

Abstract
Cigarette smoke (CS)-induced mitochondrial damage with increased reactive oxygen species (ROS) production has been implicated in COPD pathogenesis by accelerating senescence. Mitophagy may play a pivotal role for removal of CS-induced damaged mitochondria, and the PINK1 (PTEN-induced putative kinase 1)-PARK2 pathway has been proposed as a crucial mechanism for mitophagic degradation. Therefore, we sought to investigate to determine if PINK1-PARK2-mediated mitophagy is involved in the regulation of CS extract (CSE)-induced cell senescence and in COPD pathogenesis. Mitochondrial damage, ROS production, and cell senescence were evaluated in primary human bronchial epithelial cells (HBEC). Mitophagy was assessed in BEAS-2B cells stably expressing EGFP-LC3B, using confocal microscopy to measure colocalization between TOMM20-stained mitochondria and EGFP-LC3B dots as a representation of autophagosome formation. To elucidate the involvement of PINK1 and PARK2 in mitophagy, knockdown and overexpression experiments were performed. PINK1 and PARK2 protein levels in lungs from patients were evaluated by means of lung homogenate and immunohistochemistry. We demonstrated that CSE-induced mitochondrial damage was accompanied by increased ROS production and HBEC senescence. CSE-induced mitophagy was inhibited by PINK1 and PARK2 knockdown, resulting in enhanced mitochondrial ROS production and cellular senescence in HBEC. Evaluation of protein levels demonstrated decreased PARK2 in COPD lungs compared with non-COPD lungs. These results suggest that PINK1-PARK2 pathway-mediated mitophagy plays a key regulatory role in CSE-induced mitochondrial ROS production and cellular senescence in HBEC. Reduced PARK2 expression levels in COPD lung suggest that insufficient mitophagy is a part of the pathogenic sequence of COPD.
Keywords: COPD, mitophagy, PARK2, ROS, senescence
Abbreviations: Baf A1, bafilomycin A1; COPD, chronic obstructive pulmonary disease; CS, cigarette smoke; CSE, cigarette smoke extract; EM, electron microscopy; FEV1, forced expiratory volume in one second; FVC, forced vital capacity; HBEC, human bronchial epithelial cell; MAP1LC3/LC3, microtubule-associated protein 1 light chain 3; NAC, N-acetylcysteine; PCD, programmed cell death; PINK1, PTEN-induced putative kinase 1; ROS, reactive oxygen species; SA-β-Gal, senescence-associated β-galactosidase; TLR, toll-like receptor; WB, western blotting
Introduction
Advanced age is one of the most important risk factors for development of chronic obstructive pulmonary disease (COPD) and an increased number of senescent cells is a major feature of aging.1-3 Recent advances COPD have implicated acceleration of cell senescence in both alveolar and airway epithelial cells, which is functionally characterized by impaired cell regeneration and aberrant cytokine secretion of the senescence-associated secretory phenotype (SASP), in COPD pathogenesis.2,4-9 Although the molecular mechanism underlying cell senescence regulation is still unknown, reactive oxygen species (ROS) released during mitochondrial respiration has been generally implicated in the progression of cellular senescence.3,10 Mitochondrial respiratory chain dysfunction accompanying enhanced ROS production can be attributed to cigarette smoke (CS) exposure, a main cause for COPD development.11,12
Mitochondrial quality is maintained via the optimal balance between biogenesis and degradation for renewal, and mitochondria are also highly dynamic organelles undergoing continuous fusion and fission cycles.13,14 Fusion has been shown to rescue damaged mitochondria by redistributing proteins and by maintaining mitochondrial DNA integrity.15 In case of more extensive damage, segregation of irreversibly damaged mitochondria through fission is a prerequisite for appropriate engulfment and degradation via mitochondria selective autophagy known as mitophagy.16 Hence, orchestrated mitochondrial biogenesis, dynamics, and degradation by mitophagy constitute a series of quality control measures to prevent the accumulation of damaged mitochondria and excessive ROS production.3,17
Mitophagy is a highly conserved mechanism of selectively delivering unwanted mitochondria for lysosomal degradation.16 Proteins localized on the mitochondrial outer membrane, including BNIP3L, BNIP3, and FUNDC1 (FUN14 domain containing 1), are specific receptors for mitophagic recognition during red blood cell maturation, metabolic stress, and hypoxia.18-21 Thus far, the PINK1-PARK2 pathway has been largely implicated in the removal of damaged mitochondria with depolarized membranes.22 Stress-induced membrane depolarization stabilizes PINK1, resulting in recruitment of PARK2, an E3-ubiquitin ligase, to mitochondria. PARK2-mediated ubiquitination of mitochondrial substrates, including BCL2, MFNs (mitofusins), and VDAC (voltage-dependent anion channel), is required for the binding of the receptor protein SQSTM1/p62, which can recognize both ubiquitinated substrates and microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3) on phagophores, the precursor to autophagosomes.17,22 Therefore, concomitant accumulation of SQSTM1 and ubiquitinated proteins is now widely recognized as at least partly reflecting insufficient autophagy, including mitophagy.23
In addition to accumulation of ubiquitinated proteins and SQSTM1 resulting from insufficient autophagic degradation, we have recently demonstrated an increase in fragmented mitochondria in the airway epithelial cells of COPD lung.8,24,25 Furthermore, our in vitro experiments demonstrate that cigarette smoke extract (CSE)-induced accumulation of fragmented mitochondria is accompanied by increased mitochondrial ROS production and acceleration of human bronchial epithelial cell (HBEC) senescence.24 Taken together, it is likely that insufficient mitophagic elimination is involved in the accumulation of fragmented mitochondria in response to CS exposure in COPD lung. Hence, we examined the involvement of the PINK1-PARK2 pathway for mitophagic elimination of damaged mitochondria in regulation of mitochondrial ROS production and cellular senescence in HBEC in the context of COPD pathogenesis.
Results
CSE induces mitochondrial ROS production and cellular senescence in HBEC
We have recently reported that depolarized mitochondria-derived ROS is involved in the progression of cellular senescence in response to CSE exposure.24 In line with this observation, MitoTracker Red staining demonstrated that CSE induced depolarization of the mitochondrial membrane potential (Fig. 1A). This was accompanied by increased ROS production as determined by the DCFH-DA assay for total ROS and DHR123 staining for mitochondrial ROS, respectively (Fig. 1B and C). To clarify the involvement of ROS in CSE-induced HBEC senescence, we performed western blotting of CDKN2A/p16 and CDKN1A/p21 (senescence-associated cyclin dependent kinase inhibitors). Increased CDKN2A and CDKN1A expression levels indicated acceleration of cellular senescence. Both N-acetylecysteine (NAC) (an efficient antioxidant for intracellular ROS) and Mito-TEMPO (a specific antioxidant for mitochondrial ROS) efficiently inhibited the increase of mitochondrial ROS production and cellular senescence mediated by CSE exposure (Fig. 1C and D). CSE-induced acceleration of HBEC senescence and efficient inhibition by antioxidant treatments were further confirmed by means of phospho-Histone H2AFX/H2A.X (Ser139) staining of DNA damage (Fig. 1E).
Figure 1.

