

Abstract
PTEN (phosphatase and tensin homolog), a tumor suppressor frequently mutated in human cancer, has various cytoplasmic and nuclear functions. PTEN translocates to the nucleus from the cytoplasm in response to oxidative stress. However, the mechanism and function of the translocation are not completely understood. In this study, topotecan (TPT), a topoisomerase I inhibitor, and cisplatin (CDDP) were employed to induce DNA damage. The results indicate that TPT or CDDP activates ATM (ATM serine/threonine kinase), which phosphorylates PTEN at serine 113 and further regulates PTEN nuclear translocation in A549 and HeLa cells. After nuclear translocation, PTEN induces autophagy, in association with the activation of the p-JUN-SESN2/AMPK pathway, in response to TPT. These results identify PTEN phosphorylation by ATM as essential for PTEN nuclear translocation and the subsequent induction of autophagy in response to DNA damage.
Keywords: ATM, autophagy, DNA damage, PTEN, topotecan
Abbreviations: AKT/PKB, v-akt murine thymoma viral oncogene homolog; AMPK, protein kinase, AMP-activated; ATG, autophagy-related; ATM, ATM serine/threonine kinase; Baf.A1, bafilomycin A1; CASP3, caspase 3, apoptosis-related cysteine peptidase; CCND1, cyclin D1; CDDP, cisplatin; CENPC/CENP-C, centromere protein C; CITED1/p300/CBP, Cbp/p300-interacting transactivator, with Glu/Asp-rich C-terminal domain, 1; CSNK2/CK2, casein kinase 2; DSBs, DNA double-strand breaks; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GFP, green fluorescent protein; GLTSCR2/PICT-1, glioma tumor suppressor candidate region gene 2; GSK3B, glycogen synthase kinase 3 β; GST, glutathione S-transferase; H2AFX; H2A histone family; member X; JUN; jun proto-oncogene; MAP1LC3/LC3, microtubule-associated protein 1 light chain 3; MTORC1, mechanistic target of rapamycin complex 1; MVP, major vault protein; NC, normal control; NEDD4, neural precursor cell expressed, developmentally down-regulated 4, E3 ubiquitin protein ligase; PAGE, polyacrylamide gel electrophoresis; PARP, poly (ADP-ribose) polymerase 1; PI3K, phosphoinositide 3-kinase; PMSF, phenylmethanesulfonyl fluoride; PPase, protein phosphatase; PtdIns(3, 4, 5)P3, phosphatidylinositol (3, 4, 5)-trisphosphate; PTEN, phosphatase and tensin homolog; RAD51, RAD51 recombinase; RPS6KB/p70S6K; ribosomal protein S6 kinase, 70kDa; SDS, sodium dodecyl sulfate; SESN2, sestrin 2; siRNA, small interfering RNA; SQSTM1/p62, sequestosome 1; TP53, tumor protein p53; TPT, topotecan; TUBA4A, tubulin, α, 4a; WT, wild type; YFP, yellow fluorescent protein
Introduction
PTEN/MMAC1 (phosphatase and tensin homolog) has been identified as a tumor suppressor gene from a variety of cancers that display frequent deletion of its location on human chromosome 10q23, a locus that is highly susceptible to mutation in primary human cancers and cancer cell lines.1 PTEN converts phosphatidylinositol (3,4,5)-trisphosphate (PtdInsP3) to phosphatidylinositol (4,5)-bisphosphate in the cytoplasm, thereby directly antagonizing the activity of phosphoinositide 3-kinase (PI3K).2,3 PTEN inactivation results in the constitutive activation of the PI3K-AKT pathway and a subsequent increase in protein synthesis, cell cycle progression, migration, and survival.4,5
PTEN is localized to the nucleus and cytoplasm. Early immunohistochemical studies of PTEN localization found that the endogenous wild-type PTEN protein is exclusively localized in the cytoplasm of various cancer cell lines.6 Subsequently, multiple reports have identified PTEN nuclear or perinuclear PTEN in normal primary neurons and endothelial cells, PTEN-positive primary breast carcinoma samples, normal epithelial and follicular thyroid cells, follicular thyroid adenomas, and thyroid carcinomas.7,8 However, the nuclear PTEN immunostaining is relatively weak in thyroid carcinomas compared with normal thyroid follicular cells and follicular thyroid adenomas.9
Although normal islet cells exhibit predominantly nuclear PTEN immunostaining in pancreatic tissue, 19 of 23 sporadic endocrine pancreatic tumors predominantly display cytoplasmic PTEN expression patterns.10 Whiteman observes a similar phenomenon, finding increased nuclear PTEN expression in normal melanocytes and reduced nuclear PTEN expression in melanoma cells. However, PTEN protein expression is primarily localized to the cytoplasm of metastatic melanoma samples. An association between the lack of nuclear PTEN and mitotic index has been reported; nuclear PTEN is evident in quiescent cells, but PTEN primarily localizes to the cytosol of actively dividing cells.11 Generally, PTEN is primarily localized to the nucleus in normal quiescent tissues, whereas cytoplasmic PTEN is predominately found in neoplastic tissues. Thus, nuclear PTEN appears to be essential for tumor suppression.
