

Abstract
Autophagy plays a key role in the pathophysiology of schizophrenia as manifested by a 40% decrease in BECN1/Beclin 1 mRNA in postmortem hippocampal tissues relative to controls. This decrease was coupled with the deregulation of the essential ADNP (activity-dependent neuroprotector homeobox), a binding partner of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 β) another major constituent of autophagy. The drug candidate NAP (davunetide), a peptide fragment from ADNP, enhanced the ADNP-LC3B interaction. Parallel genetic studies have linked allelic variation in the gene encoding MAP6/STOP (microtubule-associated protein 6) to schizophrenia, along with altered MAP6/STOP protein expression in the schizophrenic brain and schizophrenic-like behaviors in Map6-deficient mice. In this study, for the first time, we reveal significant decreases in hippocampal Becn1 mRNA and reversal by NAP but not by the antipsychotic clozapine (CLZ) in Map6-deficient (Map6+/−) mice. Normalization of Becn1 expression by NAP was coupled with behavioral protection against hyperlocomotion and cognitive deficits measured in the object recognition test. CLZ reduced hyperlocomotion below control levels and did not significantly affect object recognition. The combination of CLZ and NAP resulted in normalized outcome behaviors. Phase II clinical studies have shown NAP-dependent augmentation of functional activities of daily living coupled with brain protection. The current studies provide a new mechanistic pathway and a novel avenue for drug development.
Keywords: activity-dependent neuroprotective protein (ADNP, MGI database), activity-dependent neuroprotector homeobox (ADNP, HUGO gene nomenclature committee database), hyperactivity; immunohistochemistry, microtubule-associated protein 6 (MAP6)/stable tubule only polypeptide (STOP) deficiency, NAP (davunetide); object recognition, real-time PCR
Abbreviations: Adnp, activity-dependent neuroprotective protein (mouse); ADNP, activity-dependent neuroprotector homeobox (human); Adnp2 (mouse), ADNP2 (human), ADNP homeobox 2; Becn1 (mouse), BECN1 (human), Beclin 1, autophagy-related; CLZ, clozapine; Hprt/Hprt1, hypoxanthine phosphoribosyl transferase; Map1lc3b (mouse), MAP1LC3B (human), microtubule-associated protein 1 light chain 3 β; Map6 (mouse), MAP6 (human), microtubule-associated protein 6
Introduction
Schizophrenia affects approximately 1% of the adult population.1 Schizophrenia patients have 3 major symptoms: positive symptoms (including delusions and hallucinations), negative symptoms (including anhedonia, the inability to begin and maintain planned activities) and cognitive symptoms (including memory deficits, difficulties in focusing, and short attention span).2 The core treatment for schizophrenia is antipsychotic medications,3 which do not address general functioning, thus maintaining cognitive impairment associated with schizophrenia as an unmet global healthcare problem.4,5
Autophagy is a cellular process that preserves the balance between the synthesis, degradation, and recycling of cellular components. Autophagy is essential for neuronal survival and function and was recently suggested to play a key role in the pathophysiology of schizophrenia.6 Several proteins control the autophagy pathway, including BECN1 and MAP1LC3 (LC3 henceforth in the text). BECN1, triggering the formation of autophagosomes via membrane recruitment,7 exhibits reduced expression in the hippocampus of postmortem schizophrenia subjects but not in peripheral lymphocytes.6 LC3, serving as a marker for autophagy, is cleaved at the C terminus to become LC3-I and is transformed by conjugation to phosphatidylethanolamine (PE) to produce LC3-II. LC3-II connects to the phagophore membrane.8 ADNP, an essential protein for brain formation,9,10 interacts with LC3. This interaction is increased in the presence of the short ADNP peptide and the therapeutic drug candidate NAP.6 In contrast to BECN1, the expression of Adnp and its family member Adnp2,11-14 which is deregulated in postmortem hippocampal samples from schizophrenia subjects,15 showed significantly increased expression in lymphocytes from related patients. Such augmentation is similar to increases in the antiapoptotic protein BCL2 (B-cell CLL/lymphoma 2), which interacts with BECN1. The increase in ADNP was associated with the initial stages of the disease, possibly reflecting a compensatory effect. The increase in ADNP2 might be a consequence of neuroleptic treatment, as observed in rats subjected to CLZ treatment.6
Genome-wide association analysis encompassing thousands of schizophrenia patients has identified risk loci for schizophrenia. Identified genes included those encoding proteins associated with calcium, glutamate, and N-methyl-D-aspartate/NMDA signaling in addition to protein degradation, cytoskeleton, and synapse assembly.16,17 In this respect, ADNP may affect schizophrenia, in part through interaction with microtubules.18,19 Adnp haploinsufficiency results in cognitive and social deficits, paralleled by microtubule-associated pathology; treatment with the ADNP snippet NAP (a microtubule-stabilizer) provides partial protection from these effects.20-22 and pipeline drug candidates.23,24 Adnp haploinsufficiency is also associated with decreased hippocampal Becn1 expression.6 It remains to be observed if NAP and/or pipeline products can protect against BECN1 deficiency.
Schizophrenia is now conceptualized as a syndrome with distinguishable symptoms, such as psychosis, cognitive dysfunction, and negative symptoms. This description has been extended to animals, thus allowing for the possibility of identifying animal models of schizophrenia and/or the aforementioned manifestations of the disorder. Map6-deficient mice fulfill the criteria of the above model. Indeed, Map6-deficient mice exhibit severe behavioral disorders that can be linked to positive and negative symptoms in addition to cognitive dysfunction.25-32 Additionally, Map6-null mice fulfill the 3 criteria: construct validity, face validity and predictive validity, all required for a valid animal model of psychiatric disorder. Concerning construct validity criteria, as in humans, map6-null mice exhibit abnormal neuronal connectivity linked to synaptic and neurotransmission defects (hypoglutamatergic neurotransmission and dopamine hyper-reactivity).32-37 With respect to face validity criteria, map6-null mice exhibit severe and numerous behavioral defects related to psychiatric symptoms, including hyperactivity, anxiety, social withdrawal, increased resignation, and working memory defects.25,28,29,32,38-40 Regarding predictive criteria, these mice demonstrate several behavioral and biological deficits that are sensitive to typical and atypical antipsychotics, as well as antidepressants.32,38,39,41
Interestingly, the taxol-like microtubule stabilizer epothilone D ameliorates synaptic impairment and schizophrenic-like behavior in Map6-deficient mice.31,42 Our recent study43 showed that either NAP or CLZ treatment significantly reduced the Map6+/− mouse positive symptoms (hyperactivity) in the open field. However, as opposed to CLZ, NAP treatment significantly ameliorated cognitive deficits in the Map6-deficient mouse model. Phase II clinical studies have shown NAP-dependent improvement of functional activities of daily living coupled to brain protection.44,45
We now ask the following questions: 1) Are autophagy-associated proteins deregulated in the Map6-deficient mouse brain, mimicking the human schizophrenia brain? 2) Will the treatment of Map6+/− mice with NAP reverse MAP6-related deficiencies, including normalization of the expression of autophagy-related proteins? 3) Will CLZ mimic the NAP effect or will a combination be beneficial?
