

Abstract
Autophagy has been implicated in the progression and chemoresistance of various cancers. In this study, we have shown that osteosarcoma Saos-2 cells lacking ATG4B, a cysteine proteinase that activates LC3B, are defective in autophagy and fail to form tumors in mouse models. By combining in silico docking with in vitro and cell-based assays, we identified small compounds that suppressed starvation-induced protein degradation, LC3B lipidation, and formation of autophagic vacuoles. NSC185058 effectively inhibited ATG4B activity in vitro and in cells while having no effect on MTOR and PtdIns3K activities. In addition, this ATG4B antagonist had a negative impact on the development of Saos-2 osteosarcoma tumors in vivo. We concluded that tumor suppression was due to a reduction in ATG4B activity, since we found autophagy suppressed within treated tumors and the compound had no effects on oncogenic protein kinases. Our findings demonstrate that ATG4B is a suitable anti-autophagy target and a promising therapeutic target to treat osteosarcoma.
Keywords: antiautophagy compounds, ATG4B, LC3B, in silico docking, osteosarcoma, and xenografts
Abbreviations: 3MA, 3-methyladenine; ACTB, actin, beta; ATG, autophagy-related; ATG4B, autophagy-related 4B, cysteine protease; AV, autophagic vacuole; BECN1, beclin 1, autophagy related; CMPase, cytidine monophosphatase; dNGLUC, Gaussia luciferase; DMEM, Dulbecco's modified Eagle medium; ECL, enhanced chemiluminescence; FYVE, zinc-finger domain named after 4 cysteine-rich proteins: FAB1, YOTB, VAC1, and EEA1; GABARAPL2, GABA(A) receptor-associated protein-like 2; GFP, green fluorescent protein; GST, glutathione S-transferase; IC50, half maximal inhibitory concentration; HRP, horseradish peroxidase; IP, intraperitoneal; MAP1LC3B/LC3B, microtubule-associated protein 1 light chain 3beta; MP, melting point; MTOR, mechanistic target of rapamycin; NCI, National Cancer Institute; NMR, nuclear magnetic resonance; PLA2, phospholipase A2; PtdIns3K, phosphatidylinositol 3-kinase class III; PtdIns3P, phosphatidylinositol 3-phosphate; PVDF, polyvinylidene difluoride; RFP, red fluorescent protein; RLU, relative luciferase units; RPS6, ribosomal protein S6; RPS6KB1, ribosomal protein S6 kinase, 70kDa, polypeptide 1; SEM, standard error of the mean; ULK1/2, unc-51-like autophagy activating kinase 1/2
Introduction
Autophagy is a universal process whereby cellular components and damaged organelles are sequestered within autophagosomes for lysosomal degradation. Autophagy has proven to be an essential pathway for cellular homeostasis. In addition to removing dysfunctional proteins and organelles, autophagy provides amino acids, monosaccharides, nucleic acids, and lipids during times of nutrient deprivation.1-3 Autophagy is a key pathway for cell survival but, if protein loss becomes excessive, cell death will result. This degradative pathway has been implicated in the progression of a number of diseased states including cancer. Suppressed autophagy can result in net protein gain and neoplastic growth, and defects in autophagy have been implicated in poor outcomes for hepatocellular carcinoma.4 To the contrary, autophagy promotes cell survival in tumors undergoing nutrient deprivation or chemotherapy. The overproduction of the autophagy protein, LC3B (microtubule-associated protein 1 light chain 3B), is associated with tumor growth and poor prognosis in aggressive pancreatic, colorectal, and breast carcinomas.5-7
During tumor development, autophagy is enhanced to promote cell survival under ischemic conditions.8-10 Autophagy can also enhance cell survival by removing organelles damaged by chemotherapy agents.9,11,12 Resistance of osteosarcoma cell lines to doxorubicin, cisplatin, and methotrexate has been shown to be due to the induction of autophagy by the DNA-binding protein HMGB1 (high mobility group box 1).13 On the other hand, autophagy is one of 3 primary venues of cell death, which also includes apoptosis and necrosis. Many existing chemotherapy drugs act by inducing apoptosis while others promote autophagy-mediated cell death of neoplastic cells.14,15 Given that autophagy can promote cell survival or cell death, its regulation is critical for the developing tumor.
There are 2 primary regulatory pathways of autophagy: MTOR (mechanistic target of rapamycin), a negative regulator, and PtdIns3K (class III phosphatidylinositol 3-kinase), a positive regulator. MTOR inhibits the ULK1/2 (mammalian orthologs of yeast Atg1) complex, which activates autophagy by stimulating PtdIns3K activity.16 The MTOR inhibitor, rapamycin, induces autophagy-mediated cell death in glioma cells.17 PtdIns3K synthesizes phosphatidylinositol 3-phosphate, which provides a docking site for ATG proteins at the sequestering membranes of the forming autophagosome.18,19 Chemoresistance is attenuated in hepatocarcinoma cells when treated with the PtdIns3K antagonist, 3MA (3-methyladenine).20 Both pathways modulate the lipidation of LC3B by presumably regulating the activities of ATG4, ATG7, or ATG3. Of the 4 autophagins (ATG4A, ATG4B, ATG4C, and ATG4D) identified, Yin and coworkers have shown that ATG4B had the highest catalytic efficiency for cleaving the C terminus of LC3B.21 Once the C-terminal glycine of LC3B is exposed by ATG4B, ATG7 in an ATP-dependent manner activates LC3B for delivery to ATG3, which conjugates LC3B to phosphatidylethanolamine. The lipidation of LC3B anchors this protein to the forming autophagosome where it promotes membrane expansion to enlarge the autophagosome thus increasing the amplitude of autophagy.22 The lipidated LC3B is either degraded within the autolysosome or cleaved by ATG4B and the LC3B recycled. ATG4B provides the cell with enough LC3B to amplify autophagy and recycles the lipidated LC3B to sustain autophagy.23
Studies suggest that antiautophagy compounds may prove effective in suppressing tumor growth and countering tumor resistance to chemotherapies.14,15,24 Chloroquine and its derivatives, which suppress autophagy by inhibiting lysosomal degradation, are currently being used in 33 clinical trials as listed on the NCI (National Cancer Institute) website to treat lung, pancreas, colorectal, renal, prostate, skin, and breast cancers. However, Maycotte et al. have recently shown that the anticancer effects of chloroquine may be independent of autophagy inhibition.25 In addition, such antilysosomal agents can ultimately alter cellular metabolism possibly leading to cell death and unfavorable side effects. In this study, we have utilized genetic and pharmaceutical means to show that ATG4B is essential for autophagy and a potential drug target for the treatment of osteosarcoma. We identified NSC185058 by in silico docking small compounds from the NCI library to the active site of ATG4B. We have shown that NSC185058 inhibits ATG4B, the lipidation of LC3B, and autophagy without affecting the MTOR or PtdIns3K pathways. Furthermore, we have demonstrated that NSC185058 is effective in suppressing autophagy in vivo and in attenuating the growth of osteosarcoma tumors. These studies substantiate ATG4B as a target and NSC185058 as an ATG4B antagonist to treat bone cancer.
Results
ATG4B is essential for starvation-induced autophagy
ATG4B, a cysteine protease, activates LC3B for lipidation and recycles lipidated LC3B to form free LC3B.23,26 LC3B does not appear to be essential for the formation of the autophagic vacuole, but does affect the size of these vacuoles and their transport along microtubules.23,27,28 ATG4B and LC3B are present in many cell lines isolated from kidney, bone, breast, liver, and colorectal cancers (Fig. S1A). In order to establish the role of ATG4B in autophagy, we utilized shRNA to knock down ATG4B. ATG4B expression was diminished by 70% in shATG4B-Saos-2 cells compared with shCon-Saos-2 cells (Fig. S1B). The autophagy response under amino acid and serum-starved conditions was assessed by GFP-LC3B localization, protein degradation, and visualization of autophagic vacuoles (AVs) by electron microscopy. Under fed conditions (Fig. 1A and C), GFP-LC3B expressed in shCon-Saos-2 and shATG4B-Saos-2 cells was found distributed throughout the cytoplasm and nucleus. In starved shCon-Saos-2 cells, GFP-LC3B was localized to structures distributed throughout the cell (Fig. 1B, arrows). This unique punctate pattern suggests the localization of GFP-LC3B to the forming AVs and is consistent with the activation of autophagy. The typical autophagy-related punctate pattern of GFP-LC3B was absent in starved cells lacking ATG4B (Fig. 1D). This is consistent with the findings of Yin and coworkers who report that ATG4B is the primary activator of LC3B.21 Next, we found that the rates of protein degradation were significantly reduced under both fed and starved conditions in shATG4B-Saos-2 cells (Fig. 1E). The absence of AVs in starved shATG4B-Saos-2 cells was substantiated by electron microscopy (Fig. 1G and H). The fractional volume of AVs (autophagosomes and autolysosomes) was substantially decreased in starved shATG4B-Saos-2 cells compared with shCon-Saos-2 (Fig. 1F). The data suggest that ATG4B is essential for the autophagy response in osteosarcoma Saos-2 cells.
Figure 1.

Starvation-induced autophagy requires ATG4B. Saos-2 cell lines stably expressing GFP-LC3B were treated with lentiviral nonspecific “scrambled” shRNA (shCon) or ATG4B-directed shRNA (shATG4B). Stable lines expressing shCon-Saos (A and B) and shATG4B-Saos-2 (C and D) were incubated in medium enriched for amino acids and serum (A and C) or starved for amino acids and serum (B and D) for 4 h and GFP-LC3B visualized by fluorescence microscopy. GFP-LC3B labeled AVs were present in starved cells that contained ATG4B, but absent from cells lacking ATG4B. Scale bar (A–D): 10 μm. (E) Protein degradation in shCon-Saos-2 and shATG4B-Saos-2 cells was measured under fed and starved conditions as described in Materials and Methods. (F–H) shCon-Saos-2 and shATG4B-Saos-2 cells were incubated under fed and starved (G and H) conditions and the fractional volumes of AVs quantified from electron micrographs randomly selected from 3 independent experiments using morphometric methods described in Materials and Methods (F). Autophagic vacuoles (arrows) were identified by their pleomorphic structure and heterogeneous and/or CMPase (electron dense reaction product) content. The insets contain higher magnifications of representative AVs. Scale bar (G–H and insets): 1 μm. The values represent the mean ± SEM *P < 0.05; ***P < 0.001.