Despite the absence of a classic nuclear localization signal, PTEN enters the nucleus by several mechanisms, including simple diffusion, export dependent on a putative cytoplasmic localization signal, and active shuttling by the RAN GTPase or MVP (major vault protein).12-15 PTEN monoubiquitination at Lys13 or Lys289 by NEDD4 increases its nuclear localization, whereas polyubiquitination by NEDD4 promotes its cytoplasmic degradation.16,17
Accumulating evidence suggests multiple mechanisms of PTEN nuclear import depending on the cell type and cellular environment. Moreover, the role of nuclear PTEN is not the same as that of cytoplasmic PTEN. Nuclear PTEN plays a crucial role in regulating cellular homeostasis and stability by downregulating the MAPK signal pathway, reducing CCND1 (cyclin D1) levels and G0-G1 arrest, upregulating RAD51 (RAD51 recombinase) levels and double-strand-break repair, and interacting with CENPC/CENP-C (centromere protein C), which specifically enhances centromere stability and overall genomic stability by promoting CITED1/p300/CBP(Cbp/p300-interacting transactivator, with Glu/Asp-rich C-terminal domain, 1)-mediated acetylation of TP53/p53 (TRP53 in mice) in response to DNA damage and apoptosis.18-23
In this study, we find that ATM (ATM serine/threonine kinase) phosphorylates PTEN at serine 113, further promoting PTEN nuclear translocation. PTEN nuclear translocation induces autophagy through activation of the p-JUN-SESN2-AMPK pathway following treatment with the DNA-damaging agent topotecan (TPT) in A549 and HeLa cells.
Results
DNA-damaging agents induced ATM phosphorylation
TPT, which causes DNA damage, is commonly used for cancer treatment in the clinic. CDDP (cisplatin) is also an anticancer drug that causes cross-linking of DNA. ATM kinase is activated in response to DNA double-strand breaks (DSBs). To investigate whether TPT or CDDP treatment induces ATM phosphorylation in A549 and HeLa cells, we determined the levels of phosphorylated ATM by immunoblotting. As shown in Fig. S1, ATM phosphorylation increased in a dose- and time-dependent manner after treatment with various concentrations of TPT or CDDP for 24 h and with 3 μg/mL TPT or 6 μg/mL CDDP for various times, respectively. These data indicated that TPT and CDDP induced ATM phosphorylation and activation in A549 and HeLa cells.
ATM regulates PTEN nuclear translocation, following exposure to DNA-damaging agents
It has been previously reported that PTEN accumulates in the nucleus of cells treated with apoptotic stimuli, including etoposide, doxorubicin,20 or H2O2, indicating that oxidative stress promotes PTEN nuclear accumulation.24 PTEN is normally found in the cytoplasm in A549 and HeLa cells. After A549 and HeLa cells were treated with 3 μg/mL TPT for 24 h, endogenous PTEN translocated from the cytoplasm to the nucleus. However, when A549 and HeLa cells were treated with 3 μg/mL TPT and 10 μM KU55933 (ATM specific inhibitor) for 24 h, PTEN nuclear translocation was clearly suppressed (Fig. 1A).
Figure 1.

ATM regulated TPT-induced PTEN nuclear translocation in A549 and HeLa cells. (A) Representative photomicrographs showed endogenous PTEN translocation (100×). A549 and HeLa cells were treated with 3 μg/mL TPT and/or 10 μM KU55933 for 24 h. (B) 3×Flag-PTEN-WT plasmids were transfected into the HeLa cells, and the transfection efficiency was confirmed by western blot analysis. (C) Representative photomicrograph images showed exogenous PTEN translocation (100×). HeLa cells were transiently transfected with 3×Flag-PTEN-WT plasmids, and 24 h after transfection, the cells were treated with 3 μg/mL TPT and/or 10 μM KU55933 and/or ATM siRNA for 24 h. (D) Representative photomicrograph images showing exogenous PTEN translocation (100×). HeLa cells were transiently transfected with 3×Flag-PTEN-WT or 3×Flag-PTENS113A plasmids, and 24 h after transfection, the cells were treated with 6 μg/mL CDDP. (E) Western blot analysis result of exogenous PTEN nuclear translocation in A549 and HeLa cells when treated with 3 μg/mL TPT and/or 10 μM KU55933 for 24 h. (F) Western blotting result of exogenous PTEN nuclear translocation in A549 and HeLa cells when treated with 3 μg/mL TPT and/or ATM siRNA for 24 h.
To detect exogenous PTEN nuclear translocation, the 3×Flag-PTEN-WT plasmid was constructed. The 3×Flag-PTEN-WT expression vector was transiently transfected into HeLa cells, and the cells were treated with TPT (3 μg/mL) and/or KU55933 (10 μM) for 24 h. The transfection and expression efficiency were determined by immunoblotting (Fig. 1B). When HeLa cells were treated with 3 μg/mL TPT for 24 h, exogenous PTEN translocated from the cytoplasm to the nucleus, consistent with the endogenous PTEN translocation pattern observed by confocal microscopy under the same conditions. When the ATM specific inhibitor KU55933 or transient transfection with ATM siRNA was used to inhibit ATM activity, the PTEN translocation was clearly suppressed (Fig. 1C). Also, when HeLa cells were treated with 6 μg/mL CDDP for 24 h, exogenous PTEN translocated from the cytoplasm to the nucleus, which was consistent with the result from TPT treatment (Fig. 1D).