We postulate that a combination of NAP and CLZ may provide not only inhibition of psychosis (CLZ) but also functional strengthening of activities of daily living and rational cognitive behavior (NAP).
Results
Map6+/− mice, similar to human schizophrenia subjects, show BECN1 deficiency, which is ameliorated by NAP + CLZ combination treatment
Our recent studies show a 40% decrease in BECN1 mRNA in the hippocampus of postmortem schizophrenic subjects compared to age-matched controls.6 It was therefore of interest to investigate if a similar reduction occurs in the Map6-deficient mouse model, thus validating the model for future preclinical testing. Our immunohistochemical studies investigating the hippocampal area showed a statistically significant decrease in the number of cells stained with BECN1 antibody in the Map6+/− mice (∼3-fold; *P < 0.05) compared to the Map6+/+ mice (Fig. 1A, Map6+/+ staining; B, Map6+/− staining and C, graphical comparison). This result was verified at the RNA expression level (Fig. 2). Hippocampal RNA expression level was measured by quantitative real-time PCR. Our results showed that Becn1 RNA levels were significantly lower in the Map6+/− mice compared to the Map6+/+ mice (40% reduction, Fig. 2A, ***P < 0.001), mimicking the reduction in the human brain.6 CLZ treatment of Map6+/− mice did not affect Becn1 RNA levels (Fig. 2B, P = 0.347). In contrast, NAP treatment significantly increased Becn1 RNA (Fig. 2C, *P < 0.05) in the Map6+/+ mouse level (Fig 2A). This increase in Becn1 transcript expression in the presence of NAP was maintained in combination with CLZ treatment (Fig. 2D, *P < 0.05).
Figure 1.

Map6+/− mice, like human schizophrenia subjects, show hippocampal Becn1 deficiency. Experiments are detailed in Materials and Methods. Panels (A–C) compare naïve Map6+/+ to naïve Map6+/− mice. A representative stained section is shown in (A) for Map6+/+ and a representative stained section is shown in (B) for Map6+/− mice (arrows denote stained cells). Panel (C, insert) shows the comparison between the stained cell number/mm2 in (A) vs. (B), showing approximately 3-fold reduction in Map6+/− mice as compared to controls (Student t test, *P < 0.05).
Figure 2.

Becn1 expression-deficiency in Map6+/− mice is ameliorated by NAP + CLZ combination treatment. Quantitative real-time PCR was performed for Adnp (white bars), Adnp2 (gray bars) and Becn1 (black bars). Results are depicted in graphs of percentage of expression of the naïve Map6+/− mice. Each transcript was compared to itself in Map6+/− mice vs. Map6+/+ mice (A) or the comparative treatment with the relevant vehicle, all in the Map6+/− mice (Saline vs. CLZ+/- (i.e. saline-treated Map6+/− mice or CLZ treated Map6+/− respectively), B; DD vs. NAP+/- (i.e., DD-treated Map6+/− mice or NAP treated Map6+/− respectively), C and DD+Saline vs. NAP+CLZ+/-, D) using the Student t test. Panel A compares Map6+/− mice to Map6+/+ mice. A significant ∼40% decrease was observed for all tested genes in the Map6+/- mice (***P < 0.001). Panel (B) shows no effect for CLZ treatment when compared to saline injection, while panel (C) shows a significant effect of NAP on Becn1 expression and Adnp2 expression (with NAP normalizing Becn1 expression) in comparison to vehicle only, (*P < 0.05). Panel (D) shows that the combination of CLZ+NAP normalized Becn1 expression (*P < 0.05).
The hippocampal expression of the NAP parent protein ADNP and the related protein ADNP2 were downregulated in Map6-deficient mice (for either ADNP, or ADNP2, ***P < 0.001). CLZ treatment was associated with a further apparent reduction of Adnp expression (Fig 2B, P = 0.07), which may be indicative of unwanted behavioral side effects (decreased cognitive performance), as measured previously in Adnp+/− mice.23 Neither NAP nor combination treatment affected Adnp RNA expression. However, NAP treatment resulted in a slight reduction in Adnp2 levels, which was not apparent in combination with CLZ (Fig. 2C and D, *P < 0.05). Interestingly, previous results have shown a deregulation of ADNP and ADNP2 in schizophrenia with ADNP2 RNA levels increasing in the hippocampus of the postmortem brains in parallel with disease duration.15 Our current results suggest that NAP treatment may protect against this deregulation.
These results prompted further experiments to evaluate and correlate behavioral outcomes to protein expression with an emphasis on autophagy.
Map6+/− mice hyperactivity in the open field is normalized by NAP + CLZ combination treatment
As previously described,43 Map6+/− mice suffer from hyperactivity compared to Map6+/+ mice. This hyperactive behavior in Map6+/− mice has previously been linked to positive symptoms in human schizophrenia.32,35,37 Our current results confirmed this finding, showing that Map6+/− mice (n = 13) were hyperactive, traveling approximately twice the distance over the same time period compared to the control Map6+/+ mice (Fig. 3A, ***P < 0.001).
Figure 3.

Map6+/− mice suffer from hyperactivity in the open field which is normalized by NAP + CLZ combination treatment. The results are shown as the mean of the path in cm per animal in the 3-min time period. Map6+/+ (Sv129) controls exhibited a significant difference from their and Map6+/− (heterozygous) littermates (A,***P < 0.001, Student t test). Panel (B) compares CLZ and saline (vehicle)-treated Map6+/− mice. CLZ significantly reduced hyperactivity in the Map6+/- mice (***P < 0.001, Student t test). Panel (C) compares intranasal NAP and intranasal DD (vehicle)-treated Map6+/− mice. NAP significantly reduced hyperactivity in the Map6+/− mice (**P < 0.01, Student t test). Panel (D) compares combination treatment of NAP and CLZ to the appropriate vehicle control showing significant protection against hyperactivity (**P < 0.01, Student t test). In (D), treatment with DD+saline significantly decreased activity, possibility associated with the repeated handling in comparison to the Map6+/− mice (from 3A, P < 0.001, Student t test).
Three wk of daily treatment of the Map6+/− mice with either CLZ compared to subcutaneous (s.c.) vehicle control (Fig. 3B) or NAP compared to intranasal vehicle control (DD43,46) (Fig. 3C) significantly reduced hyperactivity in the open field test. In the presence of CLZ, hyperactivity was reduced by ∼5-fold (***P < 0.001), and with NAP, hyperactivity was reduced by ∼2-fold (**P < 0.01). CLZ treatment dramatically reduced activity to levels even lower than those observed in the Map6+/+ mice (possibly indicating lethargy/somnolence from drug treatment as observed in patients).47 The Map6+/− mice treated with the combination of NAP and CLZ showed activity that matched the behavior of the normal naïve mice (Fig. 3A compared to 3D, **P < 0.01, placebo treatment compared to combination). Together, our results showed that CLZ, NAP and combination treatment reduced Map6+/−-deficient-related hyperactivity compared to the appropriate vehicle controls (Fig. 3B to D).
