mutation, MENE-(2R)-E/B6 was crossed to the 2R Deficiency (Df) kit (BDSC). Such complementation mapping revealed the presence of 2 lethal mutations around 51A-B and 60A. Blind recombination of MENE-(2R)-E/B6 to w-; FRT42D revealed that the lethality leading to the MENE phenotype was the one at 60A. Recombinant #21 (B6-21) was used for further characterization. Briefly, submapping by Df complementation around 60A narrowed the candidate region to 60A3-5. Independent recombination mapping75 also showed absence of recombination with 2 viable P-elements (KG01846 [BDSC #14169] and KG04017 [BDSC #13357]) mapping to 60A3 and 60A5, confirming the presence of the mutation at 60A3-5. Direct sequencing of candidate gene exons revealed a mutation in CG11173/Snap29.
For transgenic rescue experiments we used w; FRT42, GMR-Hid/CyO, eyGal4, UASFLP. Transgenic fly lines carrying mutant tagged and mutant Snap29 forms were generated by standard techniques using the attP/attB recombination system. Over expression in follicle cells was obtained using the Cy2-Gal4 (BDSC). For colocalization experiments, we used UAS GFP-Rab11 and UAS-GFP-LAMP1 (a gift of H. Kramer). The 10XSTAT-GFP and E(spl)mβ-HLH-lacZ transgenic flies were kindly provided by E. Bach and E. Lai, respectively. Detailed genotypes for all samples in figures are shown in Table S2.
Molecular biology and bioinformatics
Sequencing of Snap29 exons was performed as previously described,2 using the following primers :
Snap29 1F 5′-GATAACTCCAGACAACAACAAAG-3′
Snap29 1R 5′-CTGGGGGTTGTAGGAGAGAG-3′;
Snap29 2F 5′-CTGGACTCTACCAACAAAAGC-3′
Snap29 2R 5′-ACCGGATGATTGTCGTAGC-3′;
Snap29 3F 5′-GCCAATAGCAACATTAACC-3′
Snap29 3R 5′-CTTAATGGCCTTGTGAAGTGC-3′.
CFP-Snap29, SNARE1Δ, SNARE2Δ and NPF>AAA inserts were cloned EcoRI XbaI into a pUASattB plasmid.
To generate the CFP-tagged Snap29, the CFP and the Snap29 coding sequences were amplified from a Snap29 cDNA vector (Drosophila Genomic Research Center) with the following primers:
CFP F EcoRI: 5′-GATCGAATTCATGGTGAGCAAGGGCGAGGA-3′
CFP R : 5′-TAGTTATGGGCCTTGTGCAGCTCGT -3′
Snap29 F: 5′-ACGAGCTGTACAAGGCCCATAACTA -3′
Snap29 R XbaI: 5′-GATCTCTAGAGCTATTCTAAGCAATG -3′
The 2 PCR products were then used as template for a second PCR using CFP F EcoRI and Snap29 R XbaI as primers.
To generate the SNARE1Δ insert 2 regions (290 bp and 532 bp) of Snap29 cDNA were amplified with the following primers:
SNARE1Δ 1F EcoRI 5′-GATCGAATTCGAAGTTTCCCTCGCC-3′
SNARE1Δ 1R 5′-CAGACCAGTCAGATGAGTTCGCTGCTCAATG-3′;
SNARE1Δ 2F 5′-ATTGAGCAGCGAACTCATCTGACTGGTCTG-3′
SNARE1Δ 2R XbaI 5′-GATCTCTAGAGCTATTCTAAGCAATG-3′.
The 2 PCR products were used as templates for a second PCR using SNARE1Δ 1F EcoRI and SNARE1Δ 2R XbaI as primers.
To generate the SNARE2Δ insert was amplified with the following primers:
SNARE2Δ F 5′-GATCGAATTCGAAGTTTCCCTCGCC-3′
SNARE2Δ R 5′-GATCTCTAGAGAATGCTCGCTTGTTCACTGGTAGGTGCTGCTG-3′.
To generate the NPF>AAA insert 2 PCR were performed using the following primers: NPF>AAA 1F EcoRI 5′- GATCGAATTCGAAGTTTCCCTCGCC-3′
NPF>AAA 1R 5′-CATCCATCTCTGCGGCTGCGGTGCTCCTC-3′,
NPF>AAA 2F 5′-GAGGAGCACCGCAGCCGCAGAGATGGATG-3′
NPF>AAA 2R XbaI 5′-GATCTCTAGAGCTATTCTAAGCAATG-3′. The 2 PCR products were used as a template for a second PCR with NPF>AAA 1F EcoRI and NPF>AAA 2R XbaI as primers.
