

Abstract
Human first-trimester trophoblast cells proliferate at low O2, but survival is compromised by oxidative stress, leading to uteroplacental insufficiency. The vasoactive drug, sildenafil citrate (Viagra, Sigma, St Louis, Missouri), has proven useful in reducing adverse pregnancy outcomes. An important biological function of this pharmaceutical is its action as an inhibitor of cyclic guanosine monophosphate (cGMP) phosphodiesterase type 5 activity, which suggests that it could have beneficial effects on trophoblast survival. To investigate whether sildenafil can prevent trophoblast cell death, human first-trimester villous explants and the HTR-8/SVneo cytotrophoblast cell line were exposed to hypoxia and reoxygenation (H/R) to generate oxidative stress, which induces apoptosis. Apoptosis was optimally inhibited during H/R by 350 ng/mL sildenafil. Sildenafil-mediated survival was reversed by l-NG-nitro-l-arginine methyl ester hydrochloride or cGMP antagonist, indicating a dependence on both nitric oxide (NO) and cGMP. Indeed, either a cGMP agonist or an NO generator was cytoprotective independent of sildenafil. These findings suggest a novel intervention route for patients with recurrent pregnancy loss or obstetrical placental disorders.
Keywords: sildenafil, apoptosis, nitric oxide, cGMP
Introduction
Preeclampsia is a condition characterized by proteinuria and hypertension that can affect up to 5% to 8% of all pregnancies after 20 weeks of gestation.1 Although the exact pathophysiology for this disease is not known, it is characterized by placental hypoxia with subsequent endothelial dysfunction.2 Preeclampsia is a major health issue that produces maternal morbidity and mortality, as well as adverse fetal outcomes, including prematurity, intrauterine growth restriction (IUGR), and fetal death.3 Currently, there are no therapeutic interventions to prevent, stabilize, or reverse preeclampsia except delivery or pregnancy termination. The safety and well-being of the mother is of the utmost importance, while the managing obstetrician must also consider fetal maturity and development.4
In healthy pregnancies, invasive extravillous trophoblast cells remodel the interface between the developing placenta and the uterine decidua to facilitate high-volume blood flow into the intervillous space.2 During the first trimester, human cytotrophoblast cells develop at low O2 concentrations (˜2%) that stimulate proliferation.5,6 Fluctuations in O2 concentration can disrupt the physiological environment and generate reactive O2 species, resulting in apoptosis of embryonic tissue.7-9 Evidence suggests that nonphysiological hypoxia/reoxygenation (H/R) can occur in the first trimester and impede remodeling of the placental vasculature by trophoblast cells.9-12 The H/R exposure can cause total demise of the fetus, but if the pregnancy survives the initial insult, an adverse pregnancy outcome can occur late in the second and third trimesters, producing preeclampsia or IUGR.8,9,13 Transient exposure of cytotrophoblast cells cultured at low oxygen levels to superphysiological levels, up to ambient oxygen, generates oxidative stress that can serve as a model of the pathologic environment encountered in adverse pregnancies.7,9 Abnormal placental development could contribute to the pathophysiology of preeclampsia as well as IUGR and pregnancy loss, which are all associated with uteroplacental insufficiency.7,9 In pregnancies that later develop preeclampsia and IUGR, there is shallow trophoblast invasion and a reduction in uteroplacental blood flow.14 Consequently, antiangiogenic factors, including soluble endoglin (sENG) and soluble fms-like tyrosine kinase 1 (sFLT1), become elevated in the maternal serum, while there is a reduction in serum levels of vascular endothelia growth factor (VEGF) and placental growth factor.15
The vasodilator nitric oxide (NO) is tightly regulated during placentation. The NO activates soluble guanylyl cyclase to convert guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP), which is degraded by phosphodiesterase 5 (PDE5) to guanosine monophosphate (GMP).16 Northern blotting has demonstrated that PDE5 messenger RNA (mRNA) is highly expressed in human placenta as well as bladder, colon, and lung, while lower levels are found in uterus, skeletal muscle, and brain.17 Disruption of both NO and the second messenger cGMP has been implicated in the pathophysiology of preeclampsia.18 The pathogenesis of IUGR and preeclampsia could be associated with reduced endothelial NO release and significant lowering of cGMP concentrations in vascular smooth muscle.19,20 Sildenafil, a highly specific PDE5 inhibitor that increases cGMP concentrations in vivo, was originally used as a vasoactive drug to treat erectile dysfunction disorder.21 Sildenafil can increase uterine blood flow, reduce radial artery resistance, improve endometrial thickening, and diminish the occurrence of implantation failure.22 In pregnant women, the resultant accumulation of cGMP and vasodilation appear to have a positive impact on fetal outcome during preeclampsia23-27 and IUGR.28,29