The schizophrenic-like preference for familiar rather than novel objects typically exhibited by Map6+/− mice is normalized by NAP + CLZ combination treatment
The object recognition and discrimination test, based on visual discrimination between 2 different objects, measures object-related memory, which is deficient in schizophrenia.48 Although one may measure several schizophrenia-related behavioral deficits, as elegantly reviewed,48 we concentrated on object recognition, as our previous data showed an association of Map6-deficiency with reduced performance in the object recognition test.43 Here, we extended these studies; Map6+/− mice were not able to discriminate the familiar object from the novel object and preferred the familiar object, an observation that was in contrast to the control Map6+/+ mice (Fig. 4A, discrimination index −0.35 vs. 0.24; **P < 0.01 for genotype effect). CLZ treatment did not reach significance (P = 0.47, Fig. 4B). In contrast, the NAP-treated Map6+/− mice (Fig. 4C) and the NAP+CLZ–treated Map6+/− mice (Fig. 4D) behaved like the Map6+/+ naïve mice, while significantly differing from the respective vehicle-treated Map6+/− mice (**P < 0.01 for either of the groups, Fig. 4C and D).
Figure 4.

The Map6+/− mice schizophrenic-like preference to familiar rather than novel objects is normalized by NAP + CLZ combination treatment. Map6+/- mice showed significant deficits in recognizing the novel object and seemed to significantly prefer the familiar object in comparison to control Map6+/+ mice (A,**P < 0.01, Student t test). CLZ treatment did not significantly improve object recognition/discrimination in the Map6+/- mice (B) NAP treatment significantly improved object recognition/discrimination in the Map6+/- mice (**P < 0.01, Student t test) to the control Map6+/+ level (C) NAP+CLZ treatment significantly improved object recognition and discrimination in the Map6+/- mice (**P < 0.01, Student t test) to the control Map6+/+ mouse level (D). Although in (B), saline treatment appears to increase discrimination in comparison to the Map6+/- mice from (A), statistical analysis showed a P value of 0.073 (Student t test).
Interestingly, the NAP-treated Map6+/− mice exhibited an even greater discrimination index compared to the Map6+/+ mice (Fig. 4A versus 4C). However, with the combination treatment of NAP + CLZ, the mice showed almost identical scores compared to those of the Map6+/+ mice (normal naïve mice).
CLZ toxicity and NAP protection
Published literature links CLZ to neuronal cell death in vitro,49 which may be prevented by NAP treatment.50 Using the neuronal human SH-SY5Y neuroblastoma cell line, we set out to verify 1) whether CLZ indeed harms neuronal-like cellular viability and 2) whether NAP reduces cell death caused by CLZ treatment. Human SH-SY5Y neuroblastoma cells were exposed to NAP (10−15 M to 10−11 M) for 24 h at 37°C either in the absence or in the presence of 20 μg/ml CLZ.49 The measurement of metabolic activity/viability was performed by a cell proliferation assay (MTS). NAP treatment at concentrations ≥10−15 M prevented CLZ-associated reduced viability in the SH-SY5Y cell line (one-way ANOVA, F(3) = 103.618, P < 0.001 for treatment; the Fisher Least Significant Difference (LSD) post-hoc analysis in comparison to the CLZ only condition (see Materials and Methods section) revealed protection by NAP concentration of 10−15 M to 10−13 M: ***P < 0.001), while NAP alone did not affect cell viability, under these experimental conditions (Fig. 5A and B, respectively).
Figure 5.
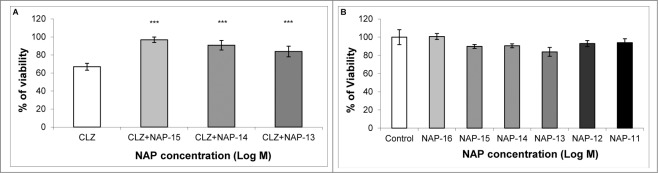
NAP protects against CLZ-associated cell death. In a neuronal cell culture model (human SH-SY5Y neuroblastoma) CLZ treatment resulted in approximately 40% decreased viability (A, CLZ vs. B, control, no CLZ). NAP treatment at 10−15M to 10−13M completely protected against CLZ-related reduction in viability. (NAP+CLZ vs. CLZ, one-way ANOVA, F(3) = 103.618, P < 0.001. The Fisher Least Significant Difference (LSD) post-hoc test revealed protection by NAP concentration of 10−15M - 10−13M: ***P < 0.001), (A). In the same neuronal cell model, without the addition of CLZ, at a wide concentration range (10−16M to 10−11M), NAP did not affect cell viability or cell division (P > 0.05), (B). However, the control group in (B) shows an ‘unacceptably large variance’, thus preventing the correct assessment of NAP effect. Experiments were repeated 3 times; one representative experiment is shown (n = 5 to 10/group/experiment).
Discussion
Standard of care in schizophrenia may give a suitable treatment against the positive symptoms but not against the negative symptoms. As a result, a large number of patients fail to lead a normal life. Therefore, it is extremely important to provide treatments addressing all aspects of the disease, encompassing both positive as well as negative symptoms and cognitive dysfunctions. A reliable animal model, imperfect as it may be, enables the prediction of treatment efficiency via mouse behavior and the potential elucidation of disease mechanisms.
CLZ, used in clinical practice, has an impact on the positive symptoms of schizophrenia. At the same time, however, cognitive and daily functional activities of schizophrenia patients remain an unmet medical need. Our results confirmed previous studies showing a beneficial effect of CLZ on the positive symptoms (hyperactivity, although with reduction below normal control levels) with limited, insignificant effects on object recognition.43 NAP treatment showed a significant reduction in hyperactivity, albeit less effective than CLZ. Furthermore, NAP showed a significant impact on the “cognitive” symptoms of the mice (differentiation between novel and familiar objects). The combination treatment resulted in the normalization of the impact on the positive symptoms (hyperactivity) in the open field and imparted a significant increase in cognitive abilities in the object recognition test relative to the CLZ treatment but not over the NAP treatment alone.
The combination of NAP in addition to traditional treatment might better affect the clinical outcome relative to traditional treatment alone. Our results in the SH-SY5Y cells led us to conclude that CLZ treatment, albeit at concentrations 30- to 60-fold higher than the optimal dose, reduced mitochondrial function, and cellular viability. NAP treatment prevented CLZ-associated decreases in cellular viability. The most recent studies have shown that CLZ reduces neuronal cell viability through the blockade of autolysosome formation,51 and our studies show that NAP increases the autophagic process.6,52
Real-time PCR RNA expression analysis revealed a significant reduction in Becn1 transcripts in the hippocampus of the Map6+/− schizophrenia-like mouse model relative to the Map6+/+ mice. Whereas all placebo-treated groups demonstrated the same reduced expression level as the Map6+/− naïve mice, the mice that were treated with NAP alone, or with NAP in combination with CLZ demonstrated higher (similar to Map6+/+ naïve) levels of Becn1 transcripts. Perhaps through autophagy or other key related processes, BECN1 might play a significant role in the Map6+/− model of schizophrenia. The decrease in Becn1 in the hippocampus of the Map6+/− mouse is in line with our recent detection of reduced BECN1 expression in the postmortem human hippocampus from schizophrenia subjects compared to that in the controls.6 These similarities between findings in mouse and human strengthen the validity of the Map6+/− model as a platform for further drug testing, with NAP as a potential lead compound.