To produce a GST-Snap29 for antibody generation, we generated an insert by PCR using the following primers:
GST BamHI 5′-GATCGGATCCGCCCATAACTACCTGC-3′
GST XhoI 5′-GATCCTCGAGGCTATTCTAAGCAATG-3′. The PCR product was inserted using BamHI and XhoI into pGEX -GST (Addgene Vector Database, 27-4597-01).
Double-stranded (ds) RNAs for intererence in S2 cells have been generated using the following primers:
T3-Snap29 5′-TAATACGACTCACTATAGGGAGA AACCCAGGAGGTGGGTAAG-3′
T7- Snap29 5′-AATTAACCCTCACTAAAGGGAGA ATGTTATCCAGCAATTCATTTTG-3′
DsRNA were in vitro transcribed with the T3 (Promega, P208C) and T7 (Promega, P207B) polymerase according to manufacturer's instructions, annealed, and incubated with the cells at a final concentration of 15ug/106 cells for 72 h.
Multiple sequence analysis was performed using ClustalX using standard parameters.
Antibody production
GST-Snap29 expression was carried out in the E. coli BL21 strain (NEB, C2530H) upon IPTG induction and gluthatione-Sepharose beads (Invitrogen, 10-1243) purification was performed under standard conditions. The purified protein was used for rabbit immunizations (Eurogentech, Liège, Belgium). Sera were affinity purified using AminoLink® Kit (Pierce Biotechnology, 44890).
Immunostainings and N trafficking analysis
Imaginal disc, ovaries, and fat bodies were fixed and stained under standard conditions. N surface staining and N trafficking assays were performed as described.2 Primary antibodies against the following antigens were used: Rabbit anti-Snap29 1:1000, rabbit anti-Syx7 1:100,76 rabbit anti-GM130 1:1000 (Abcam, ab30637), rabbit anti-ref(2)P 1:1000,36, rat anti-Atg8a 1:300,5 mouse anti-βGal 1:25 (Developmental Studies Hybridoma Bank - DSHB, 40-1a-s), mouse anti-mono- and polyubiquitinylated conjugates (FK2) 1:100 (ENZO, BML-PW8810), rabbit anti-pHis3 1:200 (Cell Signaling Technology, 3377), rabbit anti-activated decay/caspase 3 1:100 (Cell Signaling Technology, 9661), mouse anti-NECD 1:100 (DSHB, C458.2H-a), 1:100 anti-α-tubulin (AbD SeroTech, MCA78G), rabbit anti-dome 1:100 (a gift from S. Noselli), chicken anti-GFP 1:1000 (Abcam, ab13970). Appropriate Alexa Fluor 647-, Alexa Fluor 488- and Cy3-conjugated secondary antibodies were used (Jackson Immunoresearch Laboratories: Cy3 conjugate, mouse [715-165-150], rabbit [711-165-152], rat [712-165-150]; Life Technologies/Invitrogen: mouse [A-21202], rabbit [A-21206], rat [A-21203], mouse [A-31571], rabbit [A-31573], rat [A-21247]) and rhodamine-phalloidin (Sigma, P1951) were used. All images are single confocal sections taken with a confocal microscope (Leica, Heidelberg, Germany) using ×16/NA 0.5, ×40/NA 1.25, or ×63/NA 1.4 oil lenses. Images were edited with Adobe Photoshop and ImageJ and assembled with Adobe Illustrator.
Western blot, immunoprecipitation, and LC-MS/MS analysis
Third instar larva eye imaginal discs were collected, homogenized and incubated for 20 min on ice in 1 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100 (Sigma, T8787), 1% deoxycholate (Sigma, D6750) and 0.1% SDS (Sigma, L4522). Lysates were cleared by centrifugation. Supernatant fractions were recovered and quantified, separated by SDS-PAGE and transferred to nitrocellulose by standard methods. Primary antibodies used were rabbit anti-Snap29 (this study) 1:1000, anti-ref(2)P 1:1000,36 anti-ubiquitin (PD41) 1:1000 (Santa Cruz Biotechnology, sc8017), anti NICD 1:1000 (DSHB, c17.9c6-s), mouse anti-β-tubulin 1:8000 (GE Healthcare, 13-8000), anti-p6Sk (1:2000) (a gift from A. Teleman, DKFZ, Heidelberg, Germany). Secondary antibodies used were anti-rabbit and anti-mouse 1:8000 (GE Healthcare, NA934 and NXA931). Immunoblots were visualized with SuperSignal West pico/femto Chemioluminescent Substrate (Thermo Scientific, 34080-34095) using Chemidoc (Bio-Rad, Hercules, CA, USA).