Pregnancy success requires adequate endometrial growth to accommodate an implanting embryo. Low pregnancy rates in in vitro fertilization couples have been associated with a thin endometrial lining.30 Studies have indicated that the thin endometrial lining could be secondary to uterine radial artery impedance.22,31 Reduction in the endometrial thickness is associated with vascular endothelial disruption, reduced VEGF expression, reduced endometrial angiogenesis, and decreased blood flow to the endometrium.22,32-34 This pathophysiology is strikingly similar to the mechanism thought to drive uteroplacental insufficiency, leading to preeclampsia and IUGR. The PDE5 is expressed in the maternal compartment of the fetoplacental unit and is integral in the maintenance and regulation of fetal well-being.35 The NO and cGMP pathways regulate vascular adaptations occurring throughout pregnancy.
Nitric oxide is required for trophoblast cell survival and appears to be critical for endovascular invasion necessary for normal placental perfusion and the associated phenotype switching.36,37 Medical interventions that target NO and cGMP signaling pathways have been identified as therapeutic tools for pregnancies with uteroplacental insufficiencies, preeclampsia, and gestational diabetes.36 The use of sildenafil in both rodents and humans has provided a potential intervention to alleviate preeclampsia and IUGR.27 In addition to vasodiation, sildenafil might directly impact trophoblast survival through NO and cGMP signaling, which could contribute to its positive clinical effects in pregnant women.
In this study, we explored the effect of sildenafil on human first-trimester trophoblast cells using an immortalized human first-trimester cell line and first-trimester chorionic villous explant culture. The primary aims of this study were to (1) establish the effect of oxidative stress due to O2 fluctuations on survival of first-trimester trophoblast cells, (2) examine the ability of sildenafil to attenuate trophoblast apoptosis, and (3) investigate the role of signaling pathways intersecting with sildenafil in its regulation of trophoblast survival.
Material and Method
Cell Culture and O2 Regulation
HTR-8/SVneo cytotrophoblast cells were grown in Dulbecco Modified Eagle Medium (DMEM)/F12 medium containing 10% fetal bovine serum.6 Culture medium was replaced with serum-free medium 24 hours prior to all experiments. For ambient culture (20% O2), cells were cultured in a humidified 5% CO2/95% air incubator. Trophoblast exposure to H/R was performed as described previously.9 Briefly, cells were cultured at 2% O2 for 2 hours, and then culture medium was replaced with fresh medium preequilibrated to 20% O2 for an additional 6 hours of culture at ambient conditions. Cells cultured at 20% or 2% O2 for 8 hours served as controls.
Villous Explant Culture
Placental tissues were obtained with Wayne State University Institutional Review Board approval and patient informed consent from first-trimester terminations at a Michigan Family Planning Facility. Fresh tissue was placed on ice in phosphate-buffered saline (PBS) and immediately transported to the laboratory. The chorionic villi were dissected and cut into pieces of approximately 5 mg wet weight and transferred individually into DMEM/F12 culture medium supplemented with 10% donor calf serum, 100 IU penicillin, and 100 μg/mL streptomycin in a 24-well culture plate (Costar, Corning, New York). Exposure to H/R was performed as described for the cell line.
Cell Treatments
Trophoblast cells and villous explants were treated during reoxygenation by supplementing culture medium with 3 separate dosages of sildenafil citrate (35, 350, and 3500 ng/mL), with or without 10 μmol/L N2,2′-O-dibutyryl-guanosine 3′:5′-cyclic monophosphate (cGMP analog), 10 μmol/L 8-(4-chlorophenylthio)-guanosine 3′,5′-cyclic monophosphorothioate, Rp-Isomer (cGMP antagonist), 10 μmol/L (±)-S-Nitroso-N-acetylpenicillamine (SNAP; NO donor), 10 μmol/L NG-Nitro-l-arginine Methyl Ester Hydrochloride (l-NAME, NO inhibitor), or the control compound d-NAME (all from Sigma, St Louis, Missouri).