Interestingly, the loss of MAP6 protein in map6-null mice impairs peripheral olfactory neurogenesis.53 Thus, Map6-deficient olfactory neurons showed presynaptic swellings with autophagic-like structures. In olfactory and lvomeronasal epithelia, there was an increase in neuronal turnover, as shown by an increase in the number of proliferating, apoptotic, and immature cells with no changes in the number of mature neurons. Similar alterations in peripheral olfactory neurogenesis have also been described in schizophrenia patients.54 Furthermore, regeneration resulted in the abnormal organization of olfactory terminals within the olfactory glomeruli in map6-null mice.53 These complementary studies, compared to ours, suggest regional variations in neuronal regulation with abnormalities in the autophagic and apoptotic mechanisms resulting from MAP6 deficiencies, which may mimic the clinical/pathological manifestation in schizophrenia.
Our immunohistochemical data expanded upon the biochemical analyses, showing a marked decrease in the number of cells expressing BECN1 in the hippocampus of the naïve Map6+/− mice compared to that in the naïve Map6+/+ mice. The autophagic process is in a crossroad between cell viability and apoptosis; we hypothesize that the deregulation of autophagy induces cell death. The putative effects on autophagy are consistent with NAP protection against apoptosis, which includes protection against caspase activation, as observed in our previous studies55,50 and mitochondrial CYCS/cytochrome C release56 in association with the microtubule-cytoskeleton protection.
Interestingly, previous findings show an inhibitory effect of schizophrenia drugs, including CLZ, on the autophagic process51 and the inhibition of autophagy by the recruitment of BECN1 to the microtubules.57 Here, we showed that NAP alone and in combination with CLZ increased Becn1 expression. We suggest that to effectively treat schizophrenia, it is necessary for the cell to have adequate cytoskeleton, BECN1 expression and apoptosis to provide for an efficient process of autophagy and/or other BECN1-related processes. Furthermore, the schizophrenia-related deregulation of ADNP and ADNP2 expression that has previously been observed in the human hippocampus was recapitulated here to a certain degree in the Map6+/− mouse model.15 The association of ADNP and its fragment peptide NAP with the microtubule cytoskeleton58 and the autophagy pathway6,52 paves the path to replacement therapy.
Recent clinical studies have shown significant efficacy for NAP treatment on the UCSD Performance-based Skills Assessment of functional activity.44 Further studies show NAP protection of N-acetylaspartate and choline levels in dorsolateral prefrontal cortex in patients with schizophrenia, suggesting neuroprotection in vivo.45 These results support the preclinical prediction and suggest future larger clinical testing, combination treatments and further investigation of pipeline products, leading to science-based translational medicine.
Methods and Materials
Animals and treatment
Five-mo-old Map6+/− and Map6+/+ male mice were received at Tel Aviv University under a material transfer agreement from Annie Andrieux and Didier Job (INSERM). The mice were housed under controlled environmental conditions with a 12 h light and 12 h dark cycle. All experiments were conducted according to the guidelines for care and use of laboratory animals at Tel-Aviv University, under governmental permission.
We conducted an in vivo study with ∼100 Map6/− mice and 10 Map6+/+ mice. The experiment included 2 groups of naïve-untreated mice (Map6+/− mice and Map6+/+ mice; Fig. 3A) and 6 groups of treated Map6+/− mice as follows: s.c. injected 1) saline (Teva Medical, AWB 1324) and 2) CLZ (Sigma-Aldrich, C6305) in saline (Fig. 3B); intranasally administered 3) DD (7.5 mg of NaCl, 1.7 mg of citric acid monohydrate 3 mg of disodium phosphate dihydrate, and 0.2 mg of benzalkonium chloride solution [50%]/ml), the NAP vehicle43,46; 4) NAP (a kind gift from Allon Therapeutics) in DD (Fig. 3C) and combinations, each administered as above including 5) saline +DD and 6) NAP+CLZ (Fig. 3D).
Map6+/− mice were treated daily (5 d per wk) for 15 wk, and when a behavioral test was implemented, treatment was conducted 1 h prior to the experiment. The experiments were based on a large body of previous animal studies with NAP showing brain bioavailability after intranasal as well as systemic administrations and efficacy using treatment schedules of 5 d per wk.43,59-61
CLZ (3 mg/kg) was solubilized under acidic conditions (pH = ∼2.0 with HCl, Merck, 1003171000)), and the solutions titrated back to ∼pH 7.4 with NaOH (Merck, 1064981000). Map6+/− mice received daily doses of 3 mg/kg s.c. injection of CLZ (5 d a wk, 100 μl/d) for the duration of the experiment. Control Map6+/− mice were similarly injected with saline. It should be noted that most studies give 10 to 20 mg/kg/d CLZ, as we have done before.43 However, here we wanted to test for a potential synergistic effect; therefore, we reduced the CLZ dose.
DD was used as a vehicle/placebo for NAP.46,43 This placebo solution was administered to mice hand-held in a semi-supine position with nostrils facing the investigator. A pipette tip was used to administer 2.5 μl/nostril (total: 0.5 μg/5 μl/mouse/d). The mouse was hand held until the solution was fully absorbed (∼10 s).
Behavioral assessments were implemented according to our previous research.43 In short, on the 4th wk of drug application, the open field test was conducted; on the 6th wk of drug application, an object recognition test was conducted. After 15 wk of treatment, the mice were decapitated, and hippocampal tissue was extracted and stored at −80°C for biochemical analysis. A second set of brains was used for immunohistochemistry as outlined below.
Biochemical Analyses
Extractions
RNA and proteins were extracted by the NucleoSpin® RNA/Protein kit (MACHEREY-NAGEL, REF 740933.50). The quantity and purity of RNA was determined by measuring optical density at 260 nm with a spectrophotometer (NanoDrop Technologies, Wilmington, DE). RNA integrity was determined by electrophoresis on 1% agarose gel (Hispanagar, 45090001) and staining with ethidium bromide (Biolab, 05412399).
Reverse transcription and quantitative real-time PCR
Samples with the same amount of total RNA were used to synthesize single-strand cDNA using qScript™ cDNA Synthesis (Quanta BioSciences, Inc. 95047-100). Primer pairs were designed using the primer 3 web interface (http://frodo.wi.mit.edu/primer3/) and synthesized by Sigma-Genosys (The Woodlands, TX).
