

Abstract
The Parkinson disease (PD)-associated E3-ubiquitin (Ub) ligase PARK2/parkin plays a central role in many stress response pathways, and in particular, in mitochondrial quality control. Within this pathway, PARK2 activation is accompanied by a robust increase in its autoubiquitination, followed by clearance of the damaged mitochondria by selective autophagy (mitophagy). Yet, little is known about how this auto-ubiquitination is regulated during mitophagy. In our study, we demonstrate that PARK2 forms predominantly K6-linked Ub conjugates on itself. Moreover, PARK2 interacts with the deubiquitinating enzyme USP8 that preferentially removes these K6-linked conjugates, thereby regulating the activity and function of PARK2 in the pathway. When USP8 is silenced, a persistence of K6-linked Ub conjugates on PARK2 delays both its translocation to damaged mitochondria and successful completion of mitophagy. Taken together, these findings implicate a novel role for K6-linked Ub conjugates and USP8-mediated deubiquitination in the regulation of PARK2 in mitochondrial quality control.
Keywords: deubiquitination, K6 Ub-linkages, mitophagy, PARK2/Parkin, USP8
A familial form of PD is often associated with mutations in the PARK2 gene, which encodes the E3-ubiquitin (Ub) ligase PARK2. Mutations in PARK2 impede the normal function of PARK2 in a variety of cellular pathways, including mitochondrial quality control. In this pathway, PARK2 functions in concert with the PD-associated mitochondrial kinase PINK1 to promote the removal of damaged mitochondria via the autophagy pathway. In healthy mitochondria, PINK1 undergoes a constant cycle of degradation following its import into mitochondria. However, when the mitochondria become damaged, PINK1 import is stalled, causing it to accumulate on the surface of the mitochondria. As PINK1 accumulates, it phosphorylates both Ub and PARK2, leading to the activation and subsequent recruitment of PARK2 onto the mitochondria. Following its translocation from the cytosol to the mitochondria, PARK2 ubiquitinates a number of mitochondrial proteins, thereby signaling the clearance of mitochondria via autophagy.
Prior to its phosphorylation by PINK1, PARK2 exists in an auto-inhibited state. However, upon PINK1-mediated activation, translocation of PARK2 onto the mitochondria is accompanied by robust self-ubiquitination. Autoubiquitination is commonly used by many E3s to regulate their activity. Moreover, this activity is often antagonized by deubiquitinating enzymes (DUBs) that function to hydrolyze the Ub chains formed by the E3 on itself. Thus, DUBs can regulate the stability and function of their cognate E3 partners. In asking whether one or more DUBs might modulate PARK2 function in mitophagy, an unbiased siRNA knockdown screen of known DUBs was performed. From this screen, knockdown of the DUB USP8 was demonstrated to delay both the mitochondrial recruitment of PARK2 and its ability to direct mitochondria for autophagic degradation. Prior to this study, USP8 was implicated in endosomal trafficking but not mitochondrial quality control. USP8 was found to directly deubiquitinate PARK2 in vitro, thereby opposing its auto-ubiquitination. Moreover, an absence of USP8 in cells leads to an enhancement and persistence of PARK2 auto-ubiquitination upon triggering mitochondrial damage, which in turn delays PARK2 recruitment and mitophagy.
Two key findings help explain this delay in PARK2 function. First, mass spectrometry analysis reveals that PARK2 has a propensity to form predominantly K6-linked Ub chains on itself. Second, USP8 selectively hydrolyzes these K6-linked Ub conjugates on PARK2. Indeed, when using mutant ubiquitin lacking K6, USP8 is unable to deubiquitinate PARK2. Thus, in the absence of USP8 in cells, the propensity of PARK2 to assemble K6-Ub linked conjugates on itself is unopposed. Indeed, it would appear that when USP8 is knocked down, the increased accumulation of K6-linked Ub conjugates on PARK2 is at least partly responsible for the delay in PARK2 function in mitophagy. Congruent with this notion, manipulating the levels of lysine linkages within cells by overexpressing a K6R Ub mutant rescues the delay caused by knockdown of USP8. Moreover, overexpression of a mutant form of Ub that could only form K6-linkages, forcing PARK2 to form elevated levels of K6-linked Ub conjugates on itself, also causes a delay in its recruitment and mitophagy, similar to the phenotype observed with USP8 knockdown. Taken together, excessive and prolonged K6-linked Ub conjugates on PARK2 appear to prevent mitophagy from proceeding efficiently.