Cell Death Assay
Cytotrophoblast cells were cultured and treated in 96-well plates prior to assay. Cell death was detected by terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL), using a fluorescein-based kit from Roche Applied Science (Indianapolis, Indiana) as described previously.9 Cells were counterstained with 5 µg/mL 4,6-diamidino-2-phenylindole, HCl (DAPI; EMD Biosciences, Billerica, Massachusetts). Fluorescent nuclei were imaged at 400× magnification using a Leica (Wetzlar, Germany) DM IRB epifluorescence microscope. Fluorescence was imaged using a Hamamatsu Orca digital camera (Hamamatsu City, Japan). Images were processed using Simple PCI (Hamamatsu) imaging software, by counting total nuclei (DAPI) and TUNEL-positive nuclei (fluorescein) for each field. At least 150 total cells were counted in each well. The ratio of TUNEL/DAPI-labeled nuclei (TUNEL index) was calculated by averaging counts for triplicate fields in each well.
Statistical Analysis
All experiments were conducted using triplicate samples, and all experiments were repeated at least 3 times. Data were analyzed by analysis of variance (ANOVA) using SPSS version 21.0 (IBM Corp, Armonk, New York). Data assessing the effect of sildenafil concentration were transformed using square root function to satisfy the normality assumption of ANOVA. All other data were normally distributed and were not transformed. Means were separated using Tukey method. Significant interaction between status and treatment was detected for studies examining the efficacy of treatments affecting cGMP and NO pathways. Treatment effects were thus assessed separately at hypoxia and at H/R. Significance was defined as P < .05. Data are expressed as mean ± standard error of the mean.
Results
Sildenafil Prevents Apoptosis in Chorionic Villi
First-trimester chorionic villi exposed to H/R exhibited elevated cell death, as detected by TUNEL, compared to cells cultured continuously at ambient (20%) O2 (Figure 1A). However, treatment with sildenafil during H/R prevented the increase in cell death. The DAPI nuclear staining indicated equivalent amounts of tissue present for each treatment. Quantification of TUNEL demonstrated increased (P < .05) cell death in chorionic villi exposed to H/R from 0.29 ± 0.02 to 0.18 ± 0.01 and 0.12 ± 0.01, compared to culture at either 20% or 2% O2 (Figure 1B). Supplementing the medium with 350 ng/mL sildenafil during H/R exposure reduced (P < .05) TUNEL to 0.15 ± 0.01, compared to H/R alone. Addition of sildenafil to trophoblast cells cultured continuously at 2% O2 or 20% O2 had no effect on TUNEL. Inhibition of cell death by sildenafil was dose dependent at concentrations of 35, 350, and 3500 ng/mL (Figure 1C). At 35 ng/mL, sildenafil reduced (P < .05) the TUNEL index from 0.18 to 0.098 ± 0.05, with a further reduction (P < .05) to values equivalent to the vehicle control (0.016 ± 0.01) at 350 ng/mL and above, suggesting an inhibitory concentration 50 of approximately 50 ng/mL for sildenafil. There was a reduction (P < 0.05) in cell death at all sildenafil concentrations tested, compared to vehicle with a maximum at 350 ng/mL.
Figure 1.

Effect of Sildenafil on cell death in first-trimester trophoblast cells. Dissected chorionic villous explants and HTR cells were cultured at 2% O2, 20% O2, or H/R with or without 350 ng/mL sildenafil. Cell death was assessed using a TUNEL assay (A). Villi were fluorescently double labeled to visualize nuclei with DAPI (upper panel) or DNA fragmentation by TUNEL (lower panel). Cell death was quantified and TUNEL Index determined by dividing the number of TUNEL-positive cells by the total cell number (B). HTR cells were treated with sildenafil at increasing concentrations as indicated, exposed to H/R, and cell death was quantified (C). Error bars denote SEM, *P < .05 compared to 2% O2 (B) or vehicle (C). HTR indicates human trophoblast cells; H/R, hypoxia/reoxygenation; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling; SEM, standard error of the mean.