Adnp1: 5′ACGAAAAATCAGGACTATCGG3′; 5′GGACATTCCGGAAATGACTTT3′
Adnp2: 5′GGAAAGAAAGCGAGATACCG3′; 5′TCCTGGTCAGCCTCATCTTC3′
Becn1: 5′CAAATCTAAGGAGTTGCCGTT3′; 5′CTTCTTTGAACTGCTGCACAC3′
Hprt: 5′GGATTTGAATCACGTTTGTGTC3′; 5′AACTTGCGCTCATCTTAGGC3′
Real-time PCR reactions were carried out using PerfeCTa® SYBR® Green FastMix®, ROX™ (Quanta, 95073-012), and 300 nM each of forward and reverse primers. The StepOnePlus™ Real-Time PCR System Sequence Detection System instrument and software (Applied Biosystems, Foster City, CA) was utilized according to the default thermocycler program for all genes. Data were analyzed with Data Assist™ v2.0 Software (Applied Biosystems).
The comparative Ct method was used for the quantification of transcripts, comparing the Ct value of the target gene to a housekeeping gene (Hprt [hypoxanthine phosphoribosyl transferase]) for each sample. The results were calculated as percent of 2^–ΔCT in the experimental sample compared to naïve Map6+/− mice, which constituted the 100% baseline.
Tissue preparation for histology and immunohistochemistry
At the end of the drug administration period, male mice (5 to 10/group) were perfused transcardially under deep anesthesia with 4% paraformaldehyde (Merck, k42072103) in 0.1 M phosphate-buffered saline, pH 7.4 (Biological Industries, 1407753). The brains were removed and placed in the same fixative at 4°C for 4 h and then immersed in 30% sucrose in phosphate-buffered saline, pH 7.4 for 5 d at 4°C.
Embedding
Brain tissues were embedded in tissue Tek and flash-frozen with liquid nitrogen. Subsequently, 6-μm serial sections were cut using a cryostat mtc (SLEE Mainz, Germany) microtome at −23°C, and sections were placed on superfrost plus slides and kept at −80°C until use.
Immunohistochemistry
Frozen sections were air dried for 1 h and then hydrated in distilled water. Endogenous peroxidase was blocked by incubating the sections in 3% H2O2 (Merck, k2769210) in methanol for 10 min. Sections were then rinsed with distilled water and then placed in tris-buffered-saline (containing 0.05 M Tris [Duchefa biochemie,T1501.1000], 0.15 M sodium chloride [Chemlab, CL00.1429.1000], and adjusted to pH 7.6 with 1 M HCl) for 5 min. After incubation in blocking buffer of 10% fetal bovine serum (Gibco, 10108-157) for 30 min, the sections were treated with primary antibodies against BECN1 (affinity-purified polyclonal rabbit IgG; Santa Cruz Biotechnology, sc11427). Secondary antibodies were goat anti-rabbit (Vector, BA-1000) and avidin-peroxidase (Sigma, A3151). Diaminobenzidine (Sigma, D12384) was used as chromogen. Nuclear counterstaining was performed with hematoxylin (Sigma, H9627).
Measurements
Sections were examined under an optical microscope (Zeiss Axioplan-2, Carl Zeiss Microscopy, LLC Thornwood, NY) with the aid of a CCD camera (Nikon DS-5M, Nikon Inc. Melville, NY). On average, 20 optical fields were examined from each experimental group under a magnification of 40×. Measurements were performed in the prefrontal cortex (Pr. Cortex) and in the hippocampus with ImageJ (1.43u). The results are expressed as cells/mm2 and as Integrated Density (the product of Area and Mean Gray Value).
Human SH-SY5Y neuroblastoma cells as a model for CLZ toxicity and NAP protection
Cells were plated at a density of 3.6 × 105/well (96-well plates) 24 h prior to the CLZ treatment. Dulbecco's modified Eagle medium (Biological Industries, 01-170-1A) was supplemented with 15% fetal calf serum (Biological Industries, 04-102-2A), 1% penicillin-streptomycin (Biological Industries, 03-031-1), and 1% glutamine (Biological Industries, 03-020-1B) in a 5% CO2 atmosphere. CLZ was obtained from Sigma (see above). Cells were exposed to NAP (10−15 M to 10−10 M) for 24 h at 37°C either in the absence or presence of 20 μg/ml CLZ. It should be noted here that this CLZ dose represents a 30- to 60-fold higher levels than the clinically optimal CLZ blood levels (300 to 600 ng/ml).62
Drugs and control solutions were dissolved in ethanol. The measurement of metabolic activity (which assesses cell survival) was performed by a nonradioactive cell proliferation assay (MTS; Promega, CellTiter96R, G3580) and evaluated with an ELISA-reader (Bio-Tek Instruments, Highland Park, VT) at a wavelength of 490 nm.
Statistical analysis
One-way ANOVA with the Fisher least significant differences (LSD) post-hoc test on SigmaPlot software was used for multiple group comparisons. The 2-tailed Student t test was utilized when only 2 variables were compared, as outlined in the figure legends. Given the different vehicles tested, the comparisons were made between 2 different groups + vehicle (placebo) and + vehicle including treatment. Data were checked for outliers using the Grubbs test on GraphPad software. All determinations were made with a 95% confidence interval and were considered significant at the P < 0.05 level. All results are depicted as mean +/− standard error of the mean (SEM).
Acknowledgments
We thank Micah Eades and Shani Ben Moshe for their help in performing part of the experiments and Sinaya Vaisburd for the help with the revisions. Avia Merenlender–Wagner was an Eshkol Fellow, and this work constitutes part of the requirements for a PhD thesis by Tel Aviv University. Professor Gozes laboratory is supported by the AMN Foundation, CFTAU Montreal Circle of Friends and the Adams family, Adams Super Center for Brain Studies and the Lily and Avraham Gildor Chair for the Investigation of Growth Factors at Tel Aviv University. Initial studies in this research were also partially supported by Allon Therapeutics Inc. Professor Gozes is currently a Humboldt Award Recipient and a fellow at the Hanse-Wissenschftenkolleg, Germany.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1. Cho MJ, Moscicki EK, Narrow WE, Rae DS, Locke BZ, Regier DA. Concordance between two measures of depression in the hispanic health and nutrition examination survey. Soc Psychiatry Psychiatr Epidemiol 1993; 28:156-63; PMID:8235801; http://dx.doi.org/ 10.1007/BF00797317 [DOI] [PubMed] [Google Scholar]
