

Abstract
Autophagy has been intensively studied in herpes simplex virus type 1 (HSV-1), a human alphaherpesvirus. The HSV-1 genome encodes a well-known neurovirulence protein called ICP34.5. When the gene encoding this protein is deleted from the genome, the virus is markedly less virulent when injected into the brains of animal models. Subsequent characterization of ICP34.5 established that the neurovirulence protein interacts with BECN1, thereby inhibiting autophagy and facilitating viral replication in the brain. However, an ortholog of the ICP34.5 gene is lacking in the genomes of other closely related alphaherpesviruses, such as varicella-zoster virus (VZV). Further, autophagosomes are easily identified in the exanthem (rash) that is the hallmark of both VZV diseases—varicella and herpes zoster. Inhibition of autophagy leads to diminished VZV titers. Finally, no block is detected in studies of autophagic flux following VZV infection. Thus autophagy appears to be proviral during VZV infection while antiviral during HSV-1 infection. Because divergence to this degree is extremely unusual for 2 closely related herpesviruses, we postulate that VZV has accommodated its infectious cycle to benefit from autophagic flux, whereas HSV-1 has captured cellular immunomodulatory genes to inhibit autophagy.
Keywords: autophagy, Epstein-Barr virus, herpes simplex virus, lysosome, varicella-zoster virus
Altogether there are 9 human herpesviruses, separated into 3 subfamilies. Alphaherpesviruses include VZV, HSV-1, and HSV-2; betaherpesviruses include cytomegalovirus, human herpesvirus (HHV)-6A, HHV-6B, and HHV-7; gammaherpesviruses include Epstein-Barr virus (EBV) and HHV-8 (Kaposi sarcoma-associated herpesvirus). Among all 9, VZV has the smallest genome (124 kbp). In one sense, therefore, VZV is a streamlined herpesvirus in that it survives with the fewest possible genes. Depending on the definition of a viral gene, VZV lacks at least 8–9 genes found in the HSV-1 genome. Of interest, 2 of the HSV genes missing from the VZV genome include genes that modify autophagy, namely HSV ICP34.5 and US11.
The locations of the 2 genes are important with regard to evolution of VZV and HSV. Each strand of the dsDNA genome consists of a unique long (UL) and unique short (US) segment capped by repeat segments. ICP34.5 is also called RL1 because the gene is located at the terminus of the long repeat segment of the HSV genome. A portion of ICP34.5 is homologous to the PPP1R15A/GADD34 (protein phosphatase 1, regulatory subunit 15A) protein, a sensor of ER stress. Conversely, US11 is present on the short segment of the HSV genome. VZV lacks several of the HSV US genes, including US11, encoding a RNA-binding regulatory protein. With regard to evolution, the US genes are considered to be critical for establishing a niche for each herpesvirus, while the UL segment consists mainly of the core genes found in all herpes viruses. ICP34.5 is certainly a recent addition to the HSV genome, perhaps by a recombination event because of its location on the long repeat segment. In summary, the smaller VZV genome evolved without ICP34.5 and US11.
This point came to the forefront because of early observations about the presence of easily detectable autophagosomes in the vesicular skin lesions found during both human VZV diseases: varicella (chickenpox) and herpes zoster (shingles). The vesicles are the final site of virion assembly; thus the vesicles are filled with enveloped viral particles. To confirm and extend these observations, we turned to the only animal model for VZV infection in the skin, the SCID mouse with human skin xenografts. Xenografts are inserted underneath the mouse skin; they are inoculated with VZV and then removed after one, 2, and 3 wk for pathological examination. When we compared the degree of autophagosome formation in the infected xenografts with that previously observed in vesicular cells removed from patients with varicella or herpes zoster, we observed equally abundant autophagy in both.
In prior experiments, we have compared the amount of autophagy induced by VZV infection in cultured cells with autophagy induced by serum starvation or chemicals such as tunicamycin or trehalose. This experiment was carried out to show that we were not unknowingly using conditions advantageous for autophagy in our virology studies. We observed that the average number of puncta per cell during VZV infection overlapped with more traditional inducers of autophagy (Fig. 1). In subsequent viral studies, we showed that inhibition of autophagy with either 3-methyladenine or siRNA against ATG5 reduce both VZV glycoprotein biosynthesis and infectivity.