Sildenafil Rescue Requires cGMP Signaling
Using the HTR-8/SVneo cytotrophoblast cell line, the cytoprotective activity of sildenafil was observed when cells were exposed to H/R (0.029 ± 0.004), compared to vehicle treatment (0.12 ± 0.01). The H/R increased (P < .05) the TUNEL index more than 2-fold above culture at hypoxia (Figure 2). Sildenafil had no effect on the TUNEL index of trophoblast cells during continuous culture at 2% O2. A cGMP analogue replicated the inhibition of apoptosis by sildenafil in trophoblast cells exposed to H/R (0.03 ± 0.01), whereas the cGMP inhibitor antagonized the cytoprotective effect of sildenafil increasing (P < .05) cell death to 0.14 ± 0.01, during H/R. Neither the cGMP analogue nor the cGMP inhibitor affected cell death in cells cultured at 2% O2. These data show that the ability of sildenafil to inhibit PDE5 and, thereby, increase cGMP is responsible for the inhibition of apoptosis during H/R treatment.
Figure 2.
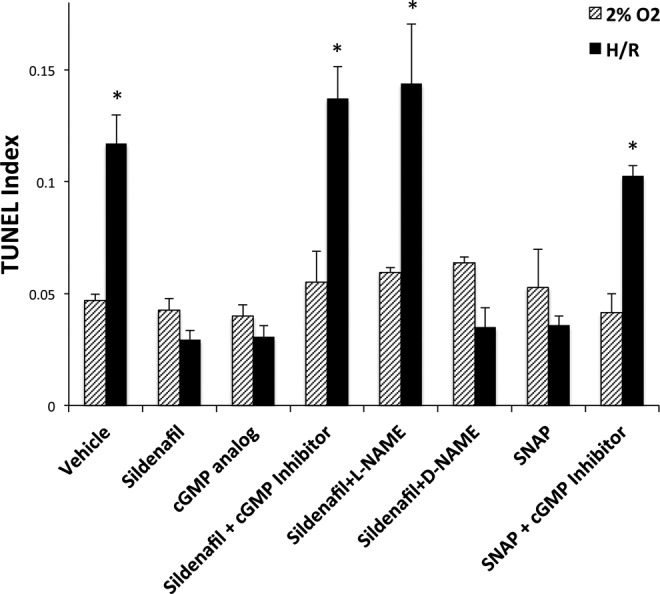
Sildenafil rescue of HTR cells through NO and cGMP signaling. HTR cells were treated with H/R and medium was supplemented as indicated with 10 μmol/L cGMP analogue, 350 ng/mL sildenafil with or without 10 μmol/L cGMP inhibitor, 10 μmol/L l-NAME, or 10 μmol/L d-NAME. Medium was also supplemented with 10 μmol/L SNAP with or without cGMP inhibitor. Cells were labeled for TUNEL and the TUNEL Index was determined. Error bars denote SEM *P < .05 compared to vehicle. HTR indicates human trophoblast cells; H/R, hypoxia/reoxygenation; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling; l-NAME, l-NG-Nitro-l-arginine Methyl Ester Hydrochloride SEM, standrd error of the mean; SNAP, S-Nitroso-N-acetylpenicillamine.
Sildenafil Rescue Requires NO Signaling
Since guanylyl cyclase is activated by NO, it was important to determine whether sildenafil requires NO to inhibit apoptosis. Trophoblast cells treated with both sildenafil and the NO antagonist l-NAME remained unprotected during H/R, increasing (P < .05) TUNEL to 0.14 ± 0.03 (Figure 2). The inactive isomer d-NAME did not interfere with the ability of sildenafil to prevent cell death, and neither treatment altered the TUNEL index of cells cultured continuously at 2% O2. Furthermore, treatment with NO donor SNAP protected trophoblast cells from H/R-induced cell death. Rescue by SNAP was dependent on cGMP downstream signaling, demonstrated by the increase (P < .05) in TUNEL observed when trophoblast cells exposed to H/R were treated with a combination of SNAP and the cGMP inhibitor.