- 2. American Psychiatric Association Desk Reference to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) American Psychiatric Publishing, United States, 2013. [Google Scholar]
- 3. Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RSE, Davis SM, Davis CE, Lebowitz BD, et al. . Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. Engl J Med 2005; 353:1209-23; PMID:16172203; http://dx.doi.org/ 10.1056/NEJMoa051688 [DOI] [PubMed] [Google Scholar]
- 4. Haro JM, Novick D, Bertsch J, Karagianis J, Dossenbach M, Jones PB. Cross-national clinical and functional remission rates: worldwide schizophrenia outpatient health outcomes (W-SOHO) study. Br J Psychiat 2011; 199:194-201; PMID:21881098; http://dx.doi.org/ 10.1192/bjp.bp.110.082065 [DOI] [PubMed] [Google Scholar]
- 5. Khanna P, Suo T, Komossa K, Ma H, Rummel-Kluge C, El-Sayeh HG, Leucht S, Xia J. Aripiprazole versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst Rev 2014; 1:CD006569; PMID:24385408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Merenlender-Wagner A, Malishkevich A, Shemer Z, Udawela M, Gibbons A, Scarr E, Dean B, Levine J, Agam G, Gozes I. Autophagy has a key role in the pathophysiology of schizophrenia. Mol Psychiat 2013; PMID:24365867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cao Y, Klionsky DJ. Physiological functions of Atg6Beclin 1: a unique autophagy-related protein. Cell Res 2007; 17:839-49; PMID:17893711; http://dx.doi.org/ 10.1038/cr.2007.78 [DOI] [PubMed] [Google Scholar]
- 8. Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861-73; PMID:18006683; http://dx.doi.org/ 10.1101/gad.1599207 [DOI] [PubMed] [Google Scholar]
- 9. Mandel S, Rechavi G, Gozes I. Activity-dependent neuroprotective protein (ADNP) differentially interacts with chromatin to regulate genes essential for embryogenesis. Dev Biol 2007; 303:814-24; PMID:17222401; http://dx.doi.org/ 10.1016/j.ydbio.2006.11.039 [DOI] [PubMed] [Google Scholar]
- 10. Pinhasov A, Mandel S, Torchinsky A, Giladi E, Pittel Z, Goldsweig AM, Servoss SJ, Brenneman DE, Gozes I. Activity-dependent neuroprotective protein: a novel gene essential for brain formation. Brain Res Dev Brain Res 2003; 144:83-90; PMID:12888219; http://dx.doi.org/ 10.1016/S0165-3806(03)00162-7 [DOI] [PubMed] [Google Scholar]
- 11. Zamostiano R, Pinhasov A, Gelber E, Steingart RA, Seroussi E, Giladi E, Bassan M, Wollman Y, Eyre HJ, Mulley JC, et al. . Cloning and characterization of the human activity-dependent neuroprotective protein. J Biol Chem 2001; 276:708-14; PMID:11013255; http://dx.doi.org/ 10.1074/jbc.M007416200 [DOI] [PubMed] [Google Scholar]
- 12. Kushnir M, Dresner E, Mandel S, Gozes I. Silencing of the ADNP-family member, ADNP2, results in changes in cellular viability under oxidative stress. J Neurochem 2008; 105:537-45; PMID:18179478; http://dx.doi.org/ 10.1111/j.1471-4159.2007.05173.x [DOI] [PubMed] [Google Scholar]
- 13. Dresner E, Malishkevich A, Arviv C, Leibman Barak S, Alon S, Ofir R, Gothilf Y, Gozes I. Novel evolutionary-conserved role for the activity-dependent neuroprotective protein (ADNP) family that is important for erythropoiesis. J Biol Chem 2012; 287:40173-85; PMID:23071114; http://dx.doi.org/ 10.1074/jbc.M112.387027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakamachi T, Li M, Shioda S, Arimura A. Signaling involved in pituitary adenylate cyclase-activating polypeptide-stimulated ADNP expression. Peptides 2006; 27:1859-64; PMID:16564114; http://dx.doi.org/ 10.1016/j.peptides.2006.01.007 [DOI] [PubMed] [Google Scholar]
- 15. Dresner E, Agam G, Gozes I. Activity-dependent neuroprotective protein (ADNP) expression level is correlated with the expression of the sister protein ADNP2: deregulation in schizophrenia. Eur Neuropsychopharmacol 2011; 21:355-61; PMID:20598862; http://dx.doi.org/ 10.1016/j.euroneuro.2010.06.004 [DOI] [PubMed] [Google Scholar]
- 16. Ripke S, O’Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S, Bergen SE, Collins AL, Crowley JJ, Fromer M, et al. . Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet 2013; 45:1150-9; PMID:23974872; http://dx.doi.org/ 10.1038/ng.2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McCarroll SA, Hyman SE. Progress in the genetics of polygenic brain disorders: significant new challenges for neurobiology. Neuron 2013; 80:578-87; PMID:24183011; http://dx.doi.org/ 10.1016/j.neuron.2013.10.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Furman S, Steingart RA, Mandel S, Hauser JM, Brenneman DE, Gozes I. Subcellular localization and secretion of activity-dependent neuroprotective protein in astrocytes. Neuron Glia Biol 2004; 1:193-9; PMID:16845437; http://dx.doi.org/ 10.1017/S1740925X05000013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mandel S, Spivak-Pohis I, Gozes I. ADNP differential nucleuscytoplasm localization in neurons suggests multiple roles in neuronal differentiation and maintenance. J Mol Neurosci 2008; 35:127-41; PMID:18286385; http://dx.doi.org/ 10.1007/s12031-007-9013-y [DOI] [PubMed] [Google Scholar]
- 20. Divinski I, Mittelman L, Gozes I. A femtomolar acting octapeptide interacts with tubulin and protects astrocytes against zinc intoxication. J Biol Chem 2004; 279:28531-8; PMID:15123709; http://dx.doi.org/ 10.1074/jbc.M403197200 [DOI] [PubMed] [Google Scholar]
- 21. Gozes I, Divinski I. The femtomolar-acting NAP interacts with microtubules: novel aspects of astrocyte protection. J Alzheimers Dis 2004; 6:S37-41; PMID:15665412 [DOI] [PubMed] [Google Scholar]
- 22. Oz S, Ivashko-Pachima Y, Gozes I. The ADNP derived peptide, NAP modulates the tubulin pool: implication for neurotrophic and neuroprotective activities. PLoS One 2012; 7:e51458; PMID:23272107; http://dx.doi.org/ 10.1371/journal.pone.0051458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vulih-Shultzman I, Pinhasov A, Mandel S, Grigoriadis N, Touloumi O, Pittel Z, Gozes I. Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J Pharmacol Exp Ther 2007; 323:438-49; PMID:17720885; http://dx.doi.org/ 10.1124/jpet.107.129551 [DOI] [PubMed] [Google Scholar]