To further confirm PTEN nuclear translocation, nuclear and cytoplasm proteins of A549 cells and HeLa cells were extracted, and the PTEN expression in these components was detected by western blotting. As expected, PTEN levels were increased in the nuclear fraction compared with the cytosolic fraction in the TPT-treated group, suggesting that TPT induces PTEN nuclear translocation. However, TPT-induced PTEN nuclear translocation was significantly inhibited upon treatment with KU55933 and ATM siRNA (Figs. 1E and F), indicating that PTEN nuclear translocation is regulated by ATM.
ATM regulates PTEN phosphorylation, following exposure to DNA-damaging agents
Phosphorylation is an important protein posttranslational modification. Given that we demonstrated that TPT induces ATM phosphorylation and that ATM further regulates TPT-induced PTEN nuclear translocation in A549 and HeLa cells, we wanted to determine whether PTEN nuclear translocation was regulated by ATM via phosphorylation.
To determine whether ATM directly phosphorylates PTEN, we first analyzed the PTEN sequence using Scansite software (http://scansite.mit.edu) and identified a potential ATM phosphorylation site at serine 113 (Ser113) (Fig. 2A). Next, we developed a phospho-specific antibody against PTEN (Ser113). To validate the Phospho-PTEN (Ser113) antibody, A549 cells were treated with: 1) 3 μg/mL TPT only; 2) 3 μg/mL TPT and λ-phosphatase; 3) 3 μg/mL TPT, λ-phosphatase, and protein phosphatase (PPase) inhibitors. The phospho-PTEN (Ser113) levels in A549 cells treated with λ-phosphatase were significantly decreased compared with the TPT group. The inhibitory effect was partly restored when PPase inhibitors were applied (Fig. 2B). The results confirmed the specificity of the phospho-PTEN (Ser113) antibody.
Figure 2.

ATM mediated TPT-induced PTEN phosphorylation. (A) Sequence analysis of PTEN was performed by Scansite software. The sequence of PTEN was retrieved from GenBank. (B) A549 cells were treated with 3 μg/mL TPT, λ-phosphatase, and PPase inhibitors as indicated. Tagged proteins were analyzed by immunoblotting. (C) HeLa cells were transiently transfected with the 3×Flag-PTEN-WT plasmid,then treated with various concentrations of TPT or CDDP. (D) A549 and HeLa cells were transiently transfected with 3×Flag-PTEN-WT plasmids, and 24 h after transfection, the cells were treated with 3 μg/mL TPT; then, immunoprecipitation was conducted with an antibody against Flag. The precipitates were subjected to SDS-PAGE and probed with antibody against phospho-PTENS113. (E) A549 cells were treated with 3 μg/mL TPT for 24 h in the absence or presence of ATM siRNA- or KU55933, and then immunoblotting analysis was performed. (F) In vitro phosphorylation assays with active ATM kinase and purified PTEN recombinant proteins. Reaction products were subjected to SDS-PAGE and probed with antibody against phospho-PTENS113.
To determine whether TPT induces PTEN phosphorylation at Ser113, we transiently transfected HeLa cells with the 3×Flag-PTEN-WT plasmid. The cells were then exposed to different concentrations of TPT or CDDP for 24 h. The results indicated that the levels of p-PTEN (Ser113) were enhanced in a concentration-dependent manner (Fig. 2C).
In addition, A549 or HeLa cells were treated with 3 μg/mL TPT for 24 h, and immunoprecipitation was performed using a Flag antibody. The precipitates were subjected to SDS-PAGE and probed with the p-PTEN (Ser113) antibody. As shown in Fig. 2D, the p-PTEN (Ser113) levels in the TPT treatment group were significantly increased compared with the control group, indicating that TPT induces PTEN (Ser113) phosphorylation.
Furthermore, we sought to investigate whether TPT-induced PTEN phosphorylation was regulated by ATM. A549 cells and HeLa cells, with reduced ATM expression by siRNA or KU55933, were treated with 3 μg/mL TPT for 24 h. As shown in Fig. 2E, the p-ATM and p-PTEN (Ser113) levels were significantly increased in the TPT treatment group. However, knockdown of ATM by siRNA or KU55933 significantly reduced the p-PTEN (Ser113) levels. These data indicate that ATM regulates TPT-induced PTEN (Ser113) phosphorylation.
The in vitro kinase assays with purified proteins was used to assess whether PTEN was directly phosphorylated by ATM. As show in Fig. 2F, the p-PTEN (Ser113) levels were significantly increased in the group treated with both ATM and PTEN. When the ATM-specific inhibitor KU55933 was used to inhibit ATM activity, the p-PTEN (Ser113) levels were decreased. The results indicated that PTEN was directly phosphorylated by ATM.