Discussion
Complete understanding of the cause and pathogenesis of preeclampsia is still not resolved. However, extravillous trophoblast invasion is inadequate,38 spiral artery remodeling is insufficient, blood flow to the placenta is compromised, and an environment that generates oxidative stress is present,39 with endothelial dysfunction prevailing.3,40,41 The NO and cGMP pathways are critical regulators of vascular endothelial functions. Increases in sFLT1 and sENG levels during oxidative stress reduces NO production42,43 and risk of preeclampsia.44 Sildenafil, by inhibiting PDE5, enhances NO signaling, an important modulator of endothelial functions, providing a potential medical therapy for preeclampsia.40
Our findings imply that cell injury as a consequence of oxidative stress can be rescued by sildenafil, through inhibition of PDE5 to activate the NO-driven cGMP signaling pathway. Sildenafil reduced apoptosis of human trophoblast cells exposed to H/R in this experiment. Cytotrophoblast survival is compromised by H/R and could contribute to preeclampsia and other adverse pregnancy outcomes.8,13 In this study, sildenafil rescued human trophoblast cells from apoptosis by activating the NO and cGMP pathways.
Other studies have found that sildenafil protects against oxidative injury in heart, brain, skeletal muscle, and pulmonary endothelial cells. A reduction in cardiac cell apoptosis with the use of sildenafil through its antioxidant effects has been observed in the hearts of diabetic mice.45 Myocardial protection and reduction in infarct size was found in rats due to upregulation of VEGF and suppression of sFLT-1 after sildenafil treatment.46 There have been some beneficial effects of sildenafil treatment in the brain of rats after oxidative injury induced by ischemia/reperfusion.47 A reduction in ischemic injury and apoptosis was also noted in rat skeletal muscle.48 Rat pulmonary endothelial cells exposed to hypoxia exhibited significantly higher cell death rates, but apoptosis and endothelial dysfunction were reduced as migration and angiogenesis increased when the cells were treated with sildenafil.49 All of these studies showed a beneficial effect of sildenafil in reducing apoptosis of muscular and vascular components in animal models. The dosage of sildenafil used in cell culture experiments was based on its administration to rodents, which was a bolus of 0.7 mg/kg body weight sildenafil administered through the tail vein 30 minutes before ischemia.50 This dose compares well with the dosage used in humans (50 mg/70 kg).46
Sildenafil rescued trophoblast cells from apoptosis in adverse and stressful environments, which required the activation of both NO and cGMP signaling pathways. Sildenafil might provide positive effects on vascular endothelial function and blood flow to the endometrium, which benefited infertile patients with thin endometrial linings during their fertility treatments. Expression of PDE5 is increased by hypoxia and its inhibitors can provide a therapeutic intervention for pulmonary hypertension as well as for preeclampsia, growth restriction, and preterm labor.51
Our findings suggest that sildenafil acts at the fetal placental interface to reduce trophoblast cell death due to a stressful environment (H/R), possibly reflecting oxidative stress encountered during pathological placentation. Sildenafil could be used as a therapeutic intervention for adverse pregnancy outcomes. The direct effects of sildenafil on trophoblast cells suggest a potential treatment platform for infertility, recurrent pregnancy loss, and pregnancy complications such as preeclampsia and IUGR.
Acknowledgments
The authors thank the Northland Family Planning Centers of Michigan for participating in this research study.
Authors’ Note: This work was performed at the Mott Center for Human Growth & Development, Wayne State University School of Medicine.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported in part by the Intramural Research Program of the National Institute for Child Health and Human Development, National Institutes of Health and grants from the NIH (HD071408) and the W.K. Kellogg Foundation.