For immunoprecipitations, S2 cells were cultured under standard conditions and incubated for 20 min on ice in lysis buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 10% glycerol, 5 mM EDTA, 1% Triton X-100, 15 mM MgCl2, 0.1 M Na4O7P2 pH 7.5, 1 mM PMSF (Sigma, P7626), 0.5 M Na3VO4, 0.5 M NaF and protease inhibitors (Sigma, P2714). Lysates were clarified by centrifugation and immunoprecipitated with 8μl of purified rabbit anti-Snap29, 3 μl of crude rat anti-Snap295 or with 4 μl of purified anti-yeast Mad2 antibody (a gift from A. Ciliberto, IFOM, Milan, Italy) as negative control, in combination with protein G-Sepharose. Precipitated immunocomplexes were washed and subjected to western blot analysis as previously described using the anti-Snap29 antibody.
For LC-MS/MS analysis, proteins were separated and processed as described.77 Peptides were analyzed by liquid chromatography on an Agilent 1100 LC system (Agilent Technologies, Santa Clara CA, USA) coupled to LTQ-FT ultra (Thermo Fisher Scientific, Waltham, MA, USA). Mass spectrometric data were analyzed for protein identification and presence of diglycine signature using Mascot Deamon and Proteome Discoverer 1.1 (1.1.0.263 Thermo Fisher Scientific, Waltham, MA, USA).
Reverse Transcription (RT)-PCR
RT-PCR was performed as described.78 The following primers were used: Snap29 F 5′ – AGCAGCGAACTCTGGACTCT – 3′, Snap29 R 5′ – TGTGATGTCTTCTCCAGTTGCT – 3′; E(Spl)mβ-HLH F 5′ –GAGTGCCTGACCCAGGAG – 3′ E(Spl)mβ-HLH R 5′ – CGGTCAGCTCCAGGATGT – 3′; os F 5′ –ATGGCCGAGTCCTGGCTACTGTT – 3′, os R 5′ – AACTGGATCGACTATCGCAACTTC – 3′; Atg8a F 5′ - GGGATGCATCGGAATGAA – 3′, Atg8a R 5′ – CGGTTTTCCTCAATTCGTTT – 3′; Atg18b F 5′ - AAAATACAATCACCAAAGCACAAA – 3′, Atg18b R 5′ – GTCCTGGTTGAAGTTCATTTGAT – 3′; Rpl32 F 5′ – CGGATCGATATGCTAAGCTGT – 3′, Rpl32 R 5′ – CGACGCACTCTGTTGTCG – 3′. Amplicon expression in each sample was normalized to its RpL32-RA mRNA content.
Electron microscopy and tomography
For morphological EM of Drosophila tissue, samples were prepared as previously described.79 Briefly, samples were fixed with 1% glutaraldehyde for 1 h and then with 1% reduced OsO4 for 1 h and embedded into Epoxy embedding medium (Sigma, 45359) or gelatin type A (Sigma, G8150). Then, constantly checking the position of the section plane with the help of stained semi-thin sections Epon or cryo-sections were prepared. Cryo-sections were then labeled with rabbit anti-ref(2)P antibody 1:100 or with rat anti-Atg8a antibody 1:40. The primary antibody was marked with 10 nm gold (a gift from G. Posthuma, University Medical Center, Utrecht, The Netherlands).
The analysis of chemically fixed samples by electron tomography was performed on 200-nm-thick sections, as described previously.79 Briefly, samples were tilted from +65° to –65° at 1° intervals, with a magnification of 26,500x or 40,000x. At least 5 tomograms were analyzed per each experimental condition.
Supplementary Material
Acknowledgments
We thank Paola Bellosta and Per Seglen for discussion, Marina Mapelli for performing the bioinformatic analysis, Angela Bachi and Paolo Soffientini for Mass Spec analysis, and Laurent Menut and Giuseppe Ossolengo for technical help. We also thank the Bach, Ciliberto, Neufeld, Ziegler, Juhasz, Noselli, Lai, Kramer, and Teleman labs for reagents. Finally, we are grateful to the BDSC, DSHB (Developmental Studies Hybridoma Bank), VDRC, TRiP (Transgenic RNAi Project at Harvard Medical School) and Drosophila Genomic Research Center centers for providing fly stock, cDNAs and antibodies.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Work in T.V. lab is supported by AIRC (Associazione Italiana Ricerca sul Cancro) grant #6118, and by Telethon Italia grant #GGP13225.