We also examined the autophagy response in MDA-MB468 cells lacking ATG4B. This was done by visualizing the changes in the cellular distribution of GFP-LC3B stably expressed in these breast cancer cells. In fed cells with or without ATG4B, GFP-LC3B is present throughout the cytosol. On the other hand, numerous GFP-LC3B labeled AVs were present in amino acid starved shCon-MDA-MB468 cells containing ATG4B, but absent in shATG4B cells (Fig. S4A). These findings show that ATG4B is essential for autophagy in these breast cancer cells.
ATG4B has a positive impact on tumor growth of Saos-2 cells
Growing tumors survive nutrient limited conditions by activating autophagy.8 We tested the ability of autophagy-defective shATG4B-Saos-2 cells to survive conditions of amino acid starvation. Saos-2 cells lacking ATG4B were more sensitive to amino acid starvation than cells containing ATG4B (Fig. 2A). We next evaluated the role of ATG4B in the growth of osteosarcoma tumors. shCon-Saos and shATG4B-Saos-2 were injected into a single subcutaneous site and tumor growth monitored over time (Fig. 2B). Autophagy-competent shCon-Saos-2 cells formed tumors, while tumor growth was attenuated using autophagy defective shATG4B-Saos-2 cells. Our findings suggest that ATG4B has a positive impact on cell growth during the formation of osteosarcoma tumors.
Figure 2.
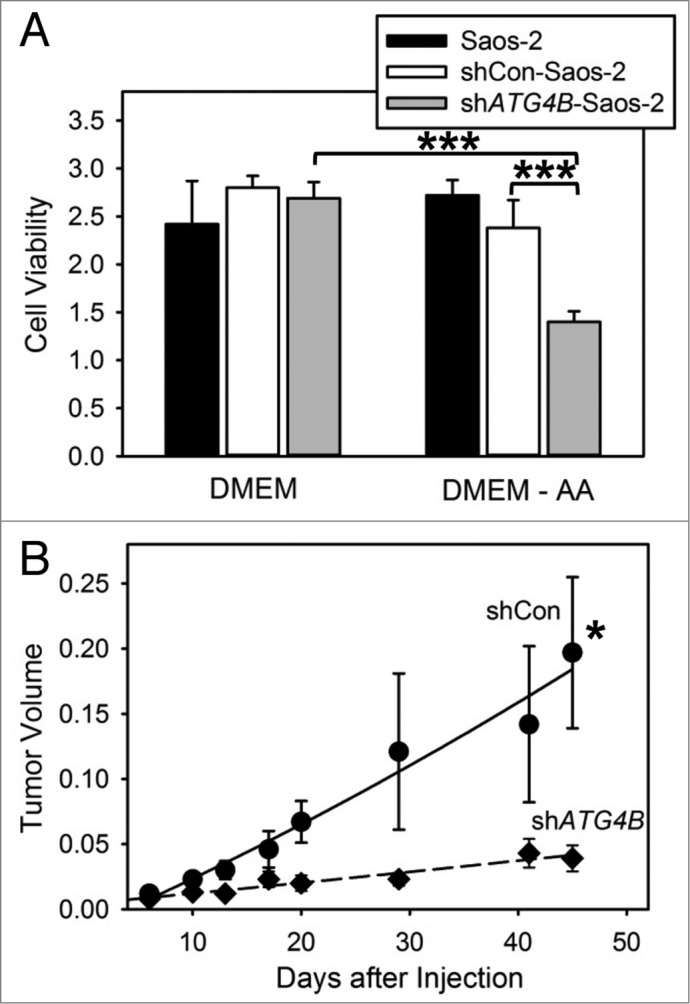
ATG4B is essential for cell survival under amino acid starved conditions and osteosarcoma tumor growth. (A) Saos-2, shCon-Saos-2, and shATG4B-Saos-2 cells were incubated in DMEM-based medium with or without amino acids. After 48 h, cell viability was quantified by MTT-based assays. The values represent the mean ± SEM (n = 8). ***P < 0.001 (B) Two groups of 5 immunodeficient nu/nu female mice were injected subcutaneously with 6 × 106 shCon-Saos-2 or shATG4B-Saos-2 cells and the tumor dimensions measure over time. The volumes (cm3) represent the mean ± SEM (n = 5). The statistical difference is based on the data points that generated the trend line. *P < 0.05.
Computer docking of compounds to the active site of ATG4B
Our data suggest that ATG4B would be an excellent drug target to inhibit autophagy, to suppress cell survival under nutrient minimal conditions, and to inhibit tumor growth. Therefore, with the crystal structure of human ATG4B established and the active site mapped, we performed in silico compound docking combined with cell-based assays to identify putative small molecule inhibitors of ATG4B and autophagy.29 Docking was done using the NCI database of 139,735 compounds that followed the Lipinski rules and fit into the active pocket identified by spheres (Fig. S2A). These compounds were scored based on their potential to utilize hydrophilic and hydrophobic binding interactions within the pocket that contained histidine 280 and aspartic acid 278, which are required for the proteolytic activity of ATG4B.29 Compounds with the highest predicted affinity for this pocket (Table S1) were obtained from the NCI and screened for their efficacy to suppress autophagy using 2 cellular models: Saos-2 osteosarcoma and MDA-MB468 breast cancer.
Effects of small compounds on autophagy
Saos-2 and MDA-MB468 cells stably expressing GFP-LC3B were used to screen the inhibitory effects of these compounds on starvation-induced autophagy. In about 80% of the fed cells, GFP-LC3B was localized in a diffuse pattern throughout the cytosol (Figs. 3A; Fig. S3A). Cells deprived of amino acids and serum showed a punctate distribution of GFP-LC3B in almost 80% of the Saos-2 cells (Figs. 3B; Fig. S3A) and 70% of the MDA-MB468 cells (Fig. S3A) consistent with ongoing autophagy. Next, we evaluated the effects of small compounds on this GFP-LC3B labeling pattern in starved Saos-2 and MDA-MB468 cells. The anti-autophagy effects of 6 representative compounds that docked at the active site of ATG4B (Fig. S2B) are presented in Figure S3A. We showed that NSC52086 had no effect on starvation-induced autophagy in either cell line, and NSC24666, NSC128761, and NSC310196 inhibited autophagy only in MDA-MB468 cells. Of all the compounds tested only NSC185058 (Fig. 3C) and NSC377071 effectively suppressed starvation-induced autophagy in both cell types (Fig. S3A). These compounds were further shown to suppress autophagy in a concentration-dependent manner (Fig. 3D). The inhibitory effects of these compounds on autophagy were substantiated by measuring the rates of protein degradation (Fig. S3B). Amino acid deprivation has been shown to activate autophagy-mediated degradation of long-lived cellular proteins. Protein degradation was enhanced 2-fold in starved Saos-2 cells. The autophagy inhibitor, 3MA, effectively inhibited starvation-induced protein degradation. Those compounds that had no effect on GFP-LC3B labeling of autophagosomes in Saos-2 cells also had no effect on protein turnover. NSC185058 reduced starvation-enhanced protein degradation by more than 50%, while NSC377071 proved more effective, comparable to 3MA.
Figure 3.

ATG4B-targeted compounds inhibit starvation-induced autophagy in Saos-2 cells. Saos-2 cells stably expressing GFP-LC3B were incubated under fed and starved conditions or under starvation conditions (St) in the presence of ATG4B-targeted compounds. After 4 h, the cells were fixed and the GFP-LC3B labeled AVs visualized by fluorescence microscopy (arrows). Fed Saos-2 cells sustained in nutrient-rich medium contained few AVs (A). Nutrient-starved cells contained numerous AVs (B), that were absent when treated with NSC185058 (C) or NSC377071 (D). Scale bar (A–D): 10 μm. (E) NSC185058 and NSC377071 inhibited autophagy in a dose response fashion. (F to I) Saos-2 cells were incubated under fed (G) or starved conditions (H) or starved conditions with NSC185058 (I) or NSC377071 (image not shown). At 4 h, the cells were fixed, processed for CMPase cytochemistry, and AVs (arrows) visualized by electron microscopy. The insets contain higher magnifications of representative AVs. Scale bar (G–I and insets H and I): 1 μm. (F) The fractional volume of AVs was quantified using morphometric methods described in Materials and Methods. The values represent the mean ± SEM (n = 3). ***P < 0.001.
If these compounds act on ATG4B, we would expect that the fractional volume of the AVs would be decreased since ATG4B and LC3B regulate autophagosome formation and size.22 To address this, cytidine monophosphatase (CMPase)-negative autophagosomes and CMPase-positive autolysosomes in fed and starved Saos-2 cells were visualized by electron microscopy (Fig. 3F–H) and fractional volumes measured using morphometric techniques (Fig. 3E). The fractional volume of AVs was greater in starved cells vs. cells incubated in fed amino acid-rich medium. Treatment with NSC185058 resulted in a dramatic decrease in the fractional volume of AVs in the starved cells (Fig. 3E and H). A similar trend was observed with NSC377071 (Fig. 3E).
We have also shown that NSC185058 and NSC377071 were equally effective on MDA-MB468 cells (Fig. S4). GFP-LC3B labeled AVs were absent in starved MDA-MB468 cells when treated with NSC185058 (Fig. S4A). The inhibitory effects of both NSC185058 and NSC377071 on starvation-induced protein turnover were comparable to 3MA (Fig. S4B). Finally, we showed that the fractional volume of the AVs was significantly reduced in starved cells treated with NSC185058 (Fig. S4F). The reduction in AV volume but not number suggested that the AVs were smaller when treated with this ATG4B antagonist.