TPT-induced PTEN translocation is dependent on PTEN (Ser113) phosphorylation
To further determine the role of phosphorylation of PTEN (Ser113) in ATM-mediated PTEN nuclear translocation in response to TPT, the mutant plasmid 3×Flag-PTENS113A was constructed. HeLa cells were transiently transfected with the 3×Flag-PTENS113A plasmid. After treatment with TPT, the cells transfected with the 3×Flag-PTEN-WT plasmid showed PTEN nuclear translocation and PTEN Ser113 phosphorylation; However, evidence for PTEN nuclear translocation and PTEN (Ser113) phosphorylation was not observed by confocal microscopy and western blotting in cells with 3×Flag-PTENS113A (Figs. 3A and B). Also, when HeLa cells expressing 3×Flag-PTEN-WT or 3×Flag-PTENS113A were treated with CDDP, similar results were obtained (Fig. 1D).
Figure 3.
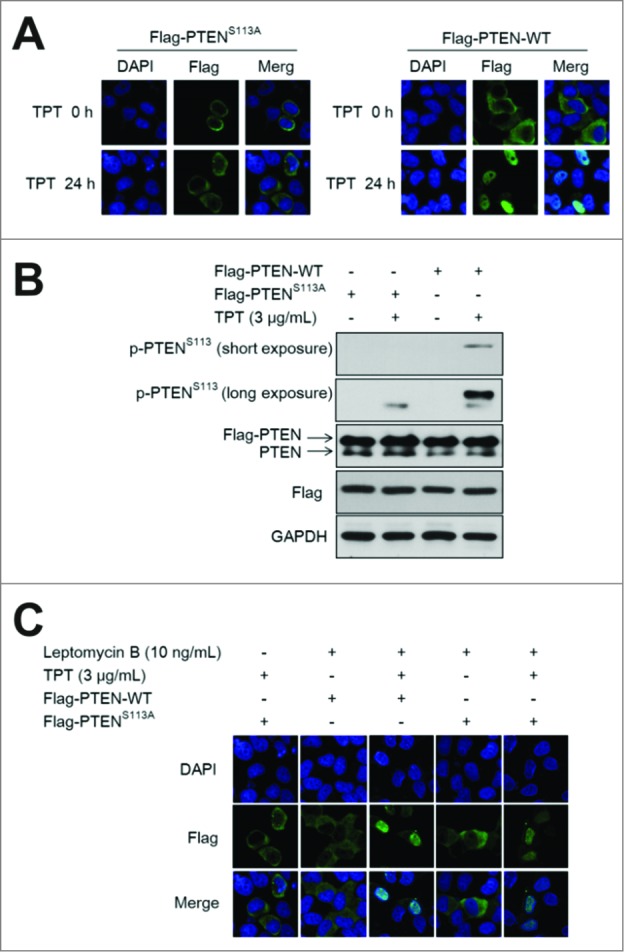
TPT-induced PTEN nuclear translocation was dependent on PTEN (S113) phosphorylation in A549 and HeLa cells. A549 cells and/or HeLa cells were transiently transfected with the 3×Flag-PTEN-WT or 3×Flag-PTENS113A plasmid, and 24 h after transfection, the cells were treated with or without 3 μg/mL TPT. (A) Representative photomicrograph images showing PTEN translocation (200×). (B) The expression levels of PTEN and p-PTENS113 were analyzed by immunoblotting. (C) Representative photomicrograph images showing PTEN translocation (200×) after treatment of 10 ng/mL of leptomycin B for 24 h.
When the cells were treated with 10ng/mL nuclear export inhibitor leptomycin B and TPT, PTENS113A localized in the nucleus (Fig. 3C). This result indicates that this mutant can enter the nucleus but is not retained there. Therefore, TPT-induced PTEN translocation is dependent on PTEN (Ser113) phosphorylation in A549 and HeLa cells.
SUMO proteins are small ubiquitin-like modifiers. Similar to protein phosphorylation, SUMOylation is also a post-translational modifier involved in various cellular processes. It was reported that SUMOylation of PTEN controls its nuclear localization.25 To investigate if PTEN (Ser113) phosphorylation was involved in PTEN SUMOylation, HeLa cells carrying 3×Flag-PTEN-WT or 3×Flag-PTENS113A were transfected with HA-SUMO1 and treated with 3 μg/mL TPT for 24 h. HA-immunoprecipitates (IP) were immunoblotted for SUMO-PTEN. As shown in Fig. S2, SUMO-PTEN was observed, and its level was increased upon TPT treatment in both cells expressing PTEN-WT and PTENS113A. The result suggests that Ser113 phosphorylation is not involved in PTEN SUMOylation.
Mutant PTENS113A enhances TPT-induced DNA damage
An initial step in the mammalian cell response to DNA double-strand breaks is the phosphorylation of histone H2AFX at serine 139 at the site of DNA breaks. Once ATM is activated at a DNA double-strand break, it immediately phosphorylates histone H2AFX at the site of the break, thereby signaling to the cell that a DSB has occurred.26 Formation of phosphorylated H2AFX at Ser139 (termed γ-H2AFX), is a rapid and sensitive cellular response to the presence of DSBs.27 TPT is a specific inhibitor of topoisomerase I in the nucleus. Immunofluorescence and confocal microscopy were used to assess whether TPT induces DNA damage in A549 and HeLa cells. A549 and HeLa cells were treated with 3 μg/mL TPT for 24 h. γ-H2AFX foci were observed in TPT-treated cells after 12 h and 24 h and increased in a time-dependent manner, whereas no detectable foci was observed in the control group (Fig. S3A). Bar graphs representing the percentage of γ-H2AFX-positive cells at different time-points were presented in Fig. S3B.