References
- 1. Vaccari T, Bilder D. At the crossroads of polarity, proliferation and apoptosis: the use of Drosophila to unravel the multifaceted role of endocytosis in tumor suppression. Mol Oncol 2009; 3:354-65; PMID:19560990; http://dx.doi.org/ 10.1016/j.molonc.2009.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vaccari T, Bilder D. The Drosophila tumor suppressor vps25 prevents nonautonomous overproliferation by regulating notch trafficking. Dev Cell 2005; 9:687-98; PMID:16256743; http://dx.doi.org/ 10.1016/j.devcel.2005.09.019 [DOI] [PubMed] [Google Scholar]
- 3. Woodfield SE, Graves HK, Hernandez JA, Bergmann A. De-regulation of JNK and JAKSTAT signaling in ESCRT-II mutant tissues cooperatively contributes to neoplastic tumorigenesis. PLoS One 2013; 8:e56021; PMID:23418496; http://dx.doi.org/ 10.1371/journal.pone.0056021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Juhasz G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev 2007; 21:3061-6; PMID:18056421; http://dx.doi.org/ 10.1101/gad.1600707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Takats S, Nagy P, Varga A, Pircs K, Karpati M, Varga K, Kovács AL, Hegedűs K, Juhász G. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J Cell Biol 2013; 201:531-9; PMID:23671310; http://dx.doi.org/ 10.1083/jcb.201211160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/ 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- 7. Suzuki K, Ohsumi Y. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett 2007; 581:2156-61; PMID:17382324; http://dx.doi.org/ 10.1016/j.febslet.2007.01.096 [DOI] [PubMed] [Google Scholar]
- 8. Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 2010; 12:747-57; PMID:20639872; http://dx.doi.org/ 10.1038/ncb2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol 2010; 12:831-5; PMID:20811355; http://dx.doi.org/ 10.1038/ncb0910-831 [DOI] [PubMed] [Google Scholar]
- 10. Gutierrez MG, Munafó DB, Berón W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci 2004; 117:2687-97; PMID:15138286; http://dx.doi.org/ 10.1242/jcs.01114 [DOI] [PubMed] [Google Scholar]
- 11. Jäger S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 2004; 117:4837-48; PMID:15340014; http://dx.doi.org/ 10.1242/jcs.01370 [DOI] [PubMed] [Google Scholar]
- 12. Berg TO, Fengsrud M, Strømhaug PE, Berg T, Seglen PO. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem 1998; 273:21883-92; PMID:9705327; http://dx.doi.org/ 10.1074/jbc.273.34.21883 [DOI] [PubMed] [Google Scholar]
- 13. Gordon PB, Seglen PO. Prelysosomal convergence of autophagic and endocytic pathways. Biochem Biophys Research Commun 1988; 151:40-7; PMID:3126737; http://dx.doi.org/ 10.1016/0006-291X(88)90556-6 [DOI] [PubMed] [Google Scholar]
- 14. Vaccari T, Rusten TE, Menut L, Nezis IP, Brech A, Stenmark H, Bilder D. Comparative analysis of ESCRT-I, ESCRT-II and ESCRT-III function in Drosophila by efficient isolation of ESCRT mutants. J Cell Sci 2009; 122:2413-23; PMID:19571114; http://dx.doi.org/ 10.1242/jcs.046391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rusten TE, Rodahl L, Pattni K, Englund C, Samakovlis C, Dove S, Brech A, Stenmark H. Fab1 phosphatidylinositol 3-phosphate 5-kinase controls trafficking but not silencing of endocytosed receptors. Mol Biol Cell 2006; 17:3989-4001; PMID:16837550; http://dx.doi.org/ 10.1091/mbc.E06-03-0239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rusten TE, Vaccari T, Lindmo K, Rodahl L, Nezis I, Sem-Jacobsen C, Wendler F, Vincent JP, Brech A, Bilder D, et al. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr Biol 2007; 17:1817-25; PMID:17935992; http://dx.doi.org/ 10.1016/j.cub.200709.032 [DOI] [PubMed] [Google Scholar]
- 17. Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerød L, Fisher EMC, Isaacs A, Brech A, Stenmark H, Simonsen A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 2007; 179:485-500; PMID:17984323;http://dx.doi.org/ 10.1083/jcb.200702115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Juhász G, Hill JH, Yan Y, Sass M, Baehrecke EH, Backer JM, Neufeld TP. The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J Cell Biol 2008; 181:655-66; PMID:18474623; http://dx.doi.org/ 10.1083/jcb.200712051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hong W. SNAREs and traffic. Biochim Biophys Acta 2005; 1744:120-44; PMID:15893389; http://dx.doi.org/ 10.1016/j.bbamcr.2005.03.014 [DOI] [PubMed] [Google Scholar]