The data show that both NSC185058 and NSC377071 inhibit starvation-induced autophagy in Saos-2 and MDA-MB468 cell lines. Furthermore, based on the inhibition of starvation-induced protein turnover combined with the decrease in the fractional volume of the autophagic vacuoles, we conclude that NSC185058 and NSC377071 negatively impact AV formation in both Saos-2 and MDA-MB468 cell lines.
NSC185058 and NSC377071 suppress the activation and lipidation of LC3B
The initial screening of the NCI compounds provided 2 chemically distinct inhibitors of autophagy. Both compounds effectively suppressed starvation-induced autophagy when measured by GFP-LC3B labeling of AVs, degradation of endogenous long-lived proteins, and morphometry of AV volumes. The next step was to show that one or both compounds act by inhibiting ATG4B. This was done by evaluating the effects of these compounds on LC3B lipidation and ATG4B activity.
We found that starvation-induced LC3B lipidation was suppressed by these compounds (Fig. 4). We suspect that this is due to the negative effects of these compounds on ATG4B activity. To test this, we used an in vitro assay developed by Li, et al.21 Our results demonstrated that NSC185058, but not NSC377071, inhibited the ATG4B cleavage of LC3B-GST (Fig. 4B) in a dose-dependent manner with an IC50 of 51 μM. Compounds NSC310196 and NSC52086 were used as negative controls, since they were ineffective on autophagy as assessed by starvation-induced protein degradation (Fig. S3B) and GFP-LC3B labeling of autophagic vacuoles (Fig. S3A). Li et al. have shown that ATG4A, but not ATG4C or ATG4D, has limited activity, albeit 103-fold lower catalytic efficiency than ATG4B, on LC3B, which may allow a compensatory response. Therefore, we also tested the efficacy of NSC185058 to inhibit ATG4A (Fig. S5A). The data suggest that NSC185058 suppressed the cleavage of GABARAPL2/GATE16-GST by ATG4A. Finally, we examined the inhibitory effects of NSC185058 using a cellular ATG4B assay (Fig. 5). This was done by quantifying the release of Gaussia luciferase (dNGLUC) from ACTB-LC3B-dNGLUC expressed in 293T (Fig. 5A) and HuH7 (Fig. 5B) cell lines. Ketteler and Seed have shown that the cleavage of this substrate and release of dNGLUC is dependent upon ATG4B levels.30 NSC185058 inhibited the C-terminal cleavage of LC3B by ATG4B in rapamycin-treated 293T cells and in amino acid starved HuH7 cells. Meanwhile, NSC377071 had a minimal effect on rapamycin-induced ATG4B activity in 293T cells as evidenced by LC3B cleavage, but inhibited starvation-induced activity in HuH7 cells. The results demonstrate that NSC185058 effectively inhibits ATG4B activity in vitro and in cells, but that NSC377071 is likely acting on a different target protein.
Figure 4.

NSC185058 effectively inhibits ATG4B activity and LC3B lipidation. (A) Saos-2 (GFP-LC3B) cells were incubated under fed and starved conditions in the presence of 3-MA (10 mM), NSC185058 (100 μM), or NSC377071 (100 μM) for 2, 4, or 6 h. Nonlipidated (GFP-LC3B-I) and lipidated (GFP-LC3B-II) forms of GFP-LC3B were separated by SDS-PAGE and identified by western blotting using anti-GFP (Sigma) antibodies. The band density was quantified and the ratios calculated. (B) The cleavage of LC3B-GST by purified ATG4B was assayed as described in Materials and Methods. (C) The concentration-dependent effect of NSC185058 on ATG4B activity as shown in panel B was quantified. The densities of the LC3B-GST, GST and LC3B bands were measured. The fraction of (GST+LC3B)/(LC3B-GST+GST+LC3B) at each time point is calculated, which correlated with the fraction of products (Fp). Using Fp at time zero (Fp-0) as the baseline, the inhibition% at each time point is calculated as [1-(Fp-t/Fp-0)] × 100%.
Figure 5.
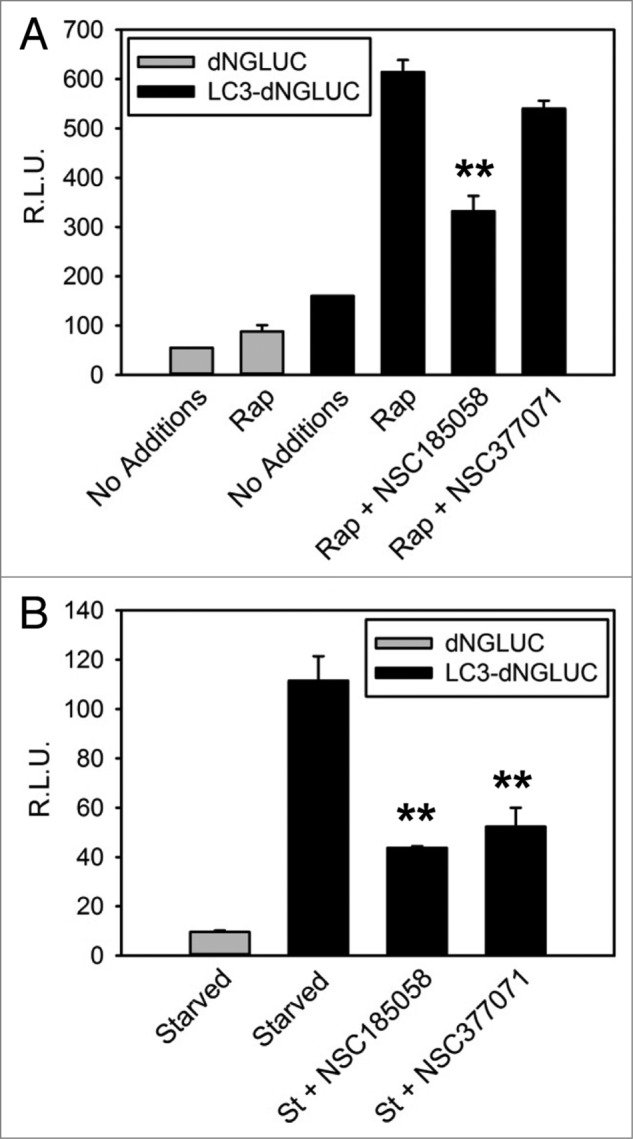
NSC185058 effectively inhibited ATG4 activity in cells. 293T (A) and HuH7 (B) cells transiently expressing ACTB-dNGLUC or ACTB-LC3B-dNGLUC were incubated in the presence of amino acids and serum. ATG4B was activated by rapamycin (Rap) or amino acid starvation (St) and the proteolytic cleavage of ACTB-LC3B-dNGLUC by ATG4B quantified by measuring dNGLUC (R.L.U.) recovered in the medium.30 The values represent the mean ± SEM (n = 3). Statistical comparisons were made to rapamycin treatment in (A) and to starvation conditions in (B) (**P < 0.01).
Effects of NSC185058 and NSC377071 on autophagy signaling pathways
Next, we examined the effects of NSC185058 and NSC377071 on the MTOR-RPS6KB1 and PtdIns3K signaling pathways, which regulate the onset of autophagy.31-33 For example, the activation of autophagy correlates with a decrease in the phosphorylation state of ribosomal protein S6 (RPS6) associated with decreased MTOR and ribosomal protein S6 kinase β 1 (RPS6KB1) activities, and the association of phosphatidylinositol 3-phosphate (PtdIns3P) with forming autophagosomes with increased PtdIns3K activity that can be visualized using RFP-tagged FYVE (Fab1-YOTB-Vac1-EEA1) zinc-finger domain that binds to PtdIns3P.34-36
Under fed conditions, RPS6 was phosphorylated and migrated more slowly than the nonphosphorylated RPS6 on SDS-PAGE (Fig. 6A, lane a). This phosphorylation was MTOR dependent as evidenced by its inhibition by rapamycin (Fig. 6A, lane c). When Saos-2 cells were switched from fed to starved conditions, RPS6 was dephosphorylated (Fig. 6A, lanes a and b). BECN1 had little effect on the phosphorylation and dephosphorylation of RPS6 in fed and starved cells (Fig. S5B, lanes d and e). The dephosphorylation of RPS6 was only marginally affected in NSC185058-treated Saos-2, shCon, and shBECN1 cells (Fig. 6A, lane h; Fig. S5B, lanes c and f). NSC310664, a putative LC3B antagonist (data not shown), was also found to have minimal effects on MTOR activity. The limited RPS6 phosphorylation in the presence of NSC185058 may be due to a feedback inhibition of autophagy, since a comparable level of phosphorylated RPS6 was observed in the presence of 3MA (Fig. 6A, lanes g and h). To the contrary, the starvation-induced dephosphorylation of RPS6 was effectively suppressed by NSC377071 resulting in a fully phosphorylated RPS6 as seen in fed cells and faster migrating partially phosphorylated RPS6 (Fig. 6A, lanes b and i). We also showed that NSC377071 partially restored RPS6 phosphorylation that had been suppressed by rapamycin (Fig. 6A, lanes c and e).
Figure 6.

Effects of NSC185058 and NSC377071 on MTOR and PtdIns3K activities. (A) Saos-2 cells were incubated under fed (lane a), starved (lane b), fed plus rapamycin (lane c), fed plus rapamycin (Rap) in the presence of NSC185058, NSC377071, and NSC310664 (lanes d to f), and starved conditions (St) in the presence of 3MA (lane g) and NSC185058, NSC377072, and NSC310664 (lanes h to j). After 4 h, the cells were solubilized, proteins separated by SDS-PAGE and transferred to PVDF membranes, and the levels of ribosomal RPS6 and phosphorylated RPS6 (p-RPS6) evaluated on western blots. (B–G) Saos-2 (B, C, E, and F), shCon-Saos-2 (D) and shATG4B-Saos-2 (G) cell lines transiently expressing FYVE-RFP were incubated under fed (B), starved (C, D, and G), and starved conditions in the presence of NSC185058 (E) or NSC377071 (F). After 4 h, the cells were fixed and the FYVE-RFP labeled vacuoles (arrows) visualized by fluorescence microscopy. Scale bar (B–G): 10 μm.