Upon treatment with 3 μg/mL TPT for 0 h to 36 h, the expression of γ-H2AFX protein increased in a time-dependent manner in A549 and HeLa cells (Fig. S3C). The results indicate that TPT induces DBS in A549 and HeLa cells.
We discovered that phospho-PTEN (Ser113) plays a key role in PTEN nuclear translocation. To further determine the role of PTEN (Ser113) in TPT-induced DNA damage, A549 and HeLa cells were transiently transfected with the 3×Flag-PTEN-WT or 3×Flag-PTENS113A plasmid 24 h prior to 3 μg/mL TPT treatment for 24 h. As shown in Fig. 4A, γ-H2AFX foci were notably increased in the cells with the 3×Flag- PTENS113A plasmid compared with the cells with the 3×Flag-PTENS113A plasmid following TPT treatment. Bar graph represents percentage of cells with γ-H2AFX-positive cells in control or TPT-treated for 24 h (Fig. 4B).
Figure 4.

The PTENS113A mutant enhanced TPT-induced DNA damage in A549 and HeLa cells. (A) A549 and HeLa cells were transiently transfected with the 3×Flag-PTEN-WT or 3×Flag-PTENS113A plasmid, and 24 h after transfection, the cells were treated with or without 3 μg/mL TPT. Representative photomicrograph images showed accumulation of γ-H2AFX as an indicator of DNA damage (100×). (B) Bar graphs representing the percentage of γ-H2AFX-positive cells upon treatment with TPT for 24 h (*P < 0.05, **P <0.001, t test, n = 3 , bars represent SEM, Cells containing more than 5 foci were scored as positive). (C) A549 and HeLa cells were transiently transfected with the 3×Flag-PTEN-WT or 3×Flag-PTENS113A plasmid, and 24 h after transfection, the ells were treated with or without 3 μg/mL TPT. The expression levels of γ-H2AFX protein were analyzed by immunoblotting.
The western blotting results indicated that the γ-H2AFX expression levels were enhanced in all TPT-treated groups, with a remarkable increase noted with the 3×Flag-PTENS113A plasmids. These results are consistent with the immunofluorescence results (Fig. 4C), suggesting that mutant PTENS113A enhances TPT-induced DNA damage in A549 and HeLa cells. The same results were obtained using A549 cells with PTEN shRNA-mediated knockdown (Fig. S3D)
Moreover, since DSB repair is affected by the cell cycle stage, we sought to determine whether the PTENS113A mutation affected the cell cycle stage using flow cytometry. A549 cells were transiently transfected with the 3×Flag-PTEN-WT or 3×Flag-PTENS113A plasmid 24 h prior to TPT treatment. As shown in Fig. S4A, the percentage of cells in G0/G1 phase increased in the vector, PTEN-WT and PTENS113A group, with no significant difference between the PTEN-WT and PTENS113 group.
TPT-stimulated apoptosis has been reported in S-phase A549 cells.28 To investigate if PTENS113A mutant affect cell apoptosis, we examinated the expression level of cleaved CASP3/caspase 3 and PARP (poly [ADP-ribose] polymerase 1) protein. The cleaved CASP3 and PARP levels were increased with 24 h TPT treatment in all the groups. However, the increase in the expression levels of cleaved CASP3 and PARP were more significant in cells with 3×Flag-PTEN-WT compared to those expressing 3×Flag-PTENS113A upon 48 h TPT treatment (Fig. S4B). These results suggested that the PTENS113A mutation did affect cell apoptosis and further confirmed that PTEN nuclear translocation played a key role in its tumor-suppressor function.
Nuclear translocation of PTEN regulates TPT-induced autophagy
The microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3), an ortholog of yeast Atg8, exists on the autophagosome and phagophore membranes. MAP1LC3-II/LC3-II (the cleaved and lipidated form of microtubule-associated protein 1) amounts correlate well with the number of autophagosomes.29 Enhancement of the conversion of MAP1LC3-I/LC3-I (soluble form of microtubule-associated protein 1 light chain 3) to MAP1LC3-II/LC3-II and upregulation of MAP1LC3/LC3 expression occurs when autophagy is induced.30 SQSTM1/p62 possesses a short region that interacts with MAP1LC3/LC3.31 SQSTM1/p62 participates in autophagy and is degraded in the autolysosome. MAP1LC3-II/LC3-II and SQSTM1/p62 are reliable markers of autophagy. To test whether TPT induces autophagy in cancer cells, we first assessed YFP-LC3 accumulation and punctate formation using confocal microscopy. YFP-LC3 plasmids were transiently transfected into A549 and HeLa cells for 24 h, and the cells were then treated with 3 μg/mL TPT for an additional 24 h. YFP-LC3 significantly accumulated in the cytoplasm, which suggested autophagosome formation (Fig. 5A). Next, western blot analysis revealed that MAP1LC3-II/LC3-II expression markedly increased but that level of SQSTM1/p62 significantly decreased due to degradation in a concentration- and time-dependent manner in TPT-treated A549 and HeLa cells (Fig. 5B). These results suggest that TPT induces the conversion of MAP1LC3-I/LC3-I to MAP1LC3-II/LC3-II and the degradation of SQSTM1/p62 in A549 and HeLa cells undergoing TPT-stimulated autophagy.