- 20. Rotem-Yehudar R, Galperin E, Horowitz M. Association of insulin-like growth factor 1 receptor with EHD1 and SNAP29. J Biol Chem 2001; 276:33054-60; PMID:11423532; http://dx.doi.org/ 10.1074/jbc.M009913200 [DOI] [PubMed] [Google Scholar]
- 21. Steegmaier M, Yang B, Yoo JS, Huang B, Shen M, Yu S, Luo Y, Scheller RH. Three novel proteins of the syntaxinSNAP-25 family. J Biol Chem 1998; 273:34171-9; PMID:9852078; http://dx.doi.org/ 10.1074/jbc.273.51.34171 [DOI] [PubMed] [Google Scholar]
- 22. Wesolowski J, Caldwell V, Paumet F. A novel function for SNAP29 (synaptosomal-associated protein of 29 kDa) in mast cell phagocytosis. PLoS One 2012; 7:e49886; PMID:23185475; http://dx.doi.org/ 10.1371/journal.pone.0049886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wong SH, Xu Y, Zhang T, Griffiths G, Lowe SL, Subramaniam VN, Seow KT, Hong W. GS32, a novel Golgi SNARE of 32 kDa, interacts preferentially with syntaxin 6. Mol Biol Cell 1999; 10:119-34; PMID:9880331; http://dx.doi.org/ 10.1091/mbc.10.1.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hohenstein AC, Roche PA. SNAP-29 is a promiscuous syntaxin-binding SNARE. Biochem Biophys Res Commun 2001; 285:167-71; PMID:11444821; http://dx.doi.org/ 10.1006/bbrc.2001.5141 [DOI] [PubMed] [Google Scholar]
- 25. Schardt A, Brinkmann BG, Mitkovski M, Sereda MW, Werner HB, Nave K-A. The SNARE protein SNAP-29 interacts with the GTPase Rab3A: Implications for membrane trafficking in myelinating glia. J Neurosci Res 2009; 87:3465-79; PMID:19170188; http://dx.doi.org/ 10.1002/jnr.22005 [DOI] [PubMed] [Google Scholar]
- 26. Su Q, Mochida S, Tian JH, Mehta R, Sheng ZH. SNAP-29: a general SNARE protein that inhibits SNARE disassembly and is implicated in synaptic transmission. Proc Natl Acad Sci USA 2001; 98:14038-43; PMID:11707603; http://dx.doi.org/ 10.1073/pnas.251532398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kang J, Bai Z, Zegarek MH, Grant BD, Lee J. Essential roles of snap-29 in C. elegans. Dev Biol 2011; 355:77-88; PMID:21545795; http://dx.doi.org/ 10.1016/j.ydbio.2011.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sato M, Saegusa K, Sato K, Hara T, Harada A, Sato K. Caenorhabditis elegans SNAP-29 is required for organellar integrity of the endomembrane system and general exocytosis in intestinal epithelial cells. Mol Biol Cell 2011; 22:2579-87; PMID:21613542; http://dx.doi.org/ 10.1091/mbc.E11-04-0279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li Q, Frank M, Akiyama M, Shimizu H, Ho S-Y, Thisse C, Thisse B, Sprecher E, Uitto J. Abca12-mediated lipid transport and Snap29-dependent trafficking of lamellar granules are crucial for epidermal morphogenesis in a zebrafish model of ichthyosis. Dis Model Mech 2011; 4:777-85; PMID:21816950; http://dx.doi.org/ 10.1242/dmm.007146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rapaport D, Lugassy Y, Sprecher E, Horowitz M. Loss of SNAP29 impairs endocytic recycling and cell motility. PLoS One 2010; 5:e9759; PMID:20305790; http://dx.doi.org/ 10.1371/journal.pone.0009759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomeslysosomes. Cell 2012; 151:1256-69; PMID:23217709; http://dx.doi.org/ 10.1016/j.cell.2012.11.001 [DOI] [PubMed] [Google Scholar]
- 32. Fuchs-Telem D, Stewart H, Rapaport D, Nousbeck J, Gat A, Gini M, Lugassy Y, Emmert S, Eckl K, Hennies HC, et al. CEDNIK syndrome results from loss-of-function mutations in SNAP29. Br J Dermatol 2011; 164:610-6; PMID:21073448 [DOI] [PubMed] [Google Scholar]