References
- 1. Roberts JM, Pearson GD, Cutler JA, Lindheimer MD, National Heart L, Blood I. Summary of the NHLBI Working Group on Research on Hypertension During Pregnancy. Hypertension Pregnancy. 2003;22 (2):109–127. [DOI] [PubMed] [Google Scholar]
- 2. George EM, Palei AC, Dent EA, Granger JP. Sildenafil attenuates placental ischemia-induced hypertension. Am J Physiol Regul Integr Comp Physiol. 2013;305 (4):R397–R403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. 2010;376 (9741):631–644. [DOI] [PubMed] [Google Scholar]
- 4. Turner JA. Diagnosis and management of pre-eclampsia: an update. Int J Womens Health. February 2010:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Genbacev O, Joslin R, Damsky CH, Polliotti BM, Fisher SJ. Hypoxia alters early gestation human cytotrophoblast differentiation/invasion in vitro and models the placental defects that occur in preeclampsia. J Clin Invest. 1996;97 (2):540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kilburn BA, Wang J, Duniec-Dmuchowski ZM, Leach RE, Romero R, Armant DR. Extracellular matrix composition and hypoxia regulate the expression of HLA-G and integrins in a human trophoblast cell line. Biol Reprod. 2000;62 (3):739–747. [DOI] [PubMed] [Google Scholar]
- 7. Hung TH, Skepper JN, Burton GJ. In vitro ischemia-reperfusion injury in term human placenta as a model for oxidative stress in pathological pregnancies. Am J Pathol. 2001;159(3):1031–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hung TH, Burton GJ. Hypoxia and reoxygenation: a possible mechanism for placental oxidative stress in preeclampsia. Taiwan J Obstet Gynecol. 2006;45 (3):189–200. [DOI] [PubMed] [Google Scholar]
- 9. Leach RE, Kilburn BA, Petkova A, Romero R, Armant DR. Diminished survival of human cytotrophoblast cells exposed to hypoxia/reoxygenation injury and associated reduction of heparin-binding epidermal growth factor-like growth factor. Am J Obstetr Gynecol. 2008;198(4):471 e471–471 e477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Burton GJ, Jauniaux E. Placental oxidative stress: from miscarriage to preeclampsia. J Soc Gynecol Investig. 2004;11 (6):342–352. [DOI] [PubMed] [Google Scholar]
- 11. Burton GJ, Jauniaux E, Watson AL. Maternal arterial connections to the placental intervillous space during the first trimester of human pregnancy: the Boyd collection revisited. Am J Obstet Gynecol. 1999;181 (3):718–724. [DOI] [PubMed] [Google Scholar]
- 12. Jauniaux E, Watson AL, Hempstock J, Bao YP, Skepper JN, Burton GJ. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am J Pathol. 2000;157 (6):2111–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hung TH, Skepper JN, Charnock-Jones DS, Burton GJ. Hypoxia-reoxygenation: a potent inducer of apoptotic changes in the human placenta and possible etiological factor in preeclampsia. Circulation Research. Jun 28 2002;90(12):1274–1281. [DOI] [PubMed] [Google Scholar]
- 14. Khong Y, Brosens I. Defective deep placentation. Best Pract Res Clin Obstet Gynaecol.2011;25 (3):301–311. [DOI] [PubMed] [Google Scholar]
- 15. Ramesar SV, Mackraj I, Gathiram P, Moodley J. Sildenafil citrate decreases sFlt-1 and sEng in pregnant l-NAME treated Sprague-Dawley rats. Eur J Obstet Gynecol Reprod Biol. 2011;157 (2):136–140. [DOI] [PubMed] [Google Scholar]
- 16. Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62 (3):525–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stacey P, Rulten S, Dapling A, Phillips SC. Molecular cloning and expression of human cGMP-binding cGMP-specific phosphodiesterase (PDE5). Biochem Biophys Res Commun. 1998;247 (2):249–254. [DOI] [PubMed] [Google Scholar]
- 18. Karasu E, Kayacan N, Sadan G, Dinc B. Endothelial dysfunction in the human umbilical artery due to preeclampsia can be prevented by sildenafil. Clin Exp Hypertens. 2012;34 (2):79–85. [DOI] [PubMed] [Google Scholar]