- 24. Shiryaev N, Pikman R, Giladi E, Gozes I. Protection against tauopathy by the drug candidates NAP (davunetide) and D-SAL: biochemical, cellular and behavioral aspects. Curr Pharm Des 2011; 17:2603-12; PMID:21728979; http://dx.doi.org/ 10.2174/138161211797416093 [DOI] [PubMed] [Google Scholar]
- 25. Volle J, Brocard J, Saoud M, Gory-Faure S, Brunelin J, Andrieux A, Suaud-Chagny MF. Reduced expression of STOPMAP6 in mice leads to cognitive deficits. Schizophr Bull 2013; 39:969-78; PMID:23002183; http://dx.doi.org/ 10.1093/schbul/sbs113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fournet V, Schweitzer A, Chevarin C, Deloulme JC, Hamon M, Giros B, Andrieux A, Martres MP. The deletion of STOPMAP6 protein in mice triggers highly altered mood and impaired cognitive performances. J Neurochem 2012; 121:99-114; PMID:22146001; http://dx.doi.org/ 10.1111/j.1471-4159.2011.07615.x [DOI] [PubMed] [Google Scholar]
- 27. Begou M, Volle J, Bertrand JB, Brun P, Job D, Schweitzer A, Saoud M, D’Amato T, Andrieux A, Suaud-Chagny MF. The stop null mice model for schizophrenia displays [corrected] cognitive and social deficits partly alleviated by neuroleptics. Neuroscience 2008; 157:29-39; PMID:18804150; http://dx.doi.org/ 10.1016/j.neuroscience.2008.07.080 [DOI] [PubMed] [Google Scholar]
- 28. Bouvrais-Veret C, Weiss S, Andrieux A, Schweitzer A, McIntosh JM, Job D, Giros B, Martres MP. Sustained increase of alpha7 nicotinic receptors and choline-induced improvement of learning deficit in STOP knock-out mice. Neuropharmacology 2007; 52:1691-700; PMID:17512560; http://dx.doi.org/ 10.1016/j.neuropharm.2007.03.015 [DOI] [PubMed] [Google Scholar]
- 29. Powell KJ, Hori SE, Leslie R, Andrieux A, Schellinck H, Thorne M, Robertson GS. Cognitive impairments in the STOP null mouse model of schizophrenia. Behav Neurosci 2007; 121:826-35; PMID:17907815; http://dx.doi.org/ 10.1037/0735-7044.121.5.826 [DOI] [PubMed] [Google Scholar]
- 30. Fradley RL, O’Meara GF, Newman RJ, Andrieux A, Job D, Reynolds DS. STOP knockout and NMDA NR1 hypomorphic mice exhibit deficits in sensorimotor gating. Behav Brain Res 2005; 163:257-64; PMID:16046005; http://dx.doi.org/ 10.1016/j.bbr.2005.05.012 [DOI] [PubMed] [Google Scholar]
- 31. Fournet V, de Lavilleon G, Schweitzer A, Giros B, Andrieux A, Martres MP. Both chronic treatments by epothilone D and fluoxetine increase the short-term memory and differentially alter the mood status of STOPMAP6 KO mice. J Neurochem 2012; 123:982-96; PMID:23013328; http://dx.doi.org/ 10.1111/jnc.12027 [DOI] [PubMed] [Google Scholar]
- 32. Andrieux A, Salin PA, Vernet M, Kujala P, Baratier J, Gory-Faure S, Bosc C, Pointu H, Proietto D, Schweitzer A, et al. . The suppression of brain cold-stable microtubules in mice induces synaptic defects associated with neuroleptic-sensitive behavioral disorders. Genes Dev 2002; 16:2350-64; PMID:12231625; http://dx.doi.org/ 10.1101/gad.223302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Charlet A, Muller AH, Laux A, Kemmel V, Schweitzer A, Deloulme JC, Stuber D, Delalande F, Bianchi E, Van Dorsselaer A, et al. . Abnormal nociception and opiate sensitivity of STOP null mice exhibiting elevated levels of the endogenous alkaloid morphine. Mol Pain 2010; 6:96; PMID:21172011; http://dx.doi.org/ 10.1186/1744-8069-6-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fournet V, Jany M, Fabre V, Chali F, Orsal D, Schweitzer A, Andrieux A, Messanvi F, Giros B, Hamon M, et al. . The deletion of the microtubule-associated STOP protein affects the serotonergic mouse brain network. J Neurochem 2010; 115:1579-94; PMID:20969568; http://dx.doi.org/ 10.1111/j.1471-4159.2010.07064.x [DOI] [PubMed] [Google Scholar]
- 35. Bouvrais-Veret C, Weiss S, Hanoun N, Andrieux A, Schweitzer A, Job D, Hamon M, Giros B, Martres MP. Microtubule-associated STOP protein deletion triggers restricted changes in dopaminergic neurotransmission. J Neurochem 2008; 104:745-56; PMID:18199119 [DOI] [PubMed] [Google Scholar]
- 36. Brenner E, Sonnewald U, Schweitzer A, Andrieux A, Nehlig A. Hypoglutamatergic activity in the STOP knockout mouse: a potential model for chronic untreated schizophrenia. J Neurosci Res 2007; 85:3487-93; PMID:17304567; http://dx.doi.org/ 10.1002/jnr.21200 [DOI] [PubMed] [Google Scholar]
- 37. Brun P, Begou M, Andrieux A, Mouly-Badina L, Clerget M, Schweitzer A, Scarna H, Renaud B, Job D, Suaud-Chagny MF. Dopaminergic transmission in STOP null mice. J Neurochem 2005; 94:63-73; PMID:15953350; http://dx.doi.org/ 10.1111/j.1471-4159.2005.03166.x [DOI] [PubMed] [Google Scholar]
- 38. Fournet V, de Lavilleon G, Schweitzer A, Giros B, Andrieux A, Martres MP. Both chronic treatments by epothilone D and fluoxetine increase the short-term memory and differentially alter the mood-status of STOPMAP6 KO mice. J Neurochem 2012; 123:982-96; PMID:23013328; http://dx.doi.org/ 10.1111/jnc.12027 [DOI] [PubMed] [Google Scholar]
- 39. Bégou M, Volle J, Bertrand JB, Brun P, Job D, Schweitzer A, Saoud M, D’Amato T, Andrieux A, Suaud-Chagny MF. The stop null mice model for schizophrenia displays cognitive and social deficits partly alleviated by neuroleptics. Neuroscience 2008; 157:29-39; PMID:18804150; http://dx.doi.org/ 10.1016/j.neuroscience.2008.07.080 [DOI] [PubMed] [Google Scholar]
- 40. Fradley RL, O’Meara G F, Newman RJ, Andrieux A, Job D, Reynolds DS. STOP knockout and NMDA NR1 hypomorphic mice exhibit deficits in sensorimotor gating. Behav Brain Res 2005; 163:257-64; PMID:16046005; http://dx.doi.org/ 10.1016/j.bbr.2005.05.012 [DOI] [PubMed] [Google Scholar]
- 41. Sokolowska P, Passemard S, Mok A, Schwendimann L, Gozes I, Gressens P. Neuroprotective effects of NAP against excitotoxic brain damage in the newborn mice: implications for cerebral palsy. Neuroscience 2010; 173:156-68; PMID:21073926; http://dx.doi.org/ 10.1016/j.neuroscience.2010.10.074 [DOI] [PubMed] [Google Scholar]
- 42. Andrieux A, Salin P, Schweitzer A, Begou M, Pachoud B, Brun P, Gory-Faure S, Kujala P, Suaud-Chagny MF, Hofle G, et al. . Microtubule stabilizer ameliorates synaptic function and behavior in a mouse model for schizophrenia. Biol Psychiat 2006; 60:1224-30; PMID:16806091; http://dx.doi.org/ 10.1016/j.biopsych.2006.03.048 [DOI] [PubMed] [Google Scholar]