- 33. Sprecher E, Ishida-Yamamoto A, Mizrahi-Koren M, Rapaport D, Goldsher D, Indelman M, Topaz O, Chefetz I, Keren H, O'brien TJ, et al. A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am J Hum Genet 2005; 77:242-51; PMID:15968592; http://dx.doi.org/ 10.1086/432556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vaccari T, Rusten TE, Menut L, Nezis IP, Brech A, Stenmark H, Bilder D. Comparative analysis of ESCRT-I, ESCRT-II and ESCRT-III function in Drosophila by efficient isolation of ESCRT mutants. J Cell Sci 2009; 122:2413-23; PMID:19571114; http://dx.doi.org/ 10.1242/jcs.046391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Menut L, Vaccari T, Dionne H, Hill J, Wu G, Bilder D. A mosaic genetic screen for Drosophila neoplastic tumor suppressor genes based on defective pupation. Genetics 2007; 177:1667-77; PMID:17947427; http://dx.doi.org/ 10.1534/genetics.107.078360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nezis IP, Simonsen A, Sagona AP, Finley K, Gaumer S, Contamine D, Rusten TE, Stenmark H, Brech A. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol 2008; 180:1065-71; PMID:18347073; http://dx.doi.org/ 10.1083/jcb.200711108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilkin M, Tongngok P, Gensch N, Clemence S, Motoki M, Yamada K, Hori K, Taniguchi-Kanai M, Franklin E, Matsuno K, et al. Drosophila HOPS and AP-3 complex genes are required for a Deltex-regulated activation of notch in the endosomal trafficking pathway. Dev Cell 2008; 15:762-72; PMID:19000840; http://dx.doi.org/ 10.1016/j.devcel.2008.09.002 [DOI] [PubMed] [Google Scholar]
- 38. Sakata T, Sakaguchi H, Tsuda L, Higashitani A, Aigaki T, Matsuno K, Hayashi S. Drosophila Nedd4 regulates endocytosis of notch and suppresses its ligand-independent activation. Curr Biol 2004; 14:2228-36; PMID:15620649; http://dx.doi.org/ 10.1016/j.cub.2004.12.028 [DOI] [PubMed] [Google Scholar]
- 39. Vaccari T, Lu H, Kanwar R, Fortini ME, Bilder D. Endosomal entry regulates Notch receptor activation in Drosophila melanogaster. J Cell Biol 2008; 180:755-62; PMID:18299346; http://dx.doi.org/ 10.1083/jcb.200708127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chang Y-Y, Neufeld TP. An Atg1Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol Biol Cell 2009; 20:2004-14; PMID:19225150; http://dx.doi.org/ 10.1091/mbc.E08-12-1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2001; 2:211-6; PMID:11265251; http://dx.doi.org/ 10.1038/35056522 [DOI] [PubMed] [Google Scholar]
- 42. Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell 2004; 7:167-78; PMID:15296714; http://dx.doi.org/ 10.1016/j.devcel.2004.07.009 [DOI] [PubMed] [Google Scholar]
- 43. Wu H, Wang MC, Bohmann D. JNK protects Drosophila from oxidative stress by trancriptionally activating autophagy. Mech Dev 2009; 126:624-37; PMID:19540338; http://dx.doi.org/ 10.1016/j.mod.2009.06.1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guruharsha KG, Rual J-F, Zhai B, Mintseris J, Vaidya P, Vaidya N, Beekman C, Wong C, Rhee DY, Cenaj O, et al. A protein complex network of Drosophila melanogaster. Cell 2011; 147:690-703; PMID:22036573; http://dx.doi.org/ 10.1016/j.cell.2011.08.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Artavanis-Tsakonas S, Rand M, Lake R. Notch signaling: cell fate control and signal integration in development. Science 1999; 284:770-6; PMID:10221902; http://dx.doi.org/ 10.1126/science.284.5415.770 [DOI] [PubMed] [Google Scholar]
- 46. Nellesen D, Lai E, Posakony J. Discrete enhancer elements mediate selective responsiveness of enhancer of split complex genes to common transcriptional activators. Dev Biol 1999; 213:33-53; PMID:10452845; http://dx.doi.org/ 10.1006/dbio.1999.9324 [DOI] [PubMed] [Google Scholar]
- 47. Bach E, Vincent S, Zeidler M, Perrimon N. A sensitized genetic screen to identify novel regulators and components of the Drosophila janus kinasesignal transducer and activator of transcription pathway. Genetics 2003; 165:1149-66; PMID:14668372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu M, Pastor-Pareja JC, Xu T. Interaction between RasV12 and scribbled clones induces tumour growth and invasion. Nature 2011:1-5; PMID:20072127; http://dx.doi.org/ 10.1038/nature08702 [DOI] [PubMed] [Google Scholar]