- 19. Ashworth JR, Warren AY, Baker PN, Johnson IR. Loss of endothelium-dependent relaxation in myometrial resistance arteries in pre-eclampsia. Br J Obstet Gynaecol. 1997;104 (10):1152–1158. [DOI] [PubMed] [Google Scholar]
- 20. McCarthy AL, Woolfson RG, Raju SK, Poston L. Abnormal endothelial cell function of resistance arteries from women with preeclampsia. Am J Obstet Gynecol. 1993;168 (4):1323–1330. [DOI] [PubMed] [Google Scholar]
- 21. Boolell M, Allen MJ, Ballard SA, et al. Sildenafil: an orally active type 5 cyclic GMP-specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction. Int J Impot Res. 1996;8 (2):47–52. [PubMed] [Google Scholar]
- 22. Takasaki A, Tamura H, Miwa I, Taketani T, Shimamura K, Sugino N. Endometrial growth and uterine blood flow: a pilot study for improving endometrial thickness in the patients with a thin endometrium. Fertil Steril. 2010;93 (6):1851–1858. [DOI] [PubMed] [Google Scholar]
- 23. Maharaj CH, O'Toole D, Lynch T, et al. Effects and mechanisms of action of sildenafil citrate in human chorionic arteries. Reprod Biol Endocrinol. 2009;7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Santos-Silva AJ, Cairrao E, Morgado M, Alvarez E, Verde I. PDE4 and PDE5 regulate cyclic nucleotides relaxing effects in human umbilical arteries. Eur J Pharmacol. 2008;582 (1-3):102–109. [DOI] [PubMed] [Google Scholar]
- 25. Wareing M, Myers JE, O'Hara M, et al. Effects of a phosphodiesterase-5 (PDE5) inhibitor on endothelium-dependent relaxation of myometrial small arteries. Am J Obstet Gynecol. 2004;190 (5):1283–1290. [DOI] [PubMed] [Google Scholar]
- 26. George EM, Granger JP. Mechanisms and potential therapies for preeclampsia. Curr Hypertens Rep. 2011;13 (4):269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Downing J. Sildenafil for the treatment of preeclampsia. Hypertens Pregnancy. 2010;29 (2):248–250. [DOI] [PubMed] [Google Scholar]
- 28. von Dadelszen P, Dwinnell S, Magee LA, et al. Sildenafil citrate therapy for severe early-onset intrauterine growth restriction. BJOG. 2011;118 (5):624–628. [DOI] [PubMed] [Google Scholar]
- 29. Ganzevoort W, Alfirevic Z, von Dadelszen P, et al. STRIDER: sildenafil Therapy In Dismal prognosis Early-onset intrauterine growth Restriction--a protocol for a systematic review with individual participant data and aggregate data meta-analysis and trial sequential analysis. Syst Rev. 2014;3:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abdalla HI, Brooks AA, Johnson MR, Kirkland A, Thomas A, Studd JW. Endometrial thickness: a predictor of implantation in ovum recipients? Hum Reprod. 1994;9 (2):363–365. [DOI] [PubMed] [Google Scholar]
- 31. Miwa I, Tamura H, Takasaki A, Yamagata Y, Shimamura K, Sugino N. Pathophysiologic features of “thin” endometrium. Fertil Steril. 2009;91 (4):998–1004. [DOI] [PubMed] [Google Scholar]
- 32. Shifren JL, Tseng JF, Zaloudek CJ, et al. Ovarian steroid regulation of vascular endothelial growth factor in the human endometrium: implications for angiogenesis during the menstrual cycle and in the pathogenesis of endometriosis. J Clin Endocrinol Metab. 1996;81 (8):3112–3118. [DOI] [PubMed] [Google Scholar]
- 33. Sugino N, Kashida S, Takiguchi S, Karube A, Kato H. Expression of vascular endothelial growth factor and its receptors in the human corpus luteum during the menstrual cycle and in early pregnancy. J Clin Endocrinol Metab. 2000;85 (10):3919–3924. [DOI] [PubMed] [Google Scholar]
- 34. Sugino N, Kashida S, Karube-Harada A, Takiguchi S, Kato H. Expression of vascular endothelial growth factor (VEGF) and its receptors in human endometrium throughout the menstrual cycle and in early pregnancy. Reproduction. 2002;123 (3):379–387. [DOI] [PubMed] [Google Scholar]