CSE induces mitochondrial damage and ROS production accompanied by cell senescence in HBEC. (A) Fluorescence microscopy detection of Hoechst 33258 (left panels), MitoTracker Red staining (middle panels), and TOMM20 staining of mitochondria (right panels). HBEC were treated with CSE (1.0%) for 24 h (lower panels). Bar: 100 μm. (B) Fluorescence intensity of DCFH-DA staining for intracellular ROS production in response to CSE exposure. The fluorescence level in the control-treated cells was designated as a 1.0. * P < 0.05. (C) Fluorescence microscopy detection of Hoechst 33258 (upper panels) and DHR123 staining (lower panels). HBEC were pretreated with NAC (1 mM) and Mito-TEMPO (100 μM) for 3 h before CSE exposure, (1% CSE for 24 h). Bar: 20 μm. (D) Western blotting (WB) using anti-CDKN2A, anti-CDKN1A, and anti-ACTB in control-treated (lane 1, 2), NAC (1 mM)-treated (lane 3, 4), and Mito-TEMPO (100 μM)-treated (lane 5, 6) HBEC. Pretreatment with NAC (1 mM) and Mito-TEMPO (100 μM) for 3 h before CSE exposure, (1% for 48 h). Middle panel is the average (±SEM) of the relative increase in CDKN2A normalized to ACTB and the right panel is the average (±SEM) of the relative increase in CDKN1A normalized to ACTB, which are taken from densitometric analysis of WB from 3 independent experiments. Open bar is no treatment, filled bar is CSE-treated. *P < 0.05. (E) Photographs of immunofluorescent staining of phospho-Histone H2AFX (Ser139) in HBEC treated with CSE (1.0%) for 48 h in the absence or presence of NAC (1 mM) and Mito-TEMPO (100 μM). Bar: 100 μm. Shown in the right panel is the percentage (±SEM) of phospho-histone H2AFX (Ser139)-positive cells from 3 independent experiments. Open bar is no treatment, filled bar is CSE-treated. *P < 0.05.
Involvement of mitophagy in regulation of CSE-induced ROS production
To examine the autophagic degradation of mitochondria (mitophagy) in response to CSE exposure, confocal microscopy was performed in BEAS-2B cells stably expressing EGFP-LC3B. Colocalization of TOMM20-stained mitochondria and EGFP-LC3B dots was used to determine autophagosome formation. Although CSE induced EGFP-LC3B dot formation, colocalization with mitochondria was barely detected in the absence of bafilomycin A1 (Baf A1), an inhibitor of autolysosomal maturation (Fig. 2A). Baf A1 treatment alone induced EGFP-LC3B dot formation accompanied by limited colocalization with TOMM20-stained mitochondria (yellow dots), indicating baseline mitophagy (Fig. 2A). CSE treatment markedly enhanced EGFP-LC3B dot formation with concomitant colocalization with TOMM20-stained mitochondria in the presence of Baf A1 (Fig. 2A), indicating CSE-induced mitophagy activation. Consistent with our recent finding, accumulation of fragmented mitochondria was also observed following CSE treatment, especially in the presence of Baf A1.24 CSE-induced mitophagy was further confirmed in HBEC by electron microscopy (EM) evaluation. CSE treatment slightly but significantly increased the number of autophagic vacuoles containing mainly deforming mitochondria, which was markedly enhanced by concomitant Baf A1 treatment (Fig. 2B; Fig. S1). CSE treatment decreased total mitochondria count with an increased ratio of fragmented mitochondria. Concomitant Baf A1 treatment significantly enhanced cytoplasmic accumulation of fragmented mitochondria (Fig. 2B; Fig. S1). In contrast to Baf A1, Torin1, an autophagy inducer, demonstrated only a slight increase in the number of autophagosomes and mitochondria, but enhanced degradation of mitochondria inside of autophagic vacuoles, which appeared to be associated with significant decrease in the number of fragmented mitochondria in the cytoplasm (Fig. 2B; Fig. S1).
Figure 2.

CSE induces mitophagy and mitochondrial ROS production in HBEC. (A) Colocalization analysis of confocal laser scanning microscopy images of TOMM20 staining and EGFP-LC3B. pEGFP-LC3B expressing BEAS-2B cells were treated with CSE (1.0%) for 48 h in the presence or absence of bafilomycin A1 (Baf A1, 200 nM). Bar: 20 μm (B) Electron microscopy detection of mitochondria and autophagosomes in HBEC. HBEC were treated with CSE (1.0%) for 48 h in the absence or presence of Baf A1 (200 nM) and Torin1 (250 nM). Bar: 2 μm. The middle panel is the average (±SEM) count of autophagosomes taken from a 10-image field (10000 X) for each sample. Open bar is total autophagosomes, filled bar is autophagosomes containing mitochondria. *P < 0.05. The lower panel is the average (±SEM) count of mitochondria taken from 10 image fields (10000 X) for each sample. Mitochondria shorter than 1 μm without fusion to other mitochondria were counted as fragmented. Open bar is total mitochondria, filled bar is fragmented mitochondria. *P < 0.05, **P < 0.001. (C) WB using anti-ubiquitin, anti-SQSTM1, and anti-TOMM20 in the presence of Baf A1 (200 nM) (lane 3, 4) and Torin1 (250 nM) (lane 5, 6). HBEC were treated with CSE (1.0%) for 48 h and protein samples from mitochondrial fractions were collected. Shown is a representative experiment of 4 showing similar results. The middle panel is the average (±SEM) relative increase in ubiquitin normalized to TOMM20 and the lower panel is the average (±SEM) relative increase in SQSTM1 normalized to TOMM20, taken from densitometric analysis of WB from 3 independent experiments. Open bar is no treatment, filled bar is CSE-treated. *P < 0.05, **P < 0.001. (D) Fluorescence intensity of DCFH-DA staining for intracellular ROS production in response to CSE exposure. HBEC were treated with CSE (1.0%, 1 h) in the presence of Baf A1 (200 nM) (lane 3, 4) and Torin1 (250 nM) (lane 5, 6). The fluorescence level in the control-treated cells was designated as 1.0 and shown in panel is the average (±SEM). Open bar is no treatment, filled bar is CSE-treated.* P < 0.05. (E) MitoSOX Red staining for mitochondrial ROS production. Fluorescence microscopy detection was performed. HBECs were treated with CSE (1.0%) for 24 h in the presence of Baf A1 (200 nM) (middle panels) and Torin1 (250 nM) (right panels). Bar: 20 μm.
PARK2-induced ubiquitination of mitochondrial substrates is a prerequisite for the binding of the autophagy receptor protein SQSTM1,17 hence accumulation of ubiquitinated proteins and SQSTM1 in the mitochondrial fraction can be interpreted as reflecting increased mitochondrial damage without sufficient elimination. Significantly increased expression levels of both ubiquitinated proteins and SQSTM1 were observed in the mitochondrial fraction following CSE treatment, which was further enhanced in the presence of Baf A1. Conversely, Torin1 reduced accumulations of ubiquitinated protein and SQSTM1. These data suggest incomplete mitophagic degradation of damaged mitochondria in the setting of CSE exposure in HBEC (Fig. 2C). Next, to evaluate the association between mitophagy and ROS production, we performed DCFH-DA assays and MitoSOX Red staining in HBEC (Fig. 2D and E). Consistent with accumulations of damaged mitochondria, Baf A1 significantly enhanced CSE-induced total and mitochondrial ROS production, which was reduced by Torin1. These data suggest a causal link between insufficient mitophagy and excessive ROS production. Taken together, mitophagy may play a key regulatory role in the elimination of CSE-induced mitochondrial damage and ROS production in HBEC.