- 49. Dominguez M, Ferres-Marco D, Gutierrez-Avino F, Speicher S, Beneyto M. Growth and specification of the eye are controlled independently by Eyegone and Eyeless in Drosophila melanogaster. Nat Genet 2004; 36:31-9; PMID:14702038; http://dx.doi.org/ 10.1038/ng1281 [DOI] [PubMed] [Google Scholar]
- 50. Brown S, Hu N, Hombría JC. Identification of the first invertebrate interleukin JAKSTAT receptor, the Drosophila gene domeless. Curr Biol 2001; 11:1700-5; PMID:11696329; http://dx.doi.org/ 10.1016/S0960-9822(01)00524-3 [DOI] [PubMed] [Google Scholar]
- 51. Vidal OM, Stec W, Bausek N, Smythe E, Zeidler MP. Negative regulation of Drosophila JAK-STAT signalling by endocytic trafficking. J Cell Sci 2010; 123:3457-66; PMID:20841381; http://dx.doi.org/ 10.1242/jcs.066902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stec W, Vidal O, Zeidler MP. Drosophila SOCS36E negatively regulates JAKSTAT pathway signaling via two separable mechanisms. Mol Biol Cell 2013; 24:3000-9; PMID:23885117; http://dx.doi.org/ 10.1091/mbc.E13-05-0275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen W-L, Griffith J, Nag S, Wang K, Moss T, et al. SNARE proteins are required for macroautophagy. Cell 2011; 146:290-302; PMID:21784249; http://dx.doi.org/ 10.1016/j.cell.2011.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moreau K, Ravikumar B, Renna M, Puri C, Rubinsztein DC. Autophagosome precursor maturation requires homotypic fusion. Cell 2011; 146:303-17; PMID:21784250; http://dx.doi.org/ 10.1016/j.cell.2011.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013; 495:389-93; PMID:23455425; http://dx.doi.org/ 10.1038/nature11910 [DOI] [PubMed] [Google Scholar]
- 56. Lu Y, Zhang Z, Sun D, Sweeney ST, Gao F-B. Syntaxin 13, a genetic modifier of Mutant CHMP2B in frontotemporal dementia, is required for autophagosome maturation. Mol Cell 2013; 52:264-71; PMID:24095276; http://dx.doi.org/ 10.1016/j.molcel.2013.08.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Moreau K, Renna M, Rubinsztein DC. Connections between SNAREs and autophagy. Trends Biochem Sci 2013; 38:57-63; PMID:23306003; http://dx.doi.org/ 10.1016/j.tibs.2012.11.004 [DOI] [PubMed] [Google Scholar]
- 58. Vaccari T, Duchi S, Cortese K, Tacchetti C, Bilder D. The vacuolar ATPase is required for physiological as well as pathological activation of the Notch receptor. Development 2010; 137:1825-32; PMID:20460366; http://dx.doi.org/ 10.1242/dev.045484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mijaljica D, Prescott M, Devenish RJ. V-ATPase engagement in autophagic processes. Autophagy 2011; 7:666-8; PMID:21494095; http://dx.doi.org/ 10.4161/auto.7.6.15812 [DOI] [PubMed] [Google Scholar]
- 60. Strasser B, Iwaszkiewicz J, Michielin O, Mayer A. The V-ATPase proteolipid cylinder promotes the lipid-mixing stage of SNARE-dependent fusion of yeast vacuoles. EMBO J 2011; 30:4126-41; PMID:21934648; http://dx.doi.org/ 10.1038/emboj.2011.335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Varlamov O. i-SNAREs: inhibitory SNAREs that fine-tune the specificity of membrane fusion. J Cell Biol 2004; 164:79-88; PMID:14699088; http://dx.doi.org/ 10.1083/jcb.200307066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Paumet F, Wesolowski J, Garcia-Diaz A, Delevoye C, Aulner N, Shuman HA, Subtil A, Rothman JE. Intracellular bacteria encode inhibitory SNARE-like proteins. PLoS Oe 2009; 4:e7375; PMID:19823575; http://dx.doi.org/ 10.1371/journal.pone.0007375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bruns C, McCaffery JM, Curwin AJ, Duran JM, Malhotra V. Biogenesis of a novel compartment for autophagosome-mediated unconventional protein secretion. J Cell Biol 2011; 195:979-92; PMID:22144692; http://dx.doi.org/ 10.1083/jcb.201106098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Duran JM, Anjard C, Stefan C, Loomis WF, Malhotra V. Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol 2010; 188:527-36; PMID:20156967; http://dx.doi.org/ 10.1083/jcb.200911154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Manjithaya R, Anjard C, Loomis WF, Subramani S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol 2010; 188:537-46; PMID:20156962; http://dx.doi.org/ 10.1083/jcb.200911149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rossiter H, König U, Barresi C, Buchberger M, Ghannadan M, Zhang C-F, Mlitz V, Gmeiner R, Sukseree S, Födinger D, et al. Epidermal keratinocytes form a functional skin barrier in the absence of Atg7 dependent autophagy. J Dermatol Sci 2013; 71:67-75; PMID:23669018; http://dx.doi.org/ 10.1016/j.jdermsci.2013.04.015 [DOI] [PubMed] [Google Scholar]