Next, we utilized FYVE-tagged RFP to localize PtdIns3P and assess PtdIns3K activity in the Saos-2 cells.36-38 When autophagy is suppressed in fed cells, FYVE-RFP distributed throughout the cytosol (Fig. 6B). Under starvation conditions, FYVE-RFP was found associated with small and large structures throughout the cytoplasm (Fig. 6C, arrows). About 30% of the fed cells contained FYVE-RFP vesicles compared with 70% present in starved cells (Fig. S5C). ATG4B (Fig. 6G) and NSC185058 (Fig. 6E) had no effect on the labeling of these vesicles, suggesting PtdIns3K activity was unaltered (Figs. 6E and G; Fig. S5C). However, we found that NSC377071 inhibited the FYVE-RFP labeling of these structures (Fig. 6F).
These findings suggest that NSC185058 has little effect on either MTOR or PtdIns3K activities, and thus, targets ATG4B (Fig. 4; Fig. 5) to inhibit autophagy (Figs. 3; Fig. S3). Meanwhile, NSC377071 appears to be suppressing ATG4B activity (Fig. 5B) and autophagy (Fig. 3; Fig. 4) by positively affecting the MTOR pathway (Fig. 6A) and/or negatively affecting the PtdIns3K pathway (Figs. 6F; Fig. S5C).
Effects of NSC185058 and NSC377071 on autophagy in vivo
We have shown that both NSC185058 and NSC377071 suppress autophagy by inactivating ATG4B directly and indirectly, respectively. Next, we will demonstrate that these compounds are effective in vivo by monitoring the cellular distribution of GFP-LC3B transiently expressed in the livers of C57BL/6 mice. Mice were injected at 2 24 h intervals with vehicle, NSC185058, or NSC377071, autophagy induced by 20 h fasting, and GFP-LC3B imaged using a multiphoton fluorescence microscope (Fig. 7). Fasting resulted in an increase of GFP-LC3B AVs compared with ad libitum fed mice. Both NSC185058 and NSC377071 significantly decreased the appearance of GFP-LC3B AVs compared with fasted mice. The results show that these compounds inhibit autophagy in vivo, and that the inhibitory effects persisted for at least 20 h after injection.
Figure 7.

NSC185058 and NSC377071 inhibit starvation-induced autophagy in vivo. C57BL/6 mice were injected IP with adenoviral GFP-LC3B (Welgen, Inc., Worcester, MA) in order to transiently express the autophagy marker in the liver. At 2 24 h intervals, the mice were injected IP with peanut oil vehicle or antiautophagy compounds (100 mg/kg mouse weight) and the GFP-LC3B dots quantified. (A) The punctate appearance of GFP-LC3B labeled AVs (arrows) in livers from fasted untreated mice suggests ongoing autophagy. (B and C) The absence of AVs in NSC185058 or NSC377071-treated mice reveals that these compounds suppressed autophagy. Scale bar (A–C): 50 μm. (D) Quantification of the GFP-LC3B dots revealed that both NSC185058 and NSC377071 significantly inhibited starvation-induced (St) autophagy (***P < 0.001). The values represent the mean ± SEM, n = 4 to 6 trials.
Effects of NSC185058 on the growth of osteosarcoma tumors
Our data suggest that ATG4B is a potential target for inhibiting autophagy and suppressing osteosarcoma growth (Fig. 1; Fig. 2). We have identified and characterized an ATG4B antagonist that effectively inhibits autophagy in vitro and in vivo. NSC185058 was found to be cytotoxic to Saos-2 cells during a 48 h exposure (Fig. 8A). Furthermore, cytotoxicity was more evident in cells deprived of amino acids. These findings are consistent with this compound inhibiting autophagy, a pathway important in cell survival under nutrient-deprived conditions.
Figure 8.
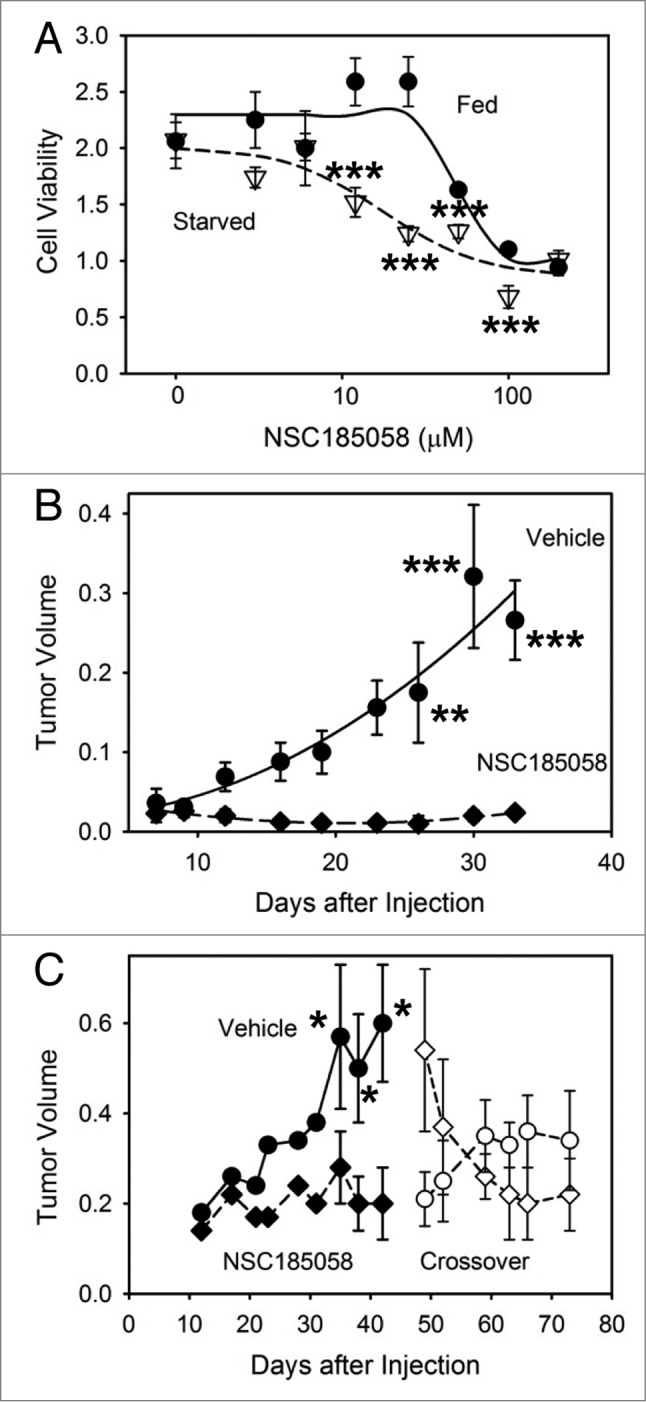
Osteosarcoma tumors treated with ATG4B antagonist NSC185058 fail to grow. (A) Saos-2 cells were incubated in nutrient-rich (closed circles) or amino acid-deprived (open triangles) medium in the presence of NSC185058 (3 to 200 μM). After 48 h, cell viability was quantified by MTT-based assays. The values represent the mean ± SEM (n = 6). The statistical differences between comparable concentrations of NSC185058 are indicated. ***P < 0.001 (B) Immunodeficient nu/nu female mice were injected subcutaneously with 6 × 106 Saos-2 (GFP-LC3B) cells. At 7 d, the mice were divided into 2 groups of 5 mice each and injected IP on Monday, Wednesday, and Friday with either peanut oil vehicle or NSC185058 (100 mg/kg body weight) in peanut oil. This is a dosage that we have shown is sufficient to suppress liver autophagy in the mouse. The values represent the mean ± SEM (n = 4) of tumor volumes (cm3). (C) A second set of immunodeficient nu/nu female mice was injected subcutaneously with 6 × 106 Saos-2 (GFP-LC3B) cells. Palpable tumors were detected at 12 d, and the mice were divided into 2 groups and injected IP on Monday, Wednesday, and Friday with either peanut oil vehicle (n = 8) or NSC185058 (100 mg/kg body weight) dissolved in peanut oil (n = 9). At 45 d, a crossover was done. The vehicle treated group was subjected to NSC185058 (open diamonds), while the NSC185058 treated group was switched to vehicle alone (open circles). NSC185058 significantly inhibited tumor growth, but the changes in tumor size after crossover failed to reach statistical significance. The values represent the mean ± SEM (n = 8 to 10) of tumor volumes (cm3 × 10−1). The statistical differences between comparable time points are indicated. *P < 0.05; **P < 0.01; ***P < 0.001
Next, we tested the efficacy of NSC185058 to suppress Saos-2 osteosarcoma growth in vivo. Saos-2 cells were injected subcutaneously into the right flank. At 7 d, when the tumors were palpable, the mice were divided into 2 groups and treated with vehicle or NSC185058. Tumor growth in vehicle treated mice was significant over 30 d (Fig. 8B). Meanwhile, tumors in mice treated with NSC185058 grew very poorly. Forty d of NSC185058 treatment did not appear to be toxic to the animals. No mice died during the treatment and, in fact, mouse body weights increased comparably to vehicle-treated mice (Fig. S7A). In addition, no pathologic changes in the liver (Fig. S7B and S7C), heart (Fig. S7D and S7E), and kidney (Fig. S7F and S7G) were evident.
Next, we determined whether NSC185058 suppressed autophagy within the tumor. This was done by removing the tumors and visualizing the autophagy response within the tumor cells by electron microscopy (Fig. 9). Tumors from vehicle treated mice contained many autophagic vacuoles suggesting ongoing autophagy (Fig. 9A, arrows). AVs were more evident at the tumor core where autophagy is the most active in growing tumors.8 Meanwhile, the autophagic vacuoles were virtually absent in those tumors isolated from treated mice suggesting that autophagy was suppressed within the cells of the osteosarcoma tumors by NSC185058. Morphometric quantification of the AVs revealed ongoing autophagy in the growing vehicle-treated tumors, which was significantly decreased in the NSC185058-treated tumors (Fig. 9C).
Figure 9.