This study demonstrates for the first time a role for K6-linked Ub conjugates in mitochondrial quality control. Yet, how do the increased K6-linked Ub chains impair PARK2 function? It is plausible that such K6-linked Ub conjugates, normally hydrolyzed by USP8, can impede interactions with PINK1 and phosphorylated Ub, both required for PARK2 activation and recruitment onto the mitochondria. Moreover, as auto-ubiquitination appears to be confined to the N-terminal Ub-like domain of PARK2, the presence of these conjugates on this domain might interfere with its binding to Ub receptors, thereby delaying its mitochondrial translocation (Fig. 1). Following its translocation onto mitochondria, a robust increase in PARK2 auto-ubiquitination is normally observed, followed by removal of these conjugates prior to mitochondrial clearance. When USP8 is silenced, these conjugates persist on PARK2. As a result, we think they may impede PARK2 from interacting with SQSTM1/p62, LC3, or other autophagic receptors, a necessary step required for PARK2 to successfully complete the mitophagy process (Fig. 1). Taken together, these findings demonstrate a novel role for K6-linked Ub conjugates in the regulation of PARK2 function within the mitophagy pathway. Moreover, unlike the DUBs USP15 and USP30 that regulate PARK2-mediated mitophagy indirectly, this study positions USP8 as the first DUB to deubiquitinate PARK2 in mitophagy, thereby regulating its function in mitochondrial quality control.
Figure 1.
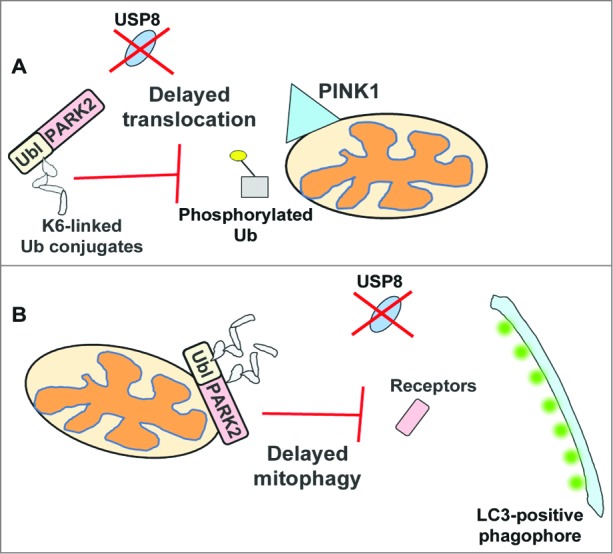
Model of how the absence of USP8 delays PARK2-mediated mitophagy. Under normal conditions, PARK2 is auto-inhibited with minimal levels of auto-ubiquitination detected. Upon mitochondrial damage, PARK2 assembles predominantly K6-linked chains, which are rapidly hydrolyzed by USP8. However, when USP8 is absent, these conjugates persist, which delays PARK2 recruitment, possibly by impeding its interaction with PINK1, phosphorylated Ub or other receptor proteins required for the mitochondrial translocation of PARK2 (A). The persistence of these conjugates may also delay PARK2-mediated clearance of mitochondria by interfering with the recruitment of autophagy receptor proteins or LC3, required for the efficient clearance of damaged mitochondria (B). Ubl, ubiquitin-like domain.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.