Figure 5.

TPT-induced autophagy in A549 and HeLa cells. (A) A549 and HeLa cells were transiently transfected with YFP-LC3 plasmids. Cells were treated with or without 3 μg/mL TPT for 24 h. Green YFP autofluorescence (YFP-LC3) was analyzed by confocal microscopy (100×). (B) A549 and HeLa cells were treated with various concentrations of TPT for 24 h or with 3 μg/mL TPT for the indicated periods. The expression levels of MAP1LC3/LC3 and SQSTM1/p62 protein were analyzed by immunoblotting.
Many studies have demonstrated that PTEN positively regulates autophagy. PTEN is the central negative regulator of the PI3K signal transduction cascade. To determine whether PTEN knockdown affects autophagy in A549 and HeLa cells, PTEN shRNA plasmids were stably transfected into these cells. A549 and HeLa cells were first treated with 3 μg/mL TPT for 24 h, and the expression of MAP1LC3/LC3 and SQSTM1/p62 was analyzed by immunoblotting. As shown in Fig. 6A, MAP1LC3-II/LC3-II expression increased in the TPT-treated groups, but MAP1LC3-II/LC3-II expression was reduced in cells transfected with the PTEN shRNA plasmid. At the same time, the SQSTM1/p62 level decreased due to degradation in the TPT-treated groups, but the effect was partly reversed when cells were transfected with the PTEN shRNA plasmid. Bar graphs represented the relative MAP1LC3-II/LC3-II protein levels normalized to that of GAPDH of different groups.
Figure 6.

Effect of PTEN expression level on TPT-induced autophagy in A549 and HeLa cells. (A) A549 and HeLa cells were stably transfected with PTEN shRNA plasmids. Cells were treated with or without 3 μg/mL TPT for 24 h. The expression levels of PTEN, MAP1LC3/LC3, and SQSTM1/p62 protein were analyzed by immunoblotting. Bar graphs represent the relative MAP1LC3/LC3-II and SQSTM1/p62 protein levels normalized to that of GAPDH of different groups (*P < 0.05, **P < 0.001, t test, n = 3 ). (B) A549 and HeLa cells were transiently transfected with the 3×Flag-PTEN-WT plasmids. Cells were treated with or without 3μg/mL TPT for 24 h. The expression levels of Flag, PTEN, MAP1LC3/LC3, and SQSTM1/p62 protein were analyzed by immunoblotting. Bar graphs represent the relative MAP1LC3/LC3-II and SQSTM1/p62 protein levels normalized to that of GAPDH of different groups. (*P < 0.05, **P < 0.001, t test, n = 3 ). (C) The effect of PTENS113 phosphorylation on autophagy in A549 and HeLa cells. The expression levels of MAP1LC3/LC3 protein were analyzed by immunoblotting. (D) HeLa cells were transiently transfected with the 3×Flag-PTEN-WT or 3×Flag-PTENS113A plasmid and YFP-LC3 plasmids. Twenty four h after transfection, the cells were treated with or without 3 μg/mL TPT. Green YFP autofluorescence (YFP-LC3) was analyzed by confocal microscopy (100×). Bar graphs represent the percentage of YFP- LC3-positive cells in control or TPT-treated group (*P < 0.05, **P < 0.001, t test, n = 3 , bars represent SEM, Cells containing more than 5 foci were scored as positive). (E) A549 and HeLa cells were treated with or without 3 μg/mL TPT for 24 h, followed by treatment with 100 μM bafilomycin A1 (Baf.A1) as indicated. The expression levels of MAP1LC3/LC3 and SQSTM1/p62 protein were analyzed by immunoblotting.
Next, we sought to determine whether PTEN overexpression regulates autophagy. A549 and HeLa cells were transiently transfected with the 3×Flag-PTEN-WT plasmids for 24 h and treated with 3 μg/mL TPT for an additional 24 h. As shown in Fig. 6B, Flag and PTEN overexpression was detected in the 3×Flag-PTEN-WT group, and MAP1LC3-II/LC3-II expression was significantly increased in all TPT-treated groups. A relatively high level of MAP1LC3-II/LC3-II was detected in the 3×Flag-PTEN-WT + TPT group in both A549 and HeLa cells. SQSTM1/p62 protein degradation was detected in all TPT-treated groups, and a relatively low level of SQSTM1/p62 was detected in the 3×Flag-PTEN-WT + TPT group in both A549 and HeLa cells. Our results demonstrate that PTEN regulates TPT-induced autophagy in A549 and HeLa cells. The same results were obtained using A549 cells with PTEN shRNA-mediated knockdown. As shown in Fig. S5A, increased MAP1LC3-II/LC3-II expression and SQSTM1/p62 degradation were observed in cells with 3×Flag-PTEN-WT (shRNA-resistant) after TPT treatment. Bar figures represented the relative MAP1LC3-II/LC3-II protein levels normalized to that of GAPDH of different groups.