PINK1 regulates mitophagy, cellular senescence, and PARK2 recruitment to mitochondria in response to CSE exposure in HBEC
To clarify the involvement of PINK1 in mitophagy and PARK2 recruitment to the mitochondrial fraction, PINK1 siRNA was employed and efficient knockdown was observed by western blotting (Fig. 3E). Confocal microscopy evaluation was performed in control and PINK1 siRNA-transfected BEAS-2B cells. PINK1 siRNA-transfected BEAS-2B cells exhibited a marked decrease in colocalization of TOMM20-stained mitochondria and EGFP-LC3B dots (autophagosome) in response to CSE exposure (Fig. 3A). PINK1 knockdown also enhanced CSE-induced mitochondrial ROS production and HBEC senescence (Fig. 3B to E). PINK1 knockdown noticeably reduced PARK2-HA levels in the mitochondrial fraction, while increased PARK2-HA levels were observed in the cytosolic fraction in PARK2-HA-transfected HBEC, supporting the notion that PINK1 is responsible for translocation of PARK2 from the cytoplasm to the mitochondria (Fig. 3F). The involvement of PINK1 in PARK2 recruitment to the mitochondria was once again demonstrated via a decrease in colocalization of TOMM20 staining of mitochondria and PARK2-HA in response to CSE exposure in PINK1 knockdown HBEC (Fig. 3G).
Figure 3 (See previous page).

PINK1 regulates CSE-induced mitophagy, ROS production, cell senescence, and PARK2 recruitment to mitochondria in HBEC. (A) Colocalization analysis of confocal laser scanning microscopy images of TOMM20 staining and EGFP-LC3B. pEGFP-LC3B expressing BEAS-2B cells were transfected with nonsilencing control siRNA and PINK1 siRNA, and CSE treatment (1.0%) was started 24 h post-siRNA transfection. After 24 h treatment with CSE, BEAS-2B cells were fixed for staining. Bar: 20 μm (B) Photographs of fluorescence staining of control or CSE (1.0%) treated HBEC with Hoechst 33258 (left panels) and with MitoSOX Red (right panels). HBEC were transfected with nonsilencing control siRNA and PINK1 siRNA, and CSE treatment (1.0%) was started 24 h post-siRNA transfection. HBEC were treated with CSE for 24 h. Bar: 100 μm (C) Fluorescence intensity of DCFH-DA staining for intracellular ROS production in response to CSE exposure. HBEC were transfected with nonsilencing control siRNA and PINK1 siRNA, and CSE treatment (1.0%) was started 24 h post-siRNA transfection. The fluorescence level in the control-treated cells was designated as 1.0 and shown in panel is the average (±SEM). Open bar is no treatment, filled bar is CSE-treated.*P < 0.05. (D) SA-β-gal staining of control or PINK1 siRNA-transfected HBEC. CSE treatment (1.0%) was started 24 h post-siRNA transfection and staining was performed after 48 h treatment. Shown is the percentage (±SEM) of SA-β-gal-positive cells from 3 independent experiments. Open bar is no treatment, filled bar is CSE-treated. *P < 0.05. (E) WB using anti-CDKN1A and anti-ACTB in nonsilencing control siRNA-transfected (lane 1, 2) and PINK1 siRNA-transfected HBEC (lane 3, 4). CSE treatment (1.0%) was started 24 h post-siRNA transfection and protein samples were collected after 48 h treatment. Shown is a representative experiment of 3 with similar results. The lower panel is the average (±SEM) relative increase in CDKN1A normalized to ACTB, taken from densitometric analysis of WB from 3 independent experiments. Open bar is no treatment, filled bar is CSE-treated. *P < 0.05, **P < 0.001. (F) WB using anti-PARK2, anti-TOMM20, and anti-ACTB in PARK2-HA vector and nonsilencing control siRNA-transfected (lane1, 2), and PARK2-HA vector and PINK1 siRNA-transfected HBEC (lane 3, 4). CSE treatment (1.0%) was started 24 h post-transfection and protein samples for mitochondrial and cytosolic fractions were collected after 48 h treatment. Shown is a representative experiment of 3 showing similar results. The middle panel is the average (±SEM) relative increase in PARK2-HA normalized to TOMM20 in mitochondrial fraction, taken from densitometric analysis of WB from 4 independent experiments. Open bar is no treatment, filled bar is CSE-treated. The right panel is the average (±SEM) relative increase in PARK2-HA normalized to ACTB in cytosolic fraction, taken from densitometric analysis of WB from 3 independent experiments. Open bar is no treatment, filled bar is CSE-treated. *P < 0.05. (G) Photographs of fluorescence staining in HBEC transfected with PARK2-HA vector and control siRNA or PARK2-HA vector and PINK1 siRNA. CSE treatment (1.0%) was started 24 h post-transfection and staining was performed after 24 h treatment. PARK2 expression was detected using an anti-HA antibody and anti-TOMM20 antibody was used for mitochondria. Pseudocolored and merged images of PARK2-HA and TOMM20 were created (Adobe photoshop) from images acquired using a digital imaging system. Bar: 10 μm.
PARK2 regulates mitophagy, ROS production, and cellular senescence in response to CSE exposure in HBEC
PARK2 expression levels were slightly increased in the mitochondrial fraction in response to CSE exposure (Fig. 4A) and PARK2 has been proposed to regulate ubiquitination of mitochondrial substrates.22 To clarify the involvement of PARK2 in regulation of ubiquitination with concomitant SQSTM1 accumulation in the mitochondrial fraction, we employed siRNA for PARK2, which resulted in an efficient knockdown that was observed by western blotting (Fig. 4A). CSE-induced accumulation of ubiquitinated proteins and SQSTM1 in the mitochondrial fraction was appreciably reduced by PARK2 knockdown (Fig. 4A). To confirm the regulatory role of PARK2 in mitophagy, confocal microscopy evaluation was performed in control and PARK2 siRNA-transfected BEAS-2B cells (Fig. 4B). Consistent with PINK1 knockdown experiments, PARK2 siRNA-transfected BEAS-2B cells exhibited a marked decrease in colocalization of TOMM20-stained mitochondria and EGFP-LC3B dots after CSE treatment. Intriguingly, CSE-induced EGFP-LC3B dot formation was also decreased by PARK2 knockdown, suggesting the potential involvement of PARK2 in not only ubiquitination for mitophagic recognition but also in autophagosome formation following CSE exposure. Involvement of PARK2 in mitophagy was further confirmed by demonstrating the colocalization of EGFP-LC3B dots and PARK2 in response to CSE exposure in PARK2-HA-transfected BEAS-2B cells (Fig. 4C). PARK2 knockdown also enhanced CSE-induced mitochondrial ROS production shown by DCFH-DA assay and MitoSOX Red staining (Fig. 4D and E) and also enhanced HBEC senescence in response to CSE exposure as measured by SA-β-gal staining and expression of CDKN1A (Fig. 4F and G). The regulatory role of PARK2 in mitophagy was further confirmed by transfection experiments of PARK2-HA. PARK2 overexpression efficiently reduced mitochondrial ROS production and HBEC senescence by CSE exposure (Fig. 4H and I).
Figure 4 (See previous page).