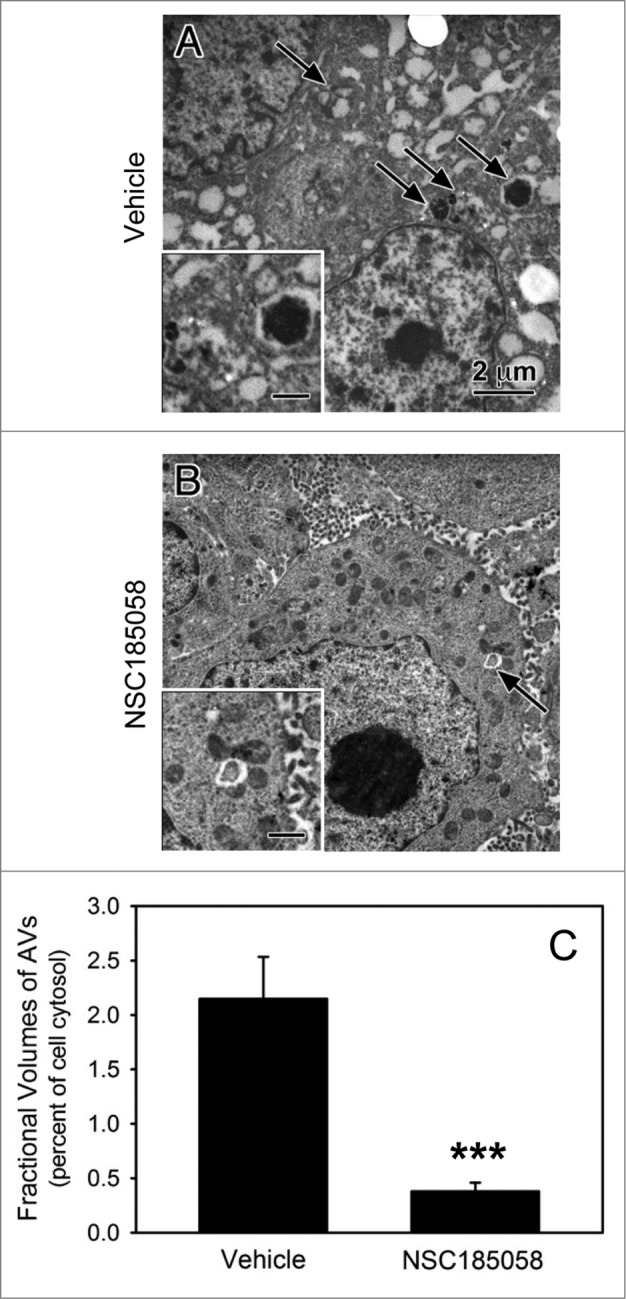
ATG4B antagonist NSC185058 inhibits autophagy within the osteosarcoma tumor. Immunodeficient nu/nu female mice were injected subcutaneously with Saos-2 cells. At 7 d, the mice were divided into 2 groups and injected IP on Monday, Wednesday, and Friday with either peanut oil (A) or NSC185058 (100 mg/kg body weight) in peanut oil (B). At the end time point, tumors were removed and then fixed and processed for electron microscopy (A and B). Pleomorphic AVs (arrows) were evident in the osteosarcomas from vehicle-treated mice, but relatively absent in the tumors from the NSC185058-treated mice. The insets contain higher magnifications of the representative AVs. The fractional volumes of the AVs were quantified by morphometrics as described in Materials and Methods (C). The values represent the mean ± SEM of 8 to 12 electron micrographs from each of 3 vehicle-treated and 4 NSC185058-treated tumors (***P < 0.001). Scale bar: 2 μm.
The antitumor effects of NSC185058 were unrelated to an inhibition of oncogenic protein kinases. Surface receptors ERBB2/HER2 and MET/c-MET and their downstream effectors AKT, MAPK/ERK, and SRC, have been implicated in the progression and metastasis of osteosarcomas.39-42 The effects of NSC185058 on kinase activities were determined by assessing the phosphorylation states of these kinases in cells growing in nutrient- and serum-rich medium (Fig. S6). Our results show that NSC185058 had no effect on the phosphorylated active states of these kinases under fed conditions. Furthermore, we have shown that the phosphorylated state of AKT was unaltered in fed cells lacking ATG4B or in starved cells treated with NSC185058 (data not shown). ERBB2 was found to be inactive under serum-starved conditions, and NSC185058 had no effect on the dephosphorylated inactive state of ERBB2. JUN phosphorylation has been implicated in the induction of autophagy and the production of metalloproteinases that modulate osteosarcoma migration.43,44 Our results show that the activated phosphorylated state of JUN was unaltered in Saos-2 cells treated with NSC185058.
Finally, we performed a crossover study to test the effects of NSC185058 on pre-existing tumors and to determine whether tumor growth is re-established upon removal of NSC185058 (Fig. 8C). As previously observed, NSC185058 suppressed osteosarcoma tumor growth when treatment began 12 d after cell injections. With the tumors established at 45 d, the animal control and treatment groups were switched. Over the next 28 d of NSC185058 treatment, the tumors decreased in size. Meanwhile, tumor growth appeared to be partially restored when NSC185058 treatment was stopped in the mice now injected with vehicle only. Our findings suggest that NSC185058 can effectively inhibit tumor growth and reduce tumor size, but that these effects are reversible.
Discussion
Autophagy is a cytoprotective pathway essential for metabolic homeostasis during exposure to the hostile tumor microenvironment and for organelle quality control during chemotherapy treatment. Thus this pathway is an ideal drug target for suppressing tumor growth and attenuating chemoresistance. Several antiautophagy drugs that target either PtdIns3K or lysosome function have been reported. 3MA and wortmannin were the first class of autophagy inhibitors that inactivated PtdIns3K and inhibited autophagosome formation.45 Spautin-1 inhibits ubiquitin-specific peptidases thereby allowing proteosome-mediated degradation of PIK3C3, the kinase subunit of PtdIns3K.36 Unfortunately, these PtdIns3K perturbants have not been proven to be effective in vivo. On the other hand, chloroquine and its derivatives are effective in vivo in suppressing the fusion of autophagosomes with lysosomes while also inactivating lysosomal proteinases. However, recent findings suggest that the anticancer effects of chloroquine may be independent of autophagy.25 That is, these lysosomotropic agents may interfere with lysosome functions possibly resulting in lysosome lysis and cell death.
In this study, we set out to identify a protein essential for autophagy and tumor growth, screen and characterize potential compounds that inhibit the protein and autophagy in vitro and in vivo, and finally, test the ability of a lead compound to inhibit tumor growth. Of the 4 autophagins, ATG4B is 1500-fold more catalytically efficient for LC3B activation than the other 3 autophagins.21 Marino et al. report a systemic reduction in autophagic activity in ATG4B-deficient mice that is not observed in ATG4C-deficient mice.46,47 ATG4B and LC3B are essential for amplifying and sustaining the autophagy response.22,23 We have shown that ATG4B knockdown in Saos-2 and MDA-MB468 cell lines are defective in starvation-induced autophagy (Fig. 1; Fig, S4). We have demonstrated that osteosarcoma Saos-2 cells lacking ATG4B fail to survive amino acid-starvation conditions and have attenuated tumor growth in mice (Fig. 2). These findings suggest that ATG4B would be good target to inhibit autophagy and tumor growth.
Autophagy proteins have been linked to tumor suppression and tumor survival. Mouse studies have shown that ATG5, ATG7, and BECN1 are tumor suppressors.48,49 Furthermore, the BECN1 gene is monoallelically deleted in a majority of sporadic human breast, ovarian, and prostate cancers.50 To the contrary, elevated levels of LC3B have been linked to poor prognosis in human pancreatic, colorectal, and breast carcinomas.5-7 Our genetic and pharmaceutical studies have shown that ATG4B has a positive impact on the growth and maintenance of osteosarcoma xenografts (Fig. 2; Fig. 8).
Reed and coworkers have used a high throughput screen to identify 17 ATG4B inhibitors.51 Two libraries of 3280 pharmacologically active compounds have been screened using an in vitro LC3B-PLA2 reporter. These purported ATG4B antagonists have IC50 values ranging between 1.1 to 5.5 μM in vitro, but the efficacy of these compounds to inhibit autophagy in cells or animals has yet to be established. Our docking protocol ranked over 139,735 compounds for subsequent screening using cell-based ATG4B and autophagy assays. Using a rigorous in vitro and in vivo testing approach, we identified 2 lead compounds that suppressed autophagy in both cell and animal models. However, upon further testing only one inhibited ATG4B directly. Our results suggest that NSC377071 indirectly suppressed ATG4B activity and autophagy by inhibiting PtdIns3K or an unknown upstream regulator. In addition, this compound inhibited ATG4B function in starved, but not rapamycin-treated cells. Further studies are needed to fully understand the inhibitory effects of this compound on rapamycin-treated autophagy. Meanwhile, we have shown that NSC185058 directly inhibited cellular ATG4B activity induced by starvation or rapamycin and suppressed the lipidation of LC3B and autophagosome formation in response to starvation. This first generation ATG4B antagonist has an IC50 of about 50 μM for ATG4B activity assayed in vitro. NSC185058 is available solely through the NCI, but can be chemically synthesized in gram amounts in a single efficient reaction. We have found that this compound had no effect on the known autophagy regulatory pathways or oncogenic protein kinases. That is, NSC185058 did not activate the MTOR pathway as determined by RPS6 phosphorylation (Fig. 6A) or inhibit PtdIns3K activity as determined by FYVE-RFP localization (Fig. 6E).