Because PTEN (Ser113) phosphorylation could affect PTEN translocation, we further assessed the relationship between PTEN Ser113 phosphorylation and TPT-induced autophagy in A549 and HeLa cells. A549 and HeLa cells were transiently transfected with vector and the 3×Flag-PTEN-WT and 3×Flag-PTENS113A plasmids and treated with TPT for an additional 24 h. The conversion of MAP1LC3-I/LC3-I to MAP1LC3-II/LC3-II was detected in all TPT-treated groups. MAP1LC3-II/LC3-II expression was increased in the cells with 3×Flag-PTEN-WT compared with the cells with 3×Flag-PTENS113A after TPT treatment. SQSTM1/p62 protein degradation was detected in all TPT-treated groups, and a relatively low level of SQSTM1/p62 was detected in the 3×Flag-PTEN-WT + TPT group in both A549 and HeLa cells (Fig. 6C). The same results were obtained using A549 cells with PTEN shRNA-mediated knockdown. As shown in Fig. S5B, increased MAP1LC3-II/LC3-II expression and SQSTM1/p62 degradation were observed in cells with 3×Flag-PTEN-WT (shRNA-resistant) compared with those in the cells expressing 3×Flag-PTENS113A (shRNA-resistant) after TPT treatment.
Immunofluorescence and confocal microscopy were used to assess the relationship between PTEN Ser113 phosphorylation and TPT-induced autophagy in HeLa cells. YFP-LC3 plasmids were transiently transfected into HeLa cells. As shown in Fig. 6D, YFP-LC3 staining was notably increased in the cells with the 3×Flag- PTENS113A plasmid compared with those in cells with 3×Flag-PTENS113A plasmid following TPT treatment. Bar graphs represented the percentage of YFP-LC3-positive HeLa cells of different groups.
Moreover, in A549 and HeLa cells transfected with the 3×Flag-PTEN-WT plasmid, MAP1LC3-II/LC3-II accumulation was markedly increased in the presence of bafilomycin A1 (Baf.A1), a lysosomal protease inhibitor, confirming that TPT activates autophagic flux.32 By contrast, MAP1LC3-II/LC3-II formation was slightly decreased in A549 and HeLa cells transfected with the 3×Flag-PTENS113A plasmid, SQSTM1/p62 protein degradation was detected in all TPT-treated groups, and a relatively low level of SQSTM1/p62 was detected in the 3×Flag-PTEN-WT + TPT group in both A549 and HeLa cells (Fig. 6E). The same results were obtained using A549 cells with PTEN shRNA-mediated knockdown. As shown in Fig. S5C, increased MAP1LC3-II/LC3-II expression and SQSTM1/p62 degradation were observed in cells with 3×Flag-PTEN-WT (shRNA-resistant) compared with those in the cells expressing 3×Flag-PTENS113A (shRNA-resistant) after TPT treatment. MAP1LC3-II/LC3-II accumulation was markedly increased in the presence of Baf.A1 in all groups. The results indicated that PTEN phosphorylation at Ser113 is required for autophagy.
Next, we want to determine how PTEN phosphorylation and nuclear translocation contribute to autophagic induction. JUN-AMPK is considered as a “stress-activated protein kinase” pathway and plays an essential role in autophagy. We have previously demonstrated that MAPK8/JNK1-MAPK9/JNK2 induces the transcriptional activation of SESN2 (sestrin2), a novel player in autophagy induction given its role in AMPK (protein kinase, AMP-activated) activation, via phosphorylation of JUN (jun proto-oncogene).33 Therefore, we assessed the phosphorylated proteins (JUN, AMPK, RPS6KB/p70S6K) and SESN2 levels in the cells with vector or 3×Flag-PTEN-WT or 3×Flag-PTENS113A after TPT treatment. Increased p-JUN-SESN2-p-AMPK and reduced p-RPS6KB expression were observed in the cells with 3×Flag-PTEN-WT compared with the cells with 3×Flag-PTENS113A following TPT treatment (Fig. 7A). The same results were obtained using A549 cells with PTEN shRNA-mediated knockdown (Fig. S6A). JUN or AMPK siRNA respectively inhibited JUN or AMPK activation. A549 cells and HeLa cells with reduced JUN or AMPK expression due to siRNA treatment were exposed to 3 μg/mL TPT for 24 h. As shown in Fig. 7B, the p-JUN, p-AMPK and SESN2 levels were significantly enhanced and p-RPS6KB reduced in the TPT treatment group. However, knockdown of JUN by siRNA significantly reduced the p-JUN, p-AMPK and SESN2 protein levels and p-RPS6KB increased. AMPK activation plays a key role in autophagic induction. As shown in Fig. 7C, the p-AMPK levels were significantly enhanced and p-RPS6KB reduced in the TPT treatment group. Then, knockdown of AMPK markedly decreased the p-AMPK protein levels and p-RPS6KB increased again. At the same time, Flag and PTEN overexpression was detected in the groups, and MAP1LC3-II/LC3-II expression was significantly increased in all TPT-treated groups. A relatively high level of MAP1LC3-II/LC3-II was detected in the 3×Flag-PTEN-WT + TPT group in both A549 and HeLa cells (Figs. 7B and C). However, there was no difference in phospho-MAPK(8/9)/JNK(1/2) levels in the cells with 3×Flag-PTEN-WT or 3×Flag-PTENS113A following TPT treatment (data not shown). The same results were obtained using A549 cells with PTEN shRNA-mediated knockdown (Fig. S6B and C). Collectively, these results suggest that PTEN nuclear translocation promotes autophagy via a mechanism associated with p-JUN-SESN2-AMPK pathway activation in A549 and HeLa cells.