PARK2 regulates CSE-induced mitochondrial ROS production and cellular senescence in HBEC. (A) WB using anti-ubiquitin, anti-SQSTM1, anti-PARK2, and anti-TOMM20 in nonsilencing control siRNA (lane 1, 2) and PARK2 siRNA-transfected HBEC (lane 3, 4). CSE treatment (1.0%) was started 24 h post-siRNA transfection and protein samples from mitochondrial fractions were collected after 48 h treatment. Shown is a representative experiment of 3 showing similar results. The middle panel is the average (±SEM) relative increase in ubiquitin normalized to TOMM20 and the right panel is the average (±SEM) relative increase in SQSTM1 normalized to TOMM20, which are taken from densitometric analysis of WB from 3 independent experiments. Open bar is no treatment, filled bar is CSE-treated. *P < 0.05. (B) Colocalization analysis of confocal laser scanning microscopy images of TOMM20 staining and EGFP-LC3B. pEGFP-LC3B -expressing BEAS-2B cells were transfected with a nonsilencing control siRNA and PARK2 siRNA, and CSE treatment (1.0%) was started 24 h post-siRNA transfection. After 24 h treatment with CSE, BEAS-2B cells were fixed for staining. Bar: 20 μm (C) Photographs of fluorescence staining of control or CSE (1.0%)-treated BEAS-2B cells expressing EGFP-LC3B in the presence of Baf A1. BEAS-2B cells were transfected with PARK2-HA expression vector and PARK2 expression was detected using an anti-HA antibody. Pseudocolored and merged images of PARK2-HA and EGFP-LC3B were created (Adobe photoshop) from images acquired using a digital imaging system. Bar: 10 μm (D) Fluorescence intensity of DCFH-DA staining for intracellular ROS production in response to CSE exposure. HBEC were transfected with nonsilencing control siRNA and PARK2 siRNA, and CSE treatment (1.0%) was started 24 h post-siRNA transfection. The fluorescence level in the control treated cells was designated as 1.0 and shown in panel is the average (±SEM). Open bar is no treatment, filled bar is CSE-treated. *P < 0.05, **P < 0.001. (E) Photographs of Hoechst 33258 (upper panels) and MitoSOX Red (lower panels) fluorescence staining in control or CSE (1.0%)-treated HBEC. HBEC were transfected with nonsilencing control siRNA and PARK2 siRNA, and CSE treatment (1.0%) was started 24 h post-siRNA transfection. HBEC were treated with CSE for 24 h. Bar: 50 μm (F) SA-β-gal staining of control or PARK2 siRNA-transfected HBEC. CSE treatment (1.0%) was started 24 h post-siRNA transfection and staining was performed after 48 h treatment. Shown is the percentage (±SEM) of SA-β-gal-positive cells from 3 independent experiments. Open bar is no treatment, filled bar is CSE-treated. *P < 0.05, **P < 0.001. (G) WB using anti-CDKN1A and anti-ACTB in nonsilencing control siRNA-transfected (lane 1, 2) and PARK2 siRNA-transfected HBEC (lane 3, 4). CSE treatment (1.0%) was started 24 h post-siRNA transfection and protein samples were collected after 48 h treatment. Shown is a representative experiment of 3 showing similar results. The right panel is the average (±SEM) of relative increase in CDKN1A normalized to ACTB, taken from densitometric analysis of WB from 3 independent experiments. Open bar is no treatment, filled bar is CSE-treated. *P < 0.05, **P < 0.001. (H) Photographs of Hoechst 33258 (upper panels) and MitoSOX Red (lower panels) fluorescence staining in control or CSE (1.0%)-treated HBEC. HBEC were transfected with control and PARK2-HA expression vector, and CSE treatment (1.0%) was started 24 h post transfection. HBEC were treated with CSE for 24 h. (I) WB using anti-PARK2, anti-CDKN1A, and anti-ACTB in control vector-transfected (lane 1, 2) and PARK2-HA expression vector-transfected HBEC (lane 3, 4). CSE treatment (1.0%) was started 24 h post-transfection and protein samples were collected after 48 h treatment. Shown is a representative experiment of 3 showing similar results. The lower panel is the average (±SEM) of relative increase in CDKN1A normalized to ACTB, taken from densitometric analysis of WB from 3 independent experiments. Open bar is no treatment, filled bar is CSE-treated. *P < 0.05, **P < 0.001.
Decreased PARK2 expression levels in lung from COPD patients
To clarify the importance of PINK1-PARK2 pathway-mediated mitophagy in COPD pathogenesis, we examined the protein expression levels of PINK1 and PARK2 in lung homogenates from nonsmokers (n = 10), non-COPD smokers (n = 10), and COPD patients (n = 10), respectively. The smoking index (pack year) was significantly different from nonsmokers but there was no difference between non-COPD smokers and COPD (Table 1). In line with recent findings of increased expression in bronchial epithelial cells from COPD patients, PINK1 tended to be highly expressed in lung homogenates from COPD patients, which may reflect accumulation of damaged mitochondria (Fig. 5A).26 However, PINK1 demonstrated no significant correlation with percentage of forced expiratory volume in one second (FEV1)/forced vital capacity (FVC) (Fig. 5B). Conversely, significantly lower expression levels of PARK2 were detected in lung homogenates from COPD patients compared to those in lung homogenates from non-COPD smokers. Interestingly, a slight increase in PARK2 expression levels was observed in lung homogenates from non-COPD smokers. Furthermore, PARK2 expression levels were positively correlated with percentage of FEV1/FVC (Fig. 5C and D).
Table 1.
Patient characteristics (for lung homogenates)
| Nonsmoker (n = 10) | Non-COPD smoker (n = 10) | COPD (n = 10) | p value | |
|---|---|---|---|---|
| Age, years | 65.0 ± 14.6 | 69.0 ± 7.9 | 69.3 ± 8.1 | NS |
| Male, % of group | 20.0 | 90.0 | 90.0 | NA |
| SI (pack year) | 0 | 56.9 ± 28.9* | 48.2 ± 25.4* | <0.001 |
| FEV1/FVC (%) | 75.3 ± 8.5 | 77.1±3.9 | 61.1 ± 7.8* | <0.001 |
COPD, chronic obstructive pulmonary disease; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; NA, not assessed; NS, not statistically significant; SI, Smoking index; Values are mean ± SD.
Figure 5.

PINK1 and PARK2 expression levels in lung homogenates. (A) WB using anti-PINK1 and anti-ACTB of lung homogenates from nonsmoker, smoker without COPD, and COPD. The lower panel is the average (±SEM) taken from densitometric analysis of WB. Open bar is nonsmoker (n = 10), filled bar is smoker without COPD (n = 10), and horizontal crosshatched bar is COPD (n = 10). (B) Shown is the relationship between relative PINK1 expression normalized to ACTB and the percentages of FEV1/FVC (n = 30). (C) WB using anti-PARK2 and anti-ACTB of lung homogenates from nonsmoker, smoker without COPD, and COPD. The lower panel is the average (±SEM) taken from densitometric analysis of WB. Open bar is nonsmoker (n = 10), filled bar is smoker without COPD (n = 10), and horizontal crosshatched bar is COPD (n = 10). *P < 0.05. (D) Shown is the relationship between relative PARK2 expression normalized to ACTB and the percentages of FEV1/FVC (n = 30).
To further elucidate the physiological relevance of PARK2 expression levels in COPD pathogenesis, we performed immunohistochemical evaluations of PARK2 in nonsmokers (Fig. 6A and B), non-COPD smokers (Fig. 6C and D), and COPD lungs (Fig. 6E to H). Patient characteristics are presented in Table 2. Airway epithelial cells are known to be representative cells in the expression of high levels of PARK2 in lung tissue. In line with results in lung homogenate, airway epithelial cells in small airways exhibited significantly decreased expression levels of PARK2 in COPD lungs compared to those in lungs from nonsmokers and non-COPD smokers (Fig. 6I).
Figure 6.