Antiautophagy drugs tested in cell culture are believed to be most effective when combined with chemotherapies. However, we have shown that antiautophagy NSC185058 can be effective as a single antitumor agent to suppress tumor growth and induce tumor regression in osteosarcoma xenografts. An intraperitoneal injection of NSC185058 (100 mg/kg body weight) effectively inhibited autophagy for at least 24 h, and administering this dosage 3 d per wk suppressed osteosarcoma growth. We observed no loss in animal weight and no abnormal morphologies in the liver, heart, and kidney (Fig. S7). These studies have been limited to 4 wk of treatment. The long-term effects of NSC185058 remain to be evaluated, but atg4b−/− mice are viable with only developmental inner ear problems and Paneth cell defects reported.46,52
Our results suggest that the antitumor effects of NSC185058 are consistent with its ability to interfere with ATG4B function and inhibit autophagy. For example, this compound had no effect on the phosphorylated states of oncogenic protein kinases (Fig. S6). Saos-2 cells lacking ATG4B or treated with NSC185058 were more sensitive to starvation conditions that mimic the tumor environment (Figs. 2A; Fig. 8A). Furthermore, the extent of NSC185058 inhibition of osteosarcoma tumor growth was comparable to that observed for tumors lacking ATG4B. However, we have observed that high concentrations of NSC185058 reduce the viability of Saos-2 cells under fed conditions while cells lacking ATG4B are not sensitive. This finding suggests that NSC185058 may have other cytotoxic effects. Although we have not identified all the possible targets of NSC185058, we have eliminated a number of likely autophagy and oncogenic suspects. Therefore, we proposed that the antitumor activity of this ATG4B antagonist is due to its efficacy to inhibit autophagy. Furthermore, we suggest that NSC185058 is toxic to the osteosarcoma cells more so than healthy tissues because the tumor cells require autophagy for surviving the nutrient deprived microenvironment of the avascular tumor while healthy vascularized tissues are rich in nutrients.
We predict that ATG4B antagonists would be effective in other cancers expressing high levels of ATG4B, such as triple-negative breast and colorectal cancer cells (Fig. S1A). Although this has not been fully tested, we have shown that NSC185058 effectively inhibits autophagy in MDA-MB468 cells (Fig. S4). Both Saos-2 and MDA-MB468 cells respond to amino acid and serum starvation conditions by activating autophagy, and NSC185058 attenuates this response by repressing AV formation. We have not yet tested the effects of NSC185058 on MDA-MB468 tumor growth, but suspect that this ATG4B antagonist will have a negative impact on tumor growth.
Osteosarcoma begins in rapidly growing long bones of children and adolescents and metastasizes most commonly to the lungs. Aggressive interventions including surgical removal of the bone and followup chemotherapy can have harsh side effects and are only partially effective, with 50% of these patients dying of this disease. Therefore, a safe and effective systemic therapy is critical to treat these young patients. Our findings suggest that ATG4B antagonists may prove useful in treating this aggressive cancer. We have demonstrated that ATG4B and autophagy have an essential role in osteosarcoma growth. We have identified a first generation ATG4B antagonist that effectively suppresses autophagy in vivo and in a single agent regimen effectively inhibits the growth of Saos-2 tumors in preclinical xenograft models. In addition, we have demonstrated that NSC185058 reduces tumor size, but its anticancer effects appear to be reversible. Although we have not adequately defined all the possible protein targets of NSC185058, these findings open new venues for antiautophagy drugs and new targets for treating osteosarcoma, breast adenocarcinoma, and other aggressive cancers.
Materials and Methods
Cell culture
Saos-2 (osteosarcoma) (ATCC, HTB-85), MDA-MB468 (triple-negative breast cancer) (ATCC, HTB132), HEK-293T (human embryonic kidney) (ATCC, CRL-11268), and HuH7 (hepatoma) (JCRB Cell Bank, JCRB0403) cell lines were grown in DMEM supplemented with 10% fetal bovine serum. Stable expression of GFP-LC3B in Saos-2 and MDA-MB468 cell lines was achieved by transfecting the pGFP-LC3B vector with Lipofectamine 2000 (Invitrogen, 52887) followed by G418 (0.5 mg/ml; RPI, G64000) selection and cloning by limiting dilutions.53 Transient expression of FYVE-RFP in Saos-2 cells was accomplished by transfecting the pFYVE-RFP vector with Lipofectamine 2000.36-38 Cell lines stably transfected with shCon, shATG4B, and shBECN1, were isolated by infection with recombinant lentiviral vectors encoding a “scrambled” shRNA, ATG4B shRNAs (Open Biosystems, RHS4533-NM_013325), or BECN1 shRNAs, (Open Biosystems, RHS4533-NM_003766) followed by selection with puromycin (5 μg/ml; Sigma, P8833). ATG4B expression was knocked down 70% in Saos-2 and 90% in MDA-MB468 cell lines (Fig. S1B) and BECN1 expression was knocked down over 90% in Saos-2 cells (Fig. S5B).
Morphological and biochemical measures of autophagy responses in vitro and in vivo
Saos-2 and MDA-MB468 cells stably expressing GFP-LC3B were incubated in DMEM medium supplemented with amino acids and serum (Fed) or Krebs-Heinseleit medium lacking amino acids and serum (Starved).54 The starved cells were incubated in the presence and absence of the NSC compounds (0.1 to 1000 μM) for 4 h, fixed with 2% paraformaldehyde and the GFP-LC3B labeled AVs examined using a Zeiss Axiophot Fluorescence Photomicroscope (Carl Zeiss Microscopy, Thornwood, NY) with ColdSnap HQ camera (PhotoMetrics, Tucson, AZ). The digital images were visualized using Photoshop or ImageJ software. The number of autophagosomes (green “dots”) per cell was quantified using ImageJ software. Taking into account basal autophagy responses, those cells with more than 5 (Saos-2) or 10 (MDA-MB468) puncta were defined as autophagy active.
Untreated and treated fed and starved cells were fixed, processed for CMPase cytochemistry and the sections examined by electron microscopy.55,56 Photographic negatives taken on a JEOL 100CX electron microscope (JEOL USA, Peabody, MA) were digitized using an Epson 4990 Scanner (Epson America, Long Beach, CA). Fractional volumes of the AVs relative to the cytosolic volume were quantified on 8 to 12 images (4000× mag) per condition per 3 or more trials by morphometric methods using ImageJ software.55,56
The degradation of long-lived proteins was measured by 14C-valine pulse-chase protocols as previously described.54,57 Cellular proteins were radiolabeled by incubating with 14C-valine for 16 h. Afterwards, the cells were switched to fed or starved media in the absence and presence of autophagy drugs and the degradation of long-lived proteins over 1 to 5 h of chase determined by measuring acid-soluble radioactivity in the medium and cells.
Autophagy responses in vivo were assessed by intraperitoneal (IP) injection of adenoviral GFP-LC3B (10 μl of 1012 particles/ml stock) (Welgen, Inc., Worcester, MA) into male C57BL/6 mice (Charles River, Wilmington, MA) providing a transient expression (2 to 5 d) of GFP-LC3B in the liver. At 2 24 h intervals, the mice were injected IP with peanut oil vehicle or ATG4B antagonists (100 mg/kg body weight). The mice were fed ad libitum or fasted for 18 to 22 h to induce autophagy. Upon euthanasia, the livers were removed and transferred directly to the stage of a Zeiss 510 NLO multiphoton laser scanning confocal microscope (Carl Zeiss Microscopy, Thornwood, NY) equipped with a META spectral detector (Carl Zeiss Microscopy, Thornwood, NY) whereby the GFP-LC3B labeled AVs were visualized and digital images recorded. The autophagy response was measured using ImageJ to quantify the GFP-LC3B dots in each of 5 fields from each trial.
In silico molecular docking of small-molecular-weight compound database
The data show that ATG4B, a cysteine proteinase, is critical for enhancing the amplitude of autophagy and sustaining its response by regulating the lipidation of LC3B. C74, W142, R229, D278, and H280 have been shown to be required for the proteolytic activation of LC3B by ATG4B.29 The crystal structure of ATG4B (Protein Data Bank code 2CY7) reveals that C74 is inaccessible to solvent whereas D278 and H280 is clustered within a surface pocket that is solvent accessible and used as a target for compound docking.29 In our effort to identify ATG4B inhibitors, which would suppress autophagy, we used in silico molecular docking to identify from a library of 139,735 chemical compounds those predicted to bind to the active site of ATG4B. Each compound screened is considered to follow the Lipinski rules of drug-likeness to focus on compounds with favorable solubility and bioavailability characteristics. Each compound was scored in 100 different orientations into the selected structural pocket and the highest scoring compounds and orientations were output. Scores were based on nonpolar interactions (based on van der Waals contacts) and polar interactions (electrostatic score). Those 20 compounds with the highest overall energy score (van der Waals + electrostatic score) were requested from the NCI DTP (http://dtp.nci.nih.gov/RequestCompounds/) and the 12 received were dissolved in DMSO and tested for their ability to inhibit starvation-induced autophagy (Table S1). All programs for in silico docking were part of the DOCK5.0 suite developed at University of California, San Francisco.58 Images of protein structure and compound docking were obtained using PyMOL software (DeLano Scientific, Palo Alto, CA).
Chemical synthesis of NSC185058
NSC185058 was synthesized from 2-picoline (46.6 g) and 2-aminopyridine (51.8 g) in the presence of elemental sulfur (47 g) by heating at 160°C for 18 h. The bulk of unreacted methylpicoline was removed by distillation under vacuum.59,60 The resulting residue was recrystallized twice from absolute ethanol taking care to filter off unreacted sulfur. The resulting light brown solid was dissolved in 150 mL of 2:1 hexane/CHCl3, decolorized with carbon, and filtered through a short 1 × 5 cm column of silica. Recrystallization from 95% ethanol afforded 8.00 g (7%) of long, yellow–orange needles, MP 81 to 82°C (lit 81 to 83°C) and fully characterized by 1H- and 13C-NMR, and high resolution mass spectrometry.61
ATG4B assays
The cleavage of LC3B-GST by purified ATG4B was assayed as previously described.21 Purified ATG4B was preincubated with the putative ATG4B-targeted compounds at different concentrations for 24 h. Purified LC3B-GST was added to the ATG4B mixture for 3 min and full-length or cleaved LC3B-GST was resolved by SDS-PAGE followed by Coomassie Blue staining. Cellular ATG4B was assayed by measuring the cellular release of an N-terminally deleted form of Gaussia luciferase (dNGLUC) as described by Ketteler, et al.30 An actin-LC3B-dNGLUC (and actin-dNGLUC control) construct in a pLKO vector was transiently expressed in 293T and HuH7 cells using Lipofectamine 2000. For unknown reasons, dNGLUC could not be detected in the medium when using the Saos-2 cell lines. The cells were incubated under fed and starved (or rapamycin) conditions in the absence or presence of the ATG4B-targeted compounds and the release of dNGLUC into the media was measured.