Figure 1.
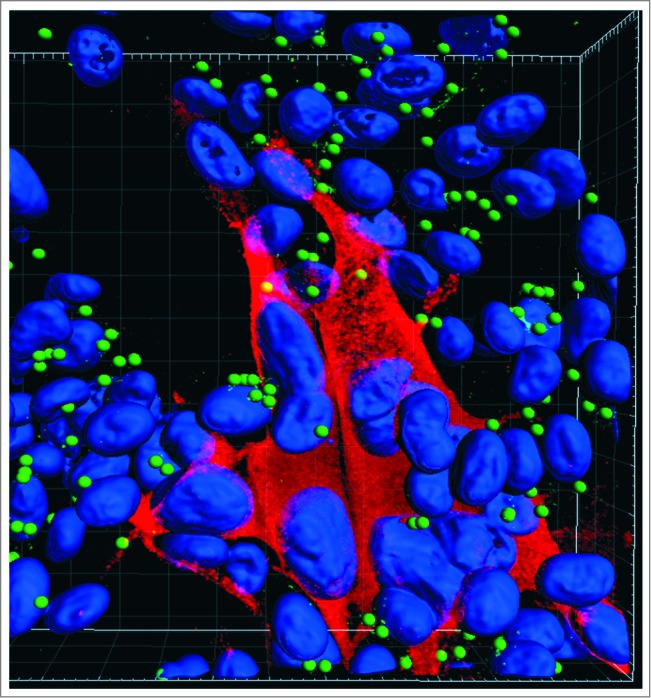
Autophagosomes (green spheres) in the cytoplasm of cultured cells infected with VZV. VZV-infected monolayers were fixed with 2% paraformaldehyde, permeabilized with 0.02% Triton X-100, and then immunolabeled with a rabbit polyclonal antibody against LC3 and a mouse monoclonal antibody against the VZV major immediate-early protein IE62. Monolayers were observed with a Zeiss LSM710 Spectral confocal microscope using 63X high numerical-aperture oil immersion objective lenses. Confocal Z-stacks comprising 40 optical slices were reconstructed into 3D animations with the aid of Imaris software. The Spot function in Imaris automatically locates autophagosomes based on size and intensity thresholds; each autophagosome is represented by a sphere 500 nm in diameter. With this imaging technology, individual autophagosomes are easily detectable within infected cells. Note: VZV infection induces fusion of cells, thus there is a large cluster of nuclei in the image. Blue, Hoechst 33342 nuclear stain; red Alexa Fluor 546, VZV protein; green Alexa Fluor 488, LC3-labeled autophagosomes.
Taking into account all the above evidence, we postulated that the abundant autophagosome formation following VZV infection was a direct effect of increased autophagy and not a block in flux. We had previously investigated autophagic flux by measurement of the level of the polyubiquitin-binding protein SQSTM1/p62. When compared with uninfected cells, SQSTM1 levels are lower in VZV-infected cells when measured by immunoblotting, a result that suggests degradation of SQSTM1 during VZV infection. In order to confirm and extend our observations, we also documented autophagic flux by a traditional flux assay that compared the time-dependent release of radiolabeled amino acids from a labeled VZV-infected monolayer and a control uninfected monolayer. In related experiments, we observed that treatment of the monolayer with 3-methyladenine virtually halts this degradation process, a result that points to autophagy as a major explanation for protein degradation in infected cells. Finally, we used the red fluorescent protein-green fluorescent protein (RFP-GFP) tandem–tagged LC3 plasmid (ptfLC3). The signal from GFP is quenched in the acidic environment of the lysosome, while the signal from RFP remains intact. Thus the green signal is lost as the autophagosome fuses with the more acidic lysosome. When fibroblasts were first transfected with ptfLC3 and subsequently infected with VZV, confocal microscopy images revealed a transition to autolysosomes.
The above experiments confirmed that autophagic flux is demonstrable in cells infected with this actively replicating alphaherpesvirus. Recent investigations with EBV have suggested a more complicated scenario for gammaherpesviruses: EBV initially appropriates nascent autophagic vesicles for its own transportation while blocking a later transition of autophagosomes to autolysosomes.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This research was supported by NIH grant AI89716.