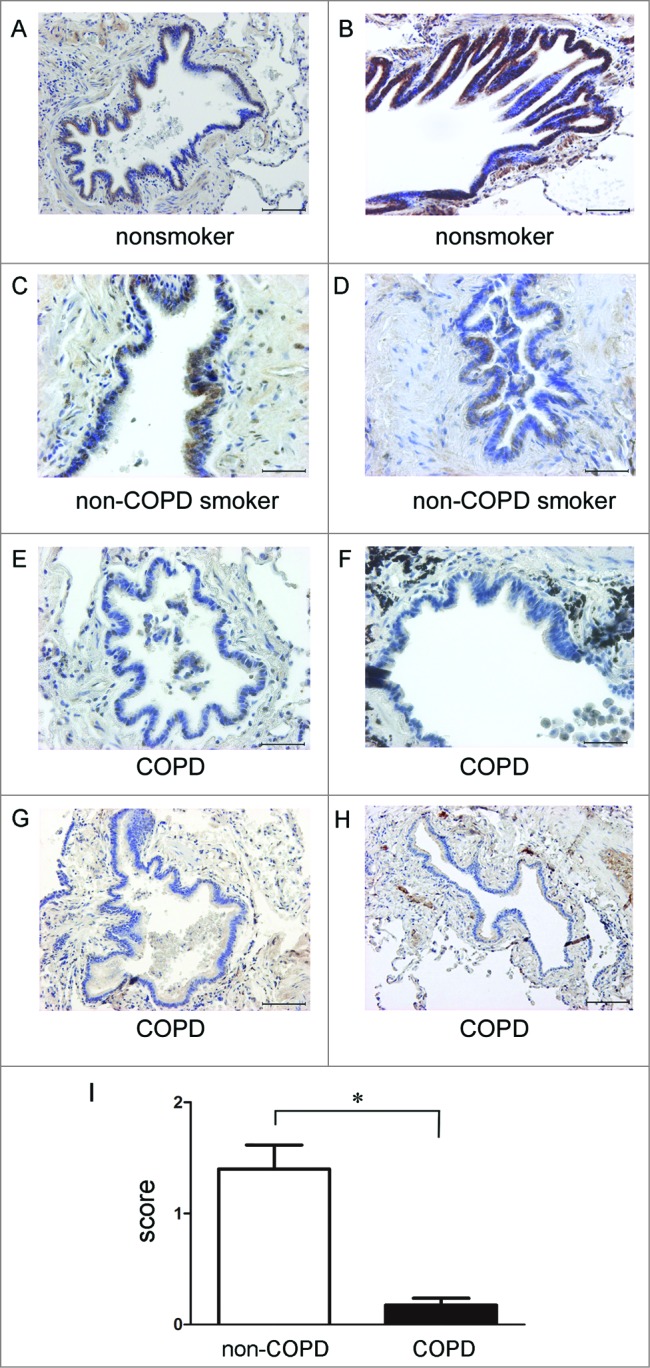
PARK2 expression in normal and COPD lung tissues. Immunohistochemical staining of PARK2 in nonsmoker, non-COPD smoker, and COPD lung tissues: Photomicrographs of small airway in nonsmoker (A and B), non-COPD smoker (C and D), and COPD lungs (E to H). Bar: 100 μm in A, B, G, H. Bar = 50 μm in C, D, E, F. (I) Shown is the average of semiquantitative score (±SEM) of PARK2-positive cells in small airway epithelial cells. Open bar is non-COPD (n = 4, total 18 small airways) and filled bar is COPD (n = 4, total 16 small airways). *P < 0.05.
Table 2.
Patient characteristics (for immunohistochemistry)
| Case | Age | Sex | Smoking index | FEV1/FVC (%) | %FEV1(%) |
|---|---|---|---|---|---|
| A | 34 | F | 0 | 95.7 | 90.8 |
| B | 48 | F | 0 | 70.7 | 97.2 |
| C | 65 | M | 50 | 77.7 | 117.8 |
| D | 77 | M | 80 | 74.8 | 121.4 |
| E | 64 | M | 64 | 68.2 | 88.9 |
| F | 61 | M | 40 | 68.6 | 84.1 |
| G | 64 | M | 44 | 57.0 | 99.6 |
| H | 60 | M | 88 | 68.1 | 81.0 |
FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; %FEV1, percentage predicted forced expiratory volume in 1 second.
Taken together, reduced PARK2 expression levels in small airway epithelial cells might be associated with insufficient mitophagy and accelerated cell senescence as a part of the pathogenic sequence of COPD development (Fig. 7).
Figure 7.

Hypothetical model of involvement of mitophagy in the regulation of cell fate by smoking stress in COPD pathogenesis. Insufficient mitophagy, which can be attributed to reduced PARK2 expression, results in accumulation of damaged mitochondria in response to cigarette smoke exposure. Accumulation of damaged mitochondria accompanied by increased reactive oxygen species production induces cellular senescence. Conversely, excessive mitophagy activation accompanied by serious mitochondrial damage in response to severe smoking stress may induce PCD, including apoptosis and necroptosis. Mitophagy may lead to either senescence or PCD depending on the amount of damage as a part of COPD pathogenesis.
Discussion
Damaged mitochondria are major sources of aberrant ROS production and mitochondrial quality control has been proposed to be a critical determinant in cell fate, including senescence.10,16,27 In this study, we demonstrate that mitophagic elimination of damaged mitochondria has a pivotal role in regulation of mitochondrial ROS production, which is involved in acceleration of HBEC senescence in response to CSE exposure. PINK1 and PARK2 knockdown experiments clarify that both PINK1 and PARK2 are essential for mitophagy in response to CSE exposure and PINK1-mediated recruitment of PARK2 to mitochondria appears to be a prerequisite for ubiquitination and mitophagic recognition (Fig. 3 and 4). Furthermore, we see positive correlation between PARK2 expression levels in lung homogenates and lung function, and immunohistochemistry shows a decrease in PARK2 expression in airway epithelial cells of COPD lungs, suggesting a clinical implication for reduced PARK2 expression in COPD pathogenesis. Conversely modestly increased PINK1 levels may reflect accumulation of damaged mitochondria, which is conferred by insufficient mitophagic degradation in COPD lung.
Improper mitophagic elimination of damaged mitochondria has been implicated in the pathogenesis of a variety of diseases,10,16,27 and we have recently demonstrated insufficient autophagy in COPD lung.8,25 Hence, we assumed that our findings of accumulations of fragmented mitochondria might result from insufficient mitophagy in the airway epithelial cells of COPD lung and also in CSE-exposed HBEC.8,24,25 Supporting this notion, autophagy and mitophagy activation induced by Torin1 resulted in enhanced degradation of autophagosomal contents including mitochondrial debris and an increase in undamaged mitochondrial number as shown by EM, and also reduced CSE-induced accumulation of ubiquitinated proteins and SQSTM1 in the mitochondrial fraction of HBEC (Fig. 2). Furthermore, Torin1 attenuated mitochondrial ROS production in response to CSE exposure (Fig. 2), suggesting the involvement of mitophagic regulation of damaged mitochondria accompanied by enhanced ROS production in cigarette-induced lung pathology.