PtdIns3K assay
Saos-2 transiently expressing FYVE-RFP (gift from Dr Junying Yuan, Harvard Medical School) was incubated in DMEM medium supplemented with amino acids and serum (Fed) or Krebs-Heinseleit medium lacking amino acids and serum (Starved). The starved cells were incubated in the presence and absence of the NSC185058 or NSC377071 for 4 h at which time the cells were examined by fluorescence microscopy. The numbers of red “dots” per cell was quantified using ImageJ software. Taking into account basal responses, those cells with 3 or more puncta were defined as PtdIns3K active.
Osteosarcoma xenograft studies
Immunodeficient nu/nu nude female mice (Charles River, Wilmington, MA) were injected subcutaneously with 6 × 106 Saos-2 (GFP-LC3B), shCon-Saos-2(GFP-LC3B) or shATG4B-Saos-2(GFP-LC3B) cells. Palpable Saos-2 (GFP-LC3B) tumors developed in 7 to 10 d, at which time, the mice were divided into 2 groups and injected IP on Monday, Wednesday, and Friday with either peanut oil vehicle or NSC185058 (100 mg/kg body weight) dissolved in peanut oil. Over time, the tumor dimensions were measured using calipers and volumes calculated using the formula: (W2 × L)/2. All animal studies were conducted in compliance with the US Department of Health and Human Services Guide and approved by the Institutional Animal Care and Use Committee and the Division of Environmental Health and Safety at the University of Florida.
Western blots
Cellular proteins were solubilized in SDS sample buffer (67 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 0.1% bromophenol blue, and 1.5% DTT) supplemented with a proteinase inhibitor cocktail (Sigma P8340) and separated on 10% or 15% SDS-PAGE. After transferring the proteins to PVDF (polyvinylidene difluoride) membranes, the membranes were blotted with anti-LC3B antiserum (prepared by Biosource, Hopkinton, MA against SEKTFKQRRTFEQRVEDV), anti-ATG4B antiserum (Cell Signaling Technology, 5299), anti-RPS6 antiserum (Cell Signaling Technology, 2317), anti-phospho-RPS6 (Ser 240/244) antiserum (Cell Signaling Technology, 2215) or mouse anti-β-tubulin 1 (Sigma, T7816). Following the secondary HRP-conjugated antibody, the PVDF membranes were treated with ECL reagent and exposed to X-ray film. The bands were quantified using ImageJ software. Activation of oncogenic kinases was determined by monitoring their phosphorylated (active) states. Cells were solubilized in SDS sample buffer containing 1 mM orthovanadate to inhibit phosphatases, the proteins separated on 10% SDS-PAGE and transferred to PVDF membranes. Antibodies specific for ERBB2/HER2 (Cell Signaling Technology, 4290), phospho-ERBB2/HER2 (Tyr1221/1222) (Cell Signaling Technology, 2243), MET (Cell Signaling Technology, 8198), phospho-MET (Tyr1234/1235) (Cell Signaling Technology, 3077), MAPK1/3) (Santa Cruz Biotechnology, SC93), phospho-MAPK1/3) (Thr202/204) (Cell Signaling Technology, 4370), AKT1–3 (Cell Signaling Technology, 9272), phospho-AKT1 (Ser473) (Cell Signaling Technology, 9271), SRC (Cell Signaling Technology, 2102), phospho-SRC (Tyr416) (Cell Signaling Technology, 6943), JUN (Cell Signaling Technology, 9165), phospho-JUN (Ser63) (Cell Signaling Technology, 2361), were used to detect the total and phosphorylated, active oncogene products on the PVDF membranes.
Acknowledgments
We would like to thank Dr Tamotsu Yoshimori (Osaka University, Osaka, Japan) for providing the pGFP-LC3B vector and Dr Junying Yuan (Harvard Medical School, Boston, MA) for providing the pFYVE-RFP expression vector. W Dunn, D Akin, and D Ostrov are the authors on a US Patent No 8,545,841 submitted by the University of Florida (UF#12579) in reference to NSC185058. The authors have no commercial affiliations or stock or equity interests that can be considered a conflict of interest.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the Ocala Royal Dames and University of Florida Office of Technology Licensing (W Dunn) and NIH grants CA95552 (W Dunn), CA93651 (B. Law), CA111456 (X-M Yin), and DK090115 and DK079879 (J-S Kim).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Mortimore GE, Pösö AR, Lardeux BR. Mechanism and regulation of protein degradation in liver. Diabetes Metab Rev 1989; 5:49-70; PMID:2649336; http://dx.doi.org/ 10.1002/dmr.5610050105 [DOI] [PubMed] [Google Scholar]
- 2. Mortimore GE, Lardeux BR, Heydrick SJ. Mechanism and control of protein and RNA degradation in the rat hepatocyte: two modes of autophagic sequestration. Revis Biol Celular 1989; 20:79-96; PMID:2484018 [PubMed] [Google Scholar]
- 3. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature 2009; 458:1131-5; PMID:19339967; http://dx.doi.org/ 10.1038/nature07976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ding ZB, Shi YH, Zhou J, Qiu SJ, Xu Y, Dai Z, Shi GM, Wang XY, Ke AW, Wu B, et al. . Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res 2008; 68:9167-75; PMID:19010888; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-1573 [DOI] [PubMed] [Google Scholar]
- 5. Yoshioka A, Miyata H, Doki Y, Yamasaki M, Sohma I, Gotoh K, Takiguchi S, Fujiwara Y, Uchiyama Y, Monden M. LC3, an autophagosome marker, is highly expressed in gastrointestinal cancers. Int J Oncol 2008; 33:461-8; PMID:18695874 [PubMed] [Google Scholar]
- 6. Fujii S, Mitsunaga S, Yamazaki M, Hasebe T, Ishii G, Kojima M, Kinoshita T, Ueno T, Esumi H, Ochiai A. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci 2008; 99:1813-9; PMID:18616529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao H, Yang M, Zhao J, Wang J, Zhang Y, Zhang Q. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med Oncol 2013; 30:475; PMID:23371253; http://dx.doi.org/ 10.1007/s12032-013-0475-1 [DOI] [PubMed] [Google Scholar]
- 8. Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al. . Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51-64; PMID:16843265; http://dx.doi.org/ 10.1016/j.ccr.2006.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007; 21:1367-81; PMID:17510285; http://dx.doi.org/ 10.1101/gad.1545107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 2007; 21:1621-35; PMID:17606641; http://dx.doi.org/ 10.1101/gad.1565707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Amaravadi R. Autophagy can contribute to cell death when combining targeted therapy. Cancer Biol Ther 2009; 8:130-3; PMID:19901522; http://dx.doi.org/ 10.4161/cbt.8.21.10416 [DOI] [PubMed] [Google Scholar]
- 12. Meijer AJ, Codogno P. Signalling and autophagy regulation in health, aging and disease. Mol Aspects Med 2006; 27:411-25; PMID:16973212; http://dx.doi.org/ 10.1016/j.mam.2006.08.002 [DOI] [PubMed] [Google Scholar]
- 13. Huang J, Ni J, Liu K, Yu Y, Xie M, Kang R, Vernon P, Cao L, Tang D. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res 2012; 72:230-8; PMID:22102692; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2001 [DOI] [PubMed] [Google Scholar]
- 14. Kung CP, Budina A, Balaburski G, Bergenstock MK, Murphy M. Autophagy in tumor suppression and cancer therapy. Crit Rev Eukaryot Gene Expr 2011; 21:71-100; PMID:21967333; http://dx.doi.org/ 10.1615/CritRevEukarGeneExpr.v21.i1.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Høyer-Hansen M, Jäättelä M. Autophagy: an emerging target for cancer therapy. Autophagy 2008; 4:574-80; PMID:18362515; http://dx.doi.org/ 10.4161/auto.5921 [DOI] [PubMed] [Google Scholar]
- 16. Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32:2-11; PMID:22025673; http://dx.doi.org/ 10.1128/MCB.06159-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iwamaru A, Kondo Y, Iwado E, Aoki H, Fujiwara K, Yokoyama T, Mills GB, Kondo S. Silencing mammalian target of rapamycin signaling by small interfering RNA enhances rapamycin-induced autophagy in malignant glioma cells. Oncogene 2007; 26:1840-51; PMID:17001313; http://dx.doi.org/ 10.1038/sj.onc.1209992 [DOI] [PubMed] [Google Scholar]
- 18. Proikas-Cezanne T, Waddell S, Gaugel A, Frickey T, Lupas A, Nordheim A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 2004; 23:9314-25; PMID:15602573; http://dx.doi.org/ 10.1038/sj.onc.1208331 [DOI] [PubMed] [Google Scholar]
- 19. Jeffries TR, Dove SK, Michell RH, Parker PJ. PtdIns-specific MPR pathway association of a novel WD40 repeat protein, WIPI49. Mol Biol Cell 2004; 15:2652-63; PMID:15020712; http://dx.doi.org/ 10.1091/mbc.E03-10-0732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Song J, Qu Z, Guo X, Zhao Q, Zhao X, Gao L, Sun K, Shen F, Wu M, Wei L. Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy 2009; 5:1131-44; PMID:19786832; http://dx.doi.org/ 10.4161/auto.5.8.9996 [DOI] [PubMed] [Google Scholar]
- 21. Li M, Hou Y, Wang J, Chen X, Shao ZM, Yin XM. Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J Biol Chem 2011; 286:7327-38; PMID:21177865; http://dx.doi.org/ 10.1074/jbc.M110.199059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007; 130:165-78; PMID:17632063; http://dx.doi.org/ 10.1016/j.cell.2007.05.021 [DOI] [PubMed] [Google Scholar]
- 23. Tanida I, Sou YS, Ezaki J, Minematsu-Ikeguchi N, Ueno T, Kominami E. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J Biol Chem 2004; 279:36268-76; PMID:15187094; http://dx.doi.org/ 10.1074/jbc.M401461200 [DOI] [PubMed] [Google Scholar]