- 35. Coppage KH, Sun X, Baker RS, Clark KE. Expression of phosphodiesterase 5 in maternal and fetal sheep. Am J Obstet Gynecol. 2005;193 (3 pt 2):1005–1010. [DOI] [PubMed] [Google Scholar]
- 36. Huang LT, Hsieh CS, Chang KA, Tain YL. Roles of nitric oxide and asymmetric dimethylarginine in pregnancy and fetal programming. Int J Mol Sci. 2012;13 (11):14606–14622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lowe DT. Nitric oxide dysfunction in the pathophysiology of preeclampsia. Nitric Oxide. 2000;4 (4):441–458. [DOI] [PubMed] [Google Scholar]
- 38. Khong TY, De Wolf F, Robertson WB, Brosens I. Inadequate maternal vascular response to placentation in pregnancies complicated by pre-eclampsia and by small-for-gestational age infants. Br J Obstet Gynaecol. 1986;93 (10):1049–1059. [DOI] [PubMed] [Google Scholar]
- 39. Redman CW, Sargent IL. Placental stress and pre-eclampsia: a revised view. Placenta. 2009;30 (suppl A):S38–S42. [DOI] [PubMed] [Google Scholar]
- 40. Johal T, Lees CC, Everett TR, Wilkinson IB. The nitric oxide pathway and possible therapeutic options in pre-eclampsia. Br J Clin Pharmacol. 2014;78 (2):244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roberts JM, Gammill HS. Preeclampsia: recent insights. Hypertension. 2005;46 (6):1243–1249. [DOI] [PubMed] [Google Scholar]
- 42. Sandrim VC, Palei AC, Metzger IF, Gomes VA, Cavalli RC, Tanus-Santos JE. Nitric oxide formation is inversely related to serum levels of antiangiogenic factors soluble fms-like tyrosine kinase-1 and soluble endogline in preeclampsia. Hypertension. 2008;52 (2):402–407. [DOI] [PubMed] [Google Scholar]
- 43. Levine RJ, Lam C, Qian C, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006;355 (10):992–1005. [DOI] [PubMed] [Google Scholar]
- 44. Levine RJ, Maynard SE, Qian C, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350 (7):672–683. [DOI] [PubMed] [Google Scholar]
- 45. Ebrahimi F, Shafaroodi H, Asadi S, et al. Sildenafil decreased cardiac cell apoptosis in diabetic mice: reduction of oxidative stress as a possible mechanism. Can J Physiol Pharmacol. 2009;87 (7):556–564. [DOI] [PubMed] [Google Scholar]
- 46. Koneru S, Varma Penumathsa S, Thirunavukkarasu M, et al. Sildenafil-mediated neovascularization and protection against myocardial ischaemia reperfusion injury in rats: role of VEGF/angiopoietin-1. J Cell Mol Med. 2008;12 (6B):2651–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ozdegirmenci O, Kucukozkan T, Akdag E, et al. Effects of sildenafil and tadalafil on ischemia/reperfusion injury in fetal rat brain. J Matern Fetal Neonatal Med. 2011;24 (2):317–323. [DOI] [PubMed] [Google Scholar]
- 48. Armstrong DM, Armstrong Ada C, Figueiredo RC, et al. Sildenafil citrate protects skeletal muscle of ischemia-reperfusion injury: immunohistochemical study in rat model. Acta Cir Bras. 2013;28 (4):282–287. [DOI] [PubMed] [Google Scholar]
- 49. Gammella E, Leuenberger C, Gassmann M, Ostergaard L. Evidence of synergistic/additive effects of sildenafil and erythropoietin in enhancing survival and migration of hypoxic endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2013;304 (4):L230–L239. [DOI] [PubMed] [Google Scholar]
- 50. Atalay B, Caner H, Cekinmez M, Ozen O, Celasun B, Altinors N. Systemic administration of phosphodiesterase V inhibitor, sildenafil citrate, for attenuation of cerebral vasospasm after experimental subarachnoid hemorrhage. Neurosurgery. 2006;59 (5):1102–1107. [DOI] [PubMed] [Google Scholar]
- 51. Sebkhi A, Strange JW, Phillips SC, Wharton J, Wilkins MR. Phosphodiesterase type 5 as a target for the treatment of hypoxia-induced pulmonary hypertension. Circulation. 2003;107 (25):3230–3235. [DOI] [PubMed] [Google Scholar]