In line with the recent findings of aging-associated neurodegenerative disorders,28,29 the PINK1-PARK2 pathway is involved in regulating the mitophagic elimination of CSE-induced damaged mitochondria that modulates ROS production and cellular senescence in HBEC (Fig. 3 and 4). Although PINK1 and PARK2 are both crucial for mitophagy,30 a recent paper shows upregulation of PINK1 in COPD lung, which is interpreted as reflecting accumulation of mitochondrial damage.26 Consistently, we observed modestly increased PINK1 levels in lung homogenates from COPD patients, whereas PARK2 levels were decreased and there was a positive correlation between PARK2 levels and percentage of FEV1/FVC (Fig. 5 and 6). Additionally, PARK2 levels in non-COPD smokers tend to be higher than those in control lung, indicating the possibility that PARK2 expression in airway epithelial cells is a critical determination of not only mitophagy activity but also predisposition to cigarette-induced COPD development. It has been reported that PARK2 overexpression increases mitochondrial activity during aging in association with life-span extension, further supporting the notion that PARK2 has a pivotal role in mitochondrial quality control in terms of regulation of senescence-associated disorders.31 However, the precise mechanism for the decrease in PARK2 levels in COPD lung remains unclear. Furthermore, several lines of evidence demonstrate the participation of other receptors during autophagosomal recognition of mitochondria, including BNIP3L, BNIP3, and FUNDC1, hence the potential involvement of those receptors in mitophagy regulation of cigarette-induced cellular senescence need to be examined in future studies.18-21
Programmed cell death (PCD) has been widely implicated in COPD pathogenesis and the involvement of autophagy, including mitophagy, has been reported in not only apoptosis but also programmed necrosis, necroptosis.32,33 The central purpose of both PCD and cell senescence is eliminating damaged cells for tissue regeneration, indicating that the extent of cell damage may be critical for determination of cell fate.34 In contrast to our finding of a protective role for mitophagy in cell senescence induced by lower concentration of CSE (1.0%), a recent paper showing involvement of PINK1-mediated mitophagy in necroptosis used higher concentration of CSE (20%),32 raising the following possibilities. First, excessive mitophagy activation by lethal stress may induce PCD, suggesting that mitophagy can lead to either PCD or senescence depending on the amount of damage. In other words, once a certain threshold of mitochondrial damage is reached, the choice is made by mitophagy between PCD and senescence (Fig. 7). Second, PINK1 has been shown to induce not only mitophagy but also mitochondrial depolarization during CSE exposure, indicating that PINK1-mediated mitochondrial damage has a dominant role in the setting of higher concentrations of CSE, resulting in PCD.32 Mitochondrial division inhibitor-1 (Mdivi-1), which was used as a mitophagy inhibitor for necroptosis reduction,32 has also been known to attenuate mitochondrial damage via inhibition of mitochondrial fission and mitochondrial outer membrane permeabilization (MOMP), suggesting that both mitophagy and mitochondrial integrity have a key regulatory role in determining cell fate in response to CSE exposure.
Although we have demonstrated the involvement of insufficient mitophagy-mediated ROS production in regulation of CSE-induced HBEC senescence, a causal link between mitochondrial ROS and cellular senescence remains controversial.3 Intriguingly, recent advances demonstrate that mitophagy also controls inflammatory reactions. Inappropriate mitophagic degradation of mitochondrial DNA induces inflammatory reactions through TLR9 (toll-like receptor 9) recognition and activation of the NALP3 inflammasome.35,36 Although a direct contribution of TLR9 and inflammasome activation to COPD pathogenesis is still unclear,37,38 IL1B/interleukin 1β, a representative senescence-associated secretory phenotype (SASP) factor involved in COPD development,39,40 is produced and activated through TLR9 signaling and inflammasome machinery.41,42 Accordingly, insufficient mitophagy-mediated cell autonomous activation of TLR9 and the inflammasome may partly explain persistent inflammation after smoking cessation, which may also be associated with inflammation-mediated acceleration of cell senescence during COPD progression.43 However, we understand the potential limitations of our in vitro experimental models using short-term CSE exposure to elucidate the mechanisms for COPD development. To clarify the clinical relevance of mitophagy in COPD pathogenesis, future studies need to be performed using appropriate animal models for COPD development in conjunction with mitophagy alteration, specifically modulation of the PINK1-PARK2 pathway.
In summary, we demonstrated that the PINK1-PARK2 pathway is involved in the modulation of CSE-induced HBEC senescence through the regulation of mitophagy and mitochondrial ROS production. Reduced PARK2 expression levels in COPD lung further support the notion that insufficient mitophagy is a part of the pathogenic sequence of COPD development, which may at least partly explain the mechanism for our recent findings of insufficient autophagy in COPD lung.8 Therefore, modalities to achieve specific and appropriate levels of mitophagy activation may be a promising therapeutic option to regulate the aging-associated pathology, COPD.
Materials and Methods
Cell culture, antibodies, and reagents
Normal airways were obtained from 1st through 4st order bronchi from pneumonectomy and lobectomy specimens for primary lung cancer. Informed consent was obtained from all surgical participants as part of an approved ongoing research protocol by the ethics committee of Jikei University School of Medicine. HBEC were isolated with protease treatment and characterized as previously described.40 HBEC were serially passaged and used for experiments until passage 3. The majority of experiments were performed with HBEC from non-COPD patients. The bronchial epithelial cell line BEAS-2B was cultured in RPMI1640 (Gibco Life Technologies, 11875-093) with 10% fetal calf serum (Gibco Life Technologies, 26140-079) and penicillin-streptomycin (Gibco Life Technologies, 15140-122).
Antibodies used were rabbit anti-CDKN1A (Cell Signaling Technology, 2947), rabbit anti-CDKN2A (Santa Cruz Biotechnology, sc-468), rabbit anti-phospho-histone H2AFX/H2A.X (Ser139) (Cell Signaling Technology, 2577), mouse anti-poly- and monoubiquitin (Enzo Life Sciences, BML-PW8810), rabbit anti-SQSTM1 (MBL, PM045), rabbit anti-PARK2 (Cell Signaling Technology, 2132), rabbit anti-PINK1 (Abcam, ab23707), rabbit anti-hemagglutinin (HA) (Cell Signaling Technology, 3724), and mouse anti-ACTB (Sigma-Aldrich, A5316). Bafilomycin A1 (Baf A1) (Sigma-Aldrich, B1793), Torin 1 (Tocris Bioscience, 4247), Hoechst 33258 (Sigma-Aldrich, 861405), Dihydrorhodamine (DHR) 123 (Molecular Probes-Life Technologies, D23806), MitoSOX Red (Molecular Probes-Life technologies, M36008), N-acetylcysteine (NAC; Wako, 017-01531), Mito-TEMPO (Enzo Life Sciences, ALX-430-150), and rat tail COL1/type-I collagen (Sigma-Aldrich, C3867) were purchased.
Plasmids, siRNA, and transfection
The LC3B cDNA was the kind gift of Dr. Mizushima (Tokyo University, Tokyo, Japan) and Dr. Yoshimori (Osaka University, Osaka, Japan), and was cloned into pEGFP-C1 vector.44 PARK2 expression vector was obtained from Addgene (Addgene, 17613). Small interfering RNA (siRNA) targeting PARK2 (Applied Biosystems Life Technologies, s10043, s10044), PINK1 (Applied Biosystems Life Technologies, s35166, s35167), and negative control siRNAs (Applied Biosystems Life Technologies, AM4635, AM4641) were purchased from Life Technologies. Specific knockdowns of PARK2 and PINK1 were validated using 2 different siRNAs, respectively. The pEGFP-LC3B plasmid was transfected into BEAS-2B cells using Lipofectamine 2000 (Invitrogen Life Technologies, 11668-027) and stably expressing clones were selected by culturing with G418 (Wako, 070-05183; 1.0 mg/ml) containing medium. Transfections of HBEC were performed using the Neon® Transfection System (Invitrogen Life Technologies, MPK5000), using matched optimized transfection kits (Invitrogen Life Technologies, MPK10096).
Preparation of cigarette smoke extract (CSE)
Cigarette smoke extract (CSE) was prepared as previously described with minor modifications.8 Forty milliliters of cigarette smoke were drawn into the syringe and slowly bubbled into sterile serum-free cell culture media in 15-ml BD falcon tubes. One cigarette was used for the preparation of 10 mL of solution. CSE solution was filtered (0.22 μm; Merck Millipore, SLGS033SS) to remove insoluble particles and was designated as a 100% CSE solution.
Mitochondria isolation
Mitochondria and cytosolic fractions were isolated from HBECs with a commercially available kit (Thermo Fisher Scientific, 89874) according to the manufacturer's instructions.