- 24. Chen HY, White E. Role of autophagy in cancer prevention. Cancer Prev Res (Phila) 2011; 4:973-83; PMID:21733821; http://dx.doi.org/ 10.1158/1940-6207.CAPR-10-0387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 2012; 8:200-12; PMID:22252008; http://dx.doi.org/ 10.4161/auto.8.2.18554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yoshimura K, Shibata M, Koike M, Gotoh K, Fukaya M, Watanabe M, Uchiyama Y. Effects of RNA interference of Atg4B on the limited proteolysis of LC3 in PC12 cells and expression of Atg4B in various rat tissues. Autophagy 2006; 2:200-8; PMID:16874114; http://dx.doi.org/ 10.4161/auto.2744 [DOI] [PubMed] [Google Scholar]
- 27. Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 2010; 29:1792-802; PMID:20418806; http://dx.doi.org/ 10.1038/emboj.2010.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abeliovich H, Dunn WA, Jr., Kim J, Klionsky DJ. Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J Cell Biol 2000; 151:1025-34; PMID:11086004; http://dx.doi.org/ 10.1083/jcb.151.5.1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sugawara K, Suzuki NN, Fujioka Y, Mizushima N, Ohsumi Y, Inagaki F. Structural basis for the specificity and catalysis of human Atg4B responsible for mammalian autophagy. J Biol Chem 2005; 280:40058-65; PMID:16183633; http://dx.doi.org/ 10.1074/jbc.M509158200 [DOI] [PubMed] [Google Scholar]
- 30. Ketteler R, Seed B. Quantitation of autophagy by luciferase release assay. Autophagy 2008; 4:801-6; PMID:18641457; http://dx.doi.org/ 10.4161/auto.6401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie 2008; 90:313-23; PMID:17928127; http://dx.doi.org/ 10.1016/j.biochi.2007.08.014 [DOI] [PubMed] [Google Scholar]
- 32. Klionsky DJ, Codogno P. The mechanism and physiological function of macroautophagy. J Innate Immun 2013; 5:427-33; PMID:23774579; http://dx.doi.org/ 10.1159/000351979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 2014; 20:460-73; PMID:23725295; http://dx.doi.org/ 10.1089/ars.2013.5371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol 2004; 36:2445-62; PMID:15325584; http://dx.doi.org/ 10.1016/j.biocel.2004.02.002 [DOI] [PubMed] [Google Scholar]
- 35. Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem 1995; 270:2320-6; PMID:7836465; http://dx.doi.org/ 10.1074/jbc.270.5.2320 [DOI] [PubMed] [Google Scholar]
- 36. Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, Furuya T, et al. . Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011; 147:223-34; PMID:21962518; http://dx.doi.org/ 10.1016/j.cell.2011.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, Zhu H, Yu AD, Xie X, Ma D, et al. . Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A 2007; 104:19023-8; PMID:18024584; http://dx.doi.org/ 10.1073/pnas.0709695104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lipinski MM, Hoffman G, Ng A, Zhou W, Py BF, Hsu E, Liu X, Eisenberg J, Liu J, Blenis J, et al. . A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell 2010; 18:1041-52; PMID:20627085; http://dx.doi.org/ 10.1016/j.devcel.2010.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mueller F, Fuchs B, Kaser-Hotz B. Comparative biology of human and canine osteosarcoma. Anticancer Res 2007; 27(1A):155-64; PMID:17352227 [PubMed] [Google Scholar]
- 40. Shor AC, Keschman EA, Lee FY, Muro-Cacho C, Letson GD, Trent JC, Pledger WJ, Jove R. Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on SRC kinase for survival. Cancer Res 2007; 67:2800-8; PMID:17363602; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-3469 [DOI] [PubMed] [Google Scholar]
- 41. Ebb D, Meyers P, Grier H, Bernstein M, Gorlick R, Lipshultz SE, Krailo M, Devidas M, Barkauskas DA, Siegal GP, et al. . Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: a report from the children's oncology group. J Clin Oncol 2012; 30:2545-51; PMID:22665540; http://dx.doi.org/ 10.1200/JCO.2011.37.4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang K, Zhuang Y, Liu C, Li Y. Inhibition of c-Met activation sensitizes osteosarcoma cells to cisplatin via suppression of the PI3K-Akt signaling. Arch Biochem Biophys 2012; 526:38-43; PMID:22820099; http://dx.doi.org/ 10.1016/j.abb.2012.07.003 [DOI] [PubMed] [Google Scholar]
- 43. Lorin S, Borges A, Ribeiro Dos Santos L, Souquère S, Pierron G, Ryan KM, Codogno P, Djavaheri-Mergny M. c-Jun NH2-terminal kinase activation is essential for DRAM-dependent induction of autophagy and apoptosis in 2-methoxyestradiol-treated Ewing sarcoma cells. Cancer Res 2009; 69:6924-31; PMID:19706754; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-1270 [DOI] [PubMed] [Google Scholar]
- 44. Kimura R, Ishikawa C, Rokkaku T, Janknecht R, Mori N. Phosphorylated c-Jun and Fra-1 induce matrix metalloproteinase-1 and thereby regulate invasion activity of 143B osteosarcoma cells. Biochim Biophys Acta 2011; 1813:1543-53; PMID:21640141; http://dx.doi.org/ 10.1016/j.bbamcr.2011.04.008 [DOI] [PubMed] [Google Scholar]
- 45. Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelárová H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem 1997; 243:240-6; PMID:9030745; http://dx.doi.org/ 10.1111/j.1432-1033.1997.0240a.x [DOI] [PubMed] [Google Scholar]
- 46. Mariño G, Fernández AF, Cabrera S, Lundberg YW, Cabanillas R, Rodríguez F, Salvador-Montoliu N, Vega JA, Germanà A, Fueyo A, et al. . Autophagy is essential for mouse sense of balance. J Clin Invest 2010; 120:2331-44; PMID:20577052; http://dx.doi.org/ 10.1172/JCI42601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mariño G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, López-Otín C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem 2007; 282:18573-83; PMID:17442669; http://dx.doi.org/ 10.1074/jbc.M701194200 [DOI] [PubMed] [Google Scholar]
- 48. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev 2011; 25:795-800; PMID:21498569; http://dx.doi.org/ 10.1101/gad.2016211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 2003; 100:15077-82; PMID:14657337; http://dx.doi.org/ 10.1073/pnas.2436255100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. . Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809-20; PMID:14638851; http://dx.doi.org/ 10.1172/JCI20039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shu CW, Madiraju C, Zhai D, Welsh K, Diaz P, Sergienko E, Sano R, Reed JC. High-throughput fluorescence assay for small-molecule inhibitors of autophagins/Atg4. J Biomol Screen 2011; 16:174-82; PMID:21245471; http://dx.doi.org/ 10.1177/1087057110392996 [DOI] [PubMed] [Google Scholar]
- 52. Cabrera S, Fernández AF, Mariño G, Aguirre A, Suárez MF, Español Y, Vega JA, Laurà R, Fueyo A, Fernández-García MS, et al. . ATG4B/autophagin-1 regulates intestinal homeostasis and protects mice from experimental colitis. Autophagy 2013; 9:1188-200; PMID:23782979; http://dx.doi.org/ 10.4161/auto.24797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/ 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Susan PP, Dunn WA, Jr. Starvation-induced lysosomal degradation of aldolase B requires glutamine 111 in a signal sequence for chaperone-mediated transport. J Cell Physiol 2001; 187:48-58; PMID:11241348; http://dx.doi.org/ [DOI] [PubMed] [Google Scholar]
- 55. Dunn WA, Jr. Studies on the mechanisms of autophagy: formation of the autophagic vacuole. J Cell Biol 1990; 110:1923-33; PMID:2351689; http://dx.doi.org/ 10.1083/jcb.110.6.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lenk SE, Dunn WA, Jr., Trausch JS, Ciechanover A, Schwartz AL. Ubiquitin-activating enzyme, E1, is associated with maturation of autophagic vacuoles. J Cell Biol 1992; 118:301-8; PMID:1321157; http://dx.doi.org/ 10.1083/jcb.118.2.301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aplin A, Jasionowski T, Tuttle DL, Lenk SE, Dunn WA, Jr. Cytoskeletal elements are required for the formation and maturation of autophagic vacuoles. J Cell Physiol 1992; 152:458-66; PMID:1506410; http://dx.doi.org/ 10.1002/jcp.1041520304 [DOI] [PubMed] [Google Scholar]
- 58. Moustakas DT, Lang PT, Pegg S, Pettersen E, Kuntz ID, Brooijmans N, Rizzo RC. Development and validation of a modular, extensible docking program: DOCK 5. J Comput Aided Mol Des 2006; 20:601-19; PMID:17149653; http://dx.doi.org/ 10.1007/s10822-006-9060-4 [DOI] [PubMed] [Google Scholar]
- 59. Porter HD. The Willgerodt reaction applied to α- and γ-alkylpyridines. J Am Chem Soc 1954; 76:127-8; http://dx.doi.org/ 10.1021/ja01630a035 [DOI] [Google Scholar]
- 60. Mazumder UK, Gupta M, Karki SS, Bhattacharya S, Rathinasamy S, Sivakumar T. Synthesis and pharmacological activities of some mononuclear Ru(II) complexes. Bioorg Med Chem 2005; 13:5766-73; PMID:15982893; http://dx.doi.org/ 10.1016/j.bmc.2005.05.047 [DOI] [PubMed] [Google Scholar]
- 61. Kinney WA, Lee NE, Blank RM, Demerson CA, Sarnella CS, Scherer NT, Mir GN, Borella LE, DiJoseph JF, Wells C. N-phenyl-2-pyridinecarbothioamides as gastric mucosal protectants. J Med Chem 1990; 33:327-36; PMID:2296028; http://dx.doi.org/ 10.1021/jm00163a053 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.