- 43. Merenlender-Wagner A, Pikman R, Giladi E, Andrieux A, Gozes I. NAP (davunetide) enhances cognitive behavior in the STOP heterozygous mouse–a microtubule-deficient model of schizophrenia. Peptides 2010; 31:1368-73; PMID:20417241; http://dx.doi.org/ 10.1016/j.peptides.2010.04.011 [DOI] [PubMed] [Google Scholar]
- 44. Javitt DC, Buchanan RW, Keefe RS, Kern R, McMahon RP, Green MF, Lieberman J, Goff DC, Csernansky JG, McEvoy JP, et al. . Effect of the neuroprotective peptide davunetide (AL-108) on cognition and functional capacity in schizophrenia. Schizophr Res 2012; 136:25-31; PMID:22169248; http://dx.doi.org/ 10.1016/j.schres.2011.11.001 [DOI] [PubMed] [Google Scholar]
- 45. Jarskog LF, Dong Z, Kangarlu A, Colibazzi T, Girgis RR, Kegeles LS, Barch DM, Buchanan RW, Csernansky JG, Goff DC, et al. . Effects of davunetide on N-acetylaspartate and choline in dorsolateral prefrontal cortex in patients with schizophrenia. Neuropsychopharmacology 2013; 38:1245-52; PMID:23325325; http://dx.doi.org/ 10.1038/npp.2013.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Alcalay RN, Giladi E, Pick CG, Gozes I. Intranasal administration of NAP, a neuroprotective peptide, decreases anxiety-like behavior in aging mice in the elevated plus maze. Neurosci Lett 2004; 361:128-31; PMID:15135910; http://dx.doi.org/ 10.1016/j.neulet.2003.12.005 [DOI] [PubMed] [Google Scholar]
- 47. Kluge M, Himmerich H, Wehmeier PM, Rummel-Kluge C, Dalal M, Hinze-Selch D, Kraus T, Dittmann RW, Pollmacher T, Schuld A. Sleep propensity at daytime as assessed by Multiple Sleep Latency Tests (MSLT) in patients with schizophrenia increases with clozapine and olanzapine. Schizophr Res 2012; 135:123-7; PMID:22257975; http://dx.doi.org/ 10.1016/j.schres.2011.12.017 [DOI] [PubMed] [Google Scholar]
- 48. Moore H, Geyer MA, Carter CS, Barch DM. Harnessing cognitive neuroscience to develop new treatments for improving cognition in schizophrenia: CNTRICS selected cognitive paradigms for animal models. Neurosci Biobehav Rev 2013; 37:2087-91; PMID:24090823; http://dx.doi.org/ 10.1016/j.neubiorev.2013.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Heiser P, Enning F, Krieg JC, Vedder H. Effects of haloperidol, clozapine and olanzapine on the survival of human neuronal and immune cells in vitro. J Psychopharmacol 2007; 21:851-6; PMID:17881431; http://dx.doi.org/ 10.1177/0269881107077221 [DOI] [PubMed] [Google Scholar]
- 50. Idan-Feldman A, Ostritsky R, Gozes I. Tau and caspase 3 as targets for neuroprotection. Int J Alzheimers Dis 2012; 2012:493670; PMID:22693678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Park J, Chung S, An H, Kim J, Seo J, Kim DH, Yoon SY. Haloperidol and clozapine block formation of autophagolysosomes in rat primary neurons. Neuroscience 2012; 209:64-73; PMID:22390943; http://dx.doi.org/ 10.1016/j.neuroscience.2012.02.035 [DOI] [PubMed] [Google Scholar]
- 52. Esteves AR, Gozes I, Cardoso SM. The rescue of microtubule-dependent traffic recovers mitochondrial function in Parkinson's disease. Biochim Biophys Acta 2014; 1842:7-21; PMID:24120997; http://dx.doi.org/ 10.1016/j.bbadis.2013.10.003 [DOI] [PubMed] [Google Scholar]
- 53. Benardais K, Kasem B, Couegnas A, Samama B, Fernandez S, Schaeffer C, Antal MC, Job D, Schweitzer A, Andrieux A, et al. . Loss of STOP protein impairs peripheral olfactory neurogenesis. PLoS One 2010; 5:e12753; PMID:20856814; http://dx.doi.org/ 10.1371/journal.pone.0012753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Turetsky BI, Hahn CG, Borgmann-Winter K, Moberg PJ. Scents and nonsense: olfactory dysfunction in schizophrenia. Schizophr Bull 2009; 35:1117-31; PMID:19793796; http://dx.doi.org/ 10.1093/schbul/sbp111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Leker RR, Teichner A, Grigoriadis N, Ovadia H, Brenneman DE, Fridkin M, Giladi E, Romano J, Gozes I. NAP, a femtomolar-acting peptide, protects the brain against ischemic injury by reducing apoptotic death. Stroke 2002; 33:1085-92; PMID:11935065; http://dx.doi.org/ 10.1161/01.STR.0000014207.05597.D7 [DOI] [PubMed] [Google Scholar]
- 56. Zemlyak I, Sapolsky R, Gozes I. NAP protects against cytochrome c release: inhibition of the initiation of apoptosis. Eur J Pharmacol 2009; 618:9-14; PMID:19619522; http://dx.doi.org/ 10.1016/j.ejphar.2009.07.013 [DOI] [PubMed] [Google Scholar]
- 57. Luo S, Garcia-Arencibia M, Zhao R, Puri C, Toh PP, Sadiq O, Rubinsztein DC. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Mol Cell 2012; 47:359-70; PMID:22742832; http://dx.doi.org/ 10.1016/j.molcel.2012.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Oz S, Kapitansky O, Ivashco-Pachima Y, Malishkevich A, Giladi E, Skalka N, Rosin-Arbesfeld R, Mittelman L, Segev O, Hirsch JA, Gozes, I.. The NAP motif of activity-dependent neuroprotective protein (ADNP) regulates dendritic spines through microtubule end binding proteins. Mol Psychiat 2014; 19:1115-24; PMID:25178163; http://dx.doi.org/ 10.1038/mp.2014.97 [DOI] [PubMed] [Google Scholar]
- 59. Gozes I, Giladi E, Pinhasov A, Bardea A, Brenneman DE. Activity-dependent neurotrophic factor: intranasal administration of femtomolar-acting peptides improve performance in a water maze. J Pharmacol Exp Ther 2000; 293:1091-8; PMID:10869414 [PubMed] [Google Scholar]
- 60. Gozes I, Morimoto BH, Tiong J, Fox A, Sutherland K, Dangoor D, Holser-Cochav M, Vered K, Newton P, Aisen PS, et al. . NAP: research and development of a peptide derived from activity-dependent neuroprotective protein (ADNP). CNS Drug Rev 2005; 11:353-68; PMID:16614735; http://dx.doi.org/ 10.1111/j.1527-3458.2005.tb00053.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Morimoto BH, de Lannoy I, Fox AW, Gozes I, Stewart AJ. Davunetide: Pharmacokinetics and distribution to brain after intravenous or intranasal administration to rat. Chimica Oggi Chem Today 2009; 27:16-20. [Google Scholar]
- 62. Hiemke C, Dragicevic A, Grunder G, Hatter S, Sachse J, Vernaleken I, Muller MJ. Therapeutic monitoring of new antipsychotic drugs. Ther Drug Monit 2004; 26:156-60; PMID:15228157; http://dx.doi.org/ 10.1097/00007691-200404000-00012 [DOI] [PubMed] [Google Scholar]