Electron microscopy (EM) and immunocytochemistry
HBEC were treated with CSE (1.0%) in the presence or absence of Baf A1 and Torin1 for 48 h. After treatment, HBEC were fixed with 2% glutaraldehyde/0.1 M phosphate buffer (pH7.4) and in 1% osmium tetroxide/0.1 M phosphate buffer (pH7.4), and dehydrated with a graded series of ethanol. Fixed HBEC were then embedded in epoxy resin. Ultrathin sections were stained with uranyl acetate and lead citrate and observed with the Hitachi H-7500 transmission electron microscope (Hitachi, Tokyo, Japan). For quantitative evaluation of autophagosomes and mitochondria in HBEC, 10 image fields (10000 X) were selected for each sample. Mitochondria shorter than 1 μm without fusion to other mitochondria were counted as fragmented.
Immunohistochemistry and Immunofluorescence staining
Immunohistochemical staining was performed as previously described with minor modification on the paraffin-embedded lung tissues.7,45 PARK2 immunostaining were assessed by counting total and positively staining cells in small airways at a magnification of ×400 and percentage of positive staining was scored in a semi-quantitative fashion; 0 (less than 10%), 1 (11 to 49%), and 2 (more than 50%). Immunofluorescence staining was also performed as previously described.7,45 BEAS-2B cells expressing EGFP-LC3B grown on 8-well culture slides were fixed with 4% paraformaldehyde for 15 min followed by permeabilization with 0.03% Triton X-100 (Wako, 160-24751) for 60 min. After blocking with 0.1% BSA (Sigma-Aldrich, A2153) for 60 min, the primary and secondary antibodies were applied according to the manufacturer's instructions. Confocal laser scanning microscopy analysis of mitochondria was performed by TOMM20 staining, assessed with a confocal microscope (Carl Zeiss LSM510, Tokyo, Japan). Fluorescence microscopy analysis of phospho-histone H2AFX, and TOMM20 staining were performed in HBEC (Olympus, Tokyo, Japan). MitoTracker Red CMXRos (MTR; Molecular Probes-Life Technologies, M-7512) staining (200 nM, 30 min at 37°C) was also performed to evaluate the integrity of the mitochondrial membrane potential.
SA-β-Gal Staining
Senescence-associated β-galactosidase (SA-β-Gal) staining was performed using HBEC grown on 24-well culture plates according to the manufacturer's instructions (BioVision, K802-250).
Measurement of ROS production
HBEC, at a density of 3 × 104 per well, were seeded in a 96-well microplate (Thermo Fisher Scientific, 237105). DCFH-DA (Cell Biolabs, STA-342) was used to measure total cellular ROS according to the manufacturer's instructions. After incubation with DCFH-DA (10 μM) for 10 min at 37°C, fluorescence of DCF was measured at an excitation wavelength of 485 nm and an emission wavelength of 535 nm with a fluorescence microplate reader (Infinite F 200) (Tecan Japan, Kanagawa, Japan). Mitochondrial ROS production was analyzed by dihydrorhodamine 123 (DHR 123) and MitoSOX Red staining according to the manufacturer's instructions. The staining was evaluated with a fluorescence microscope (Olympus BX60, Tokyo, Japan).
Western blotting
HBEC grown on 6-well culture plates were lysed in RIPA buffer (Thermo Fisher Scientific, 89900) with protease inhibitor cocktail (Roche Diagnostics, 11697498001) and 1 mM sodium orthovanadate. Western blotting was performed as previously described.8,45 For each experiment, equal amounts of total protein were resolved by 10–15% SDS-PAGE. After SDS-PAGE, proteins were transferred to polyvinylidene difluoride membrane (Millipore, ISEQ00010), and incubation with specific primary antibody was performed for 1 h at room temperature. After washing several times with PBST (0.1% polyoxyethylene [20] sorbitan monolaurate [Wako, 166-21213] in PBS [Wako, 041-20211]), the membrane was incubated by anti-rabbit IgG, HRP-linked secondary antibody (Cell Signaling Technology, 7074) or anti-mouse IgG, HRP-linked secondary antibody (Cell Signaling Technology, 7076), followed by chemiluminescence detection (GE Healthcare, RPN2232) with LAS-4000 UVmini system (Fujifilm, Tokyo, Japan). To compare the PINK1 and PARK2 expression levels in lung homogenates between different experimental sets, one homogenate sample from a normal patient was selected as a loading control for all western blotting and used for further normalization of densitometric data analysis.
Statistics
Data are shown as the average (±SEM) taken from at least 3 independent experiments. The Student t test was used for comparison of 2 data sets and analysis of variance (one-way ANOVA with Bonferroni post hoc test) for multiple data sets. Linear regression analysis was used to compare PARK2 and PINK1 expression levels in lung homogenates to percentage of forced expiratory volume in 1 s/forced vital capacity (FEV1/FVC). Significance was defined as P < 0.05. Statistical software used was Prism v.5 (GraphPad Software, Inc., San Diego, CA).
Acknowledgments
We wish to thank Stephanie Cambier (University of Washington, Seattle, USA), Emi Kikuchi and Dr. Toshiaki Tachibana (Jikei University School of Medicine, Tokyo, Japan) for technical support. Dr. Noboru Mizushima (Tokyo University, Tokyo, Japan) and Dr. Tamotsu Yoshimori (Osaka University, Osaka, Japan) for providing LC3B cDNA.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the Jikei University Graduate Research Grant to S.I., K.Kobayashi, and N.T., a Grant-In-Aid for Scientific Research from the Ministry of Education of Japan to J.A., H.H., J.K., K.S., K.N., and K.Kuwano, and Health and Labor Sciences Research Grants from the Ministry of Health Labor and Welfare of Japan to J.A. and K.Kuwano.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Vestbo J, Hurd SS, Agusti AG, Jones PW, Vogelmeier C, Anzueto A, Barnes PJ, Fabbri LM, Martinez FJ, Nishimura M, et al. . Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2013; 187(4):347-65; PMID:22878278; http://dx.doi.org/ 10.1164/rccm.201204-0596PP [DOI] [PubMed] [Google Scholar]
- 2. Chilosi M, Carloni A, Rossi A, Poletti V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl Res 2013; 162(3):156-73; PMID:23831269; http://dx.doi.org/ 10.1016/j.trsl.2013.06.004 [DOI] [PubMed] [Google Scholar]
- 3. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013; 153(6):1194-217; PMID:23746838; http://dx.doi.org/ 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Campisi J. Cellular senescence: putting the paradoxes in perspective. Curr Opin Genet Dev 2011; 21(1):107-12; PMID:21093253; http://dx.doi.org/ 10.1016/j.gde.2010.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010; 5:99-118; PMID:20078217; http://dx.doi.org/ 10.1146/annurev-pathol-121808-102144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aoshiba K, Nagai A: Senescence hypothesis for the pathogenetic mechanism of chronic obstructive pulmonary disease. Proc Am Thorac Soc 2009; 6(7):596-601; PMID:19934355; http://dx.doi.org/ 10.1513/pats.200904-017RM [DOI] [PubMed] [Google Scholar]
- 7. Hara H, Araya J, Takasaka N, Fujii S, Kojima J, Yumino Y, Shimizu K, Ishikawa T, Numata T, Kawaishi M, et al. . Involvement of creatine kinase B in cigarette smoke-induced bronchial epithelial cell senescence. Am J Respir Cell Mol Biol 2012; 46(3):306-12; PMID:21980054; http://dx.doi.org/ 10.1165/rcmb.2011-0214OC [DOI] [PubMed] [Google Scholar]