- 67. Scherfer C, Han VC, Wang Y, Anderson AE, Galko MJ. Autophagy drives epidermal deterioration in a Drosophila model of tissue aging. Aging 2013; 5:276-87; PMID:23599123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jiang P, Mizushima N. Autophagy and human diseases. Cell Research 2014; 24:69-79; PMID:24323045; http://dx.doi.org/ 10.1038/cr.2013.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Akbar MA, Ray S, Kramer H. The SM protein CarVps33A regulates SNARE-mediated trafficking to lysosomes and lysosome-related organelles. Mol Biol Cell 2009; 20:1705-14; PMID:19158398; http://dx.doi.org/ 10.1091/mbc.E08-03-0282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sevrioukov E, He J, Moghrabi N, Sunio A, Kramer H. A role for the deep orange and carnation eye color genes in lysosomal delivery in Drosophila. Mol Cell 1999; 4:479-86; PMID:10549280; http://dx.doi.org/ 10.1016/S1097-2765(00)80199-9 [DOI] [PubMed] [Google Scholar]
- 71. Hu B, Castillo E, Harewood L, Ostano P, Reymond A, Dummer R, Raffoul W, Hoetzenecker W, Hofbauer GF, Dotto GP. Multifocal epithelial tumors and field cancerization from loss of mesenchymal CSL signaling. Cell 2012; 149:1207-20; PMID:22682244; http://dx.doi.org/ 10.1016/j.cell.2012.03.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Windler SL, Bilder D. Endocytic internalization routes required for DeltaNotch signaling. Curr Biol 2010; 20:538-43; PMID:20226669; http://dx.doi.org/ 10.1016/j.cub.2010.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tapon N, Ito N, Dickson BJ, Treisman J, Hariharan I. The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell 2001; 105:345-55; PMID:11348591; http://dx.doi.org/ 10.1016/S0092-8674(01)00332-4 [DOI] [PubMed] [Google Scholar]
- 74. Newsome T, Asling B, Dickson BJ. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development 2000; 127:851-60; PMID:10648243 [DOI] [PubMed] [Google Scholar]
- 75. Zhai RG, Hiesinger PR, Koh T-W, Verstreken P, Schulze KL, Cao Y, Jafar-Nejad H, Norga KK, Pan H, Bayat V, et al. Mapping Drosophila mutations with molecularly defined P element insertions. Proc Natl Acad Sci USA 2003; 100:10860-5; PMID:12960394; http://dx.doi.org/ 10.1073/pnas.1832753100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lu H, Bilder D. Endocytic control of epithelial polarity and proliferation in Drosophila. Nat Cell Biol 2005; 7:1132-9; PMID:16258546; http://dx.doi.org/ 10.1038/ncb1324 [DOI] [PubMed] [Google Scholar]
- 77. Rappsilber J, Ishihama Y, Mann M. Stop and go extraction tips for matrix-assisted laser desorptionionization, nanoelectrospray, and LCMS sample pretreatment in proteomics. Anal Chem 2003; 75:663-70; PMID:12585499; http://dx.doi.org/ 10.1021/ac026117i [DOI] [PubMed] [Google Scholar]
- 78. Petzoldt AG, Gleixner EM, Fumagalli A, Vaccari T, Simons M. Elevated expression of the V-ATPase C subunit triggers JNK-dependent cell invasion and overgrowth in a Drosophila epithelium. Dis Mod Mech 2013; 6:689-700; PMID:23335205; http://dx.doi.org/ 10.1242/dmm.010660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Beznoussenko GV, Dolgikh VV, Seliverstova EV, Semenov PB, Tokarev YS, Trucco A, Micaroni M, Di Giandomenico D, Auinger P, Senderskiy IV, et al. Analogs of the Golgi complex in microsporidia: structure and avesicular mechanisms of function. J Cell Sci 2007; 120:1288-98; PMID:17356068; http://dx.doi.org/ 10.1242/jcs.03402 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.