- 8. Fujii S, Hara H, Araya J, Takasaka N, Kojima J, Ito S, Minagawa S, Yumino Y, Ishikawa T, Numata T, et al. . Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology 2012; 1(5):630-41; PMID:22934255; http://dx.doi.org/ 10.4161/onci.20297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou F, Onizawa S, Nagai A, Aoshiba K: Epithelial cell senescence impairs repair process and exacerbates inflammation after airway injury. Respir Res 2011; 12:78; PMID:21663649; http://dx.doi.org/ 10.1186/1465-9921-12-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marzetti E, Csiszar A, Dutta D, Balagopal G, Calvani R, Leeuwenburgh C. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: from mechanisms to therapeutics. Am J Physiol Heart Circ Physiol 2013; 305(4):H459-76; PMID:23748424; http://dx.doi.org/ 10.1152/ajpheart.00936.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van der Toorn M, Slebos DJ, de Bruin HG, Leuvenink HG, Bakker SJ, Gans RO, Koëter GH, van Oosterhout AJ, Kauffman HF. Cigarette smoke-induced blockade of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis. Am J Physiol Lung Cell Mol Physiol 2007; 292(5):L1211-8; PMID:17209140; http://dx.doi.org/ 10.1152/ajplung.00291.2006 [DOI] [PubMed] [Google Scholar]
- 12. van der Toorn M, Rezayat D, Kauffman HF, Bakker SJ, Gans RO, Koeter GH, Choi AM, van Oosterhout AJ, Slebos DJ. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 2009; 297(1):L109-14; PMID:19411310; http://dx.doi.org/ 10.1152/ajplung.90461.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Passos JF, Zglinicki T. Mitochondrial dysfunction and cell senescence-skin deep into mammalian aging. Aging (Albany NY) 2012; 4(2):74-5; PMID:22337807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seo AY, Joseph AM, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci 2010; 123(Pt 15):2533-42; PMID:20940129; http://dx.doi.org/ 10.1242/jcs.070490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 2010; 11(12):872-84; PMID:21102612; http://dx.doi.org/ 10.1038/nrm3013 [DOI] [PubMed] [Google Scholar]
- 16. Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell 2011; 146(5):682-95; PMID:21884931; http://dx.doi.org/ 10.1016/j.cell.2011.07.030 [DOI] [PubMed] [Google Scholar]
- 17. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011; 333(6046):1109-12; PMID:21868666; http://dx.doi.org/ 10.1126/science.1201940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for BNIP3L in autophagic maturation of erythroid cells. Nature 2008; 454(7201):232-5; PMID:18454133; http://dx.doi.org/ 10.1038/nature07006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 2012; 287(23):19094-104; PMID:22505714; http://dx.doi.org/ 10.1074/jbc.M111.322933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al. . Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 2012; 14(2):177-85; PMID:22267086; http://dx.doi.org/ 10.1038/ncb2422 [DOI] [PubMed] [Google Scholar]
- 21. Feng D, Liu L, Zhu Y, Chen Q. Molecular signaling toward mitophagy and its physiological significance. Exp Cell Res 2013; 319(12):1697-705; PMID:23603281; http://dx.doi.org/ 10.1016/j.yexcr.2013.03.034 [DOI] [PubMed] [Google Scholar]
- 22. Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12(2):119-31; PMID:20098416; http://dx.doi.org/ 10.1038/ncb2012 [DOI] [PubMed] [Google Scholar]
- 23. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al. . The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 2010; 12(3):213-23; PMID:20173742 [DOI] [PubMed] [Google Scholar]
- 24. Hara H, Araya J, Ito S, Kobayashi K, Takasaka N, Yoshii Y, Wakui H, Kojima J, Shimizu K, Numata T, et al. . Mitochondrial fragmentation in cigarette smoke induced-bronchial epithelial cell senescence. Am J Physiol Lung Cell Mol Physiol 2013; 305(10):L737-46; PMID:24056969; http://dx.doi.org/ 10.1152/ajplung.00146.2013 [DOI] [PubMed] [Google Scholar]
- 25. Takasaka N, Araya J, Hara H, Ito S, Kobayashi K, Kurita Y, Wakui H, Yoshii Y, Yumino Y, Fujii S, et al. . Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J Immunol 2014, 192(3):958-68; PMID:24367027; http://dx.doi.org/ 10.4049/jimmunol.1302341 [DOI] [PubMed] [Google Scholar]
- 26. Hoffmann RF, Zarrintan S, Brandenburg SM, Kol A, de Bruin HG, Jafari S, Dijk F, Kalicharan D, Kelders M, Gosker HR, et al. . Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir Res 2013; 14(1):97; PMID:24088173; http://dx.doi.org/ 10.1186/1465-9921-14-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147(4):728-41; PMID:22078875; http://dx.doi.org/ 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- 28. Narendra DP, Youle RJ. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid Redox Signal 2011; 14(10):1929-38; PMID:21194381; http://dx.doi.org/ 10.1089/ars.2010.3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oliveras-Salva M, Van Rompuy AS, Heeman B, Van den Haute C, Baekelandt V. Loss-of-function rodent models for Parkin and PINK1. J Parkinsons Dis 2011; 1(3):229-51; PMID:23939304 [DOI] [PubMed] [Google Scholar]
- 30. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183(5):795-803; PMID:19029340; http://dx.doi.org/ 10.1083/jcb.200809125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rana A, Rera M, Walker DW. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc Natl Acad Sci U S A 2013; 110(21):8638-43; PMID:23650379; http://dx.doi.org/ 10.1073/pnas.1216197110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, et al. . Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 2014; 124(9):3987-4003; PMID:25083992; http://dx.doi.org/ 10.1172/JCI74985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ, Yousem SA, et al. . Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One 2008; 3(10):e3316; PMID:18830406; http://dx.doi.org/ 10.1371/journal.pone.0003316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 2014; 15(7):482-96; PMID:24954210; http://dx.doi.org/ 10.1038/nrm3823 [DOI] [PubMed] [Google Scholar]
- 35. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, et al. . Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012; 485(7397):251-5; PMID:22535248; http://dx.doi.org/ 10.1038/nature10992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. . Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12(3):222-30; PMID:21151103; http://dx.doi.org/ 10.1038/ni.1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mortaz E, Adcock IM, Ito K, Kraneveld AD, Nijkamp FP, Folkerts G. Cigarette smoke induces CXCL8 production by human neutrophils via activation of TLR9 receptor. Eur Respir J 2010; 36(5):1143-54; PMID:19840968; http://dx.doi.org/ 10.1183/09031936.00062209 [DOI] [PubMed] [Google Scholar]
- 38. De Nardo D, De Nardo CM, Latz E. New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am J Pathol 2013; 184(1):42-54; PMID:24183846; http://dx.doi.org/ 10.1016/j.ajpath.2013.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol 2005; 32(4):311-8; PMID:15668323; http://dx.doi.org/ 10.1165/rcmb.2004-0309OC [DOI] [PubMed] [Google Scholar]
- 40. Araya J, Cambier S, Markovics JA, Wolters P, Jablons D, Hill A, Finkbeiner W, Jones K, Broaddus VC, Sheppard D, et al. . Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J Clin Invest 2007; 117(11):3551-62; PMID:17965775; http://dx.doi.org/ 10.1172/JCI32526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010; 139(1):323-34 e327; PMID:20347818; http://dx.doi.org/ 10.1053/j.gastro.2010.03.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. dos Santos G, Kutuzov MA, Ridge KM. The inflammasome in lung diseases. Am J Physiol Lung Cell Mol Physiol 2012; 303(8):L627-33; PMID:22904168; http://dx.doi.org/ 10.1152/ajplung.00225.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Birrell MA, Eltom S. The role of the NLRP3 inflammasome in the pathogenesis of airway disease. Pharmacol Ther 2011; 130(3):364-70; PMID:21421008; http://dx.doi.org/ 10.1016/j.pharmthera.2011.03.007 [DOI] [PubMed] [Google Scholar]
- 44. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J 2000; 19(21):5720-8; PMID:11060023; http://dx.doi.org/ 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi M, et al. . Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2013; 304(1):L56-69; PMID:23087019; http://dx.doi.org/ 10.1152/ajplung.00213.2012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.