Figure 7.

PTEN promoted TPT-induced autophagy via activation of the p-JUN-SESN2-AMPK pathway in A549 and HeLa cells. (A) The cells were treated with or without 3 μg/mL TPT, and the expression levels of JUN, p-JUN, SESN2, AMPK, p-AMPK, RPS6KB, and p-RPS6KB protein were analyzed by immunoblotting. (B) The cells were treated with or without 3 μg/mL TPT and/or JUN siRNA for 24 h, and the expression levels of JUN, p-JUN, SESN2, AMPK, p-AMPK, RPS6KB, p-RPS6KB, and LC3 protein were analyzed by immunoblotting. Bar graphs represent the relative MAP1LC3/LC3-II protein levels normalized to that of GAPDH of different groups. (*P < 0.05, **P < 0.001, t test, n = 3 ). (C) The cells were treated with or without 3 μg/mL TPT and/or AMPK siRNA for 24 h, and the expression levels of p-AMPK, AMPK, RPS6KB, p-RPS6KB, and MAP1LC3/LC3 protein were analyzed by immunoblotting. Bar graphs represent the relative MAP1LC3/LC3-II protein levels normalized to that of GAPDH of different groups. (*P <0.05, **P < 0.001, t test, n = 3 ). (D) The cells were treated with or without 3 μg/mL TPT, and the expression levels of p-GSK3B, GSK3B, p-AKT, and AKT protein were analyzed by immunoblotting.
Moreover, many studies have demonstrated that PTEN is a known inhibitor of AKT, which negatively regulates autophagy. We sought to determine whether PTEN phosphorylation affected the AKT pathway. A549 cells were transiently transfected with vector and the 3×Flag-PTEN-WT and 3×Flag-PTENS113A plasmids and treated with TPT for an additional 24 h. As shown in Fig. 7D, p-GSK3B and p-AKT levels were not different in the cells with vector or 3×Flag-PTEN-WT or 3×Flag-PTENS113A following TPT treatment. The results suggested that PTEN phosphorylation has no effect on the AKT pathway. PTEN inactivation results in the constitutive activation of the PI3K-AKT pathway and a subsequent increase in protein synthesis, cell cycle progression, migration, and survival.4,5
Discussion
This study demonstrates for the first time that ATM activation by TPT directly phosphorylates PTEN at Ser113 to induce PTEN nuclear translocation. PTEN nuclear translocation triggers autophagy induction through activation of the p-JUN-SESN2-AMPK pathway in response to TPT treatment.
ATM, a phospholipid acyl enzyme-3-kinase that directly responds to DNA damage, is mutated in the disease ataxia telangiectasia. ATM is activated in response to DNA damage, particularly DNA double-strand breaks (DSBs).34 ATM is involved in various cell signaling events, including the acetylation, phosphorylation, and activation of proteins related to cell cycle and DNA damage signaling. In this study, we found that ATM kinase was activated by TPT or CDDP treatment in a concentration- and time-dependent manner. Also, we found that endogenous and exogenous PTEN translocates to the nucleus after TPT or CDDP treatment, and PTEN translocation is clearly suppressed when ATM activity is inhibited by the ATM specific inhibitor KU55933 or ATM siRNA. These results indicate that DNA damaging agents-induced PTEN translocation is dependent of ATM kinase activation.
In this study, we first identified a possible ATM phosphorylation site at PTEN serine 113 (Ser113) using Scansite software. Next, we generated a phospho-specific antibody against PTEN Ser113. Coimmunoprecipitation and λ-phosphatase were employed to evaluate the specificity of the phospho-PTEN Ser113 antibody. In addition, we found that p-PTEN (Ser113) levels increased in a dose-dependent manner in A549 cells and HeLa cells after TPT or CDDP exposure. When ATM expression was reduced by ATM siRNA, TPT-induced PTEN (Ser113) phosphorylation was inhibited. To illustrate the role of PTEN (Ser113) phosphorylation in PTEN nuclear translocation, we transfected the mutated 3×Flag-PTENS113A plasmid and the wild-type 3×Flag-PTEN-WT plasmid into HeLa cells. After treatment with TPT, we found that PTEN did not translocate to the nucleus in the cells harboring the 3×Flag-PTENS113A plasmids. Furthermore,the in vitro phosphorylation assay with purified proteins demonstrated that PTEN was directly phosphorylated by ATM.
PTEN activity, localization and stability are regulated by acetylation, ubiquitination, oxidation, and phosphorylation. CSNK2/CK2 (casein kinase 2) primarily phosphorylates PTEN at Ser370 and Ser385, and GSK3B (glycogen synthase kinase 3 β) phosphorylates PTEN at Ser362 and Thr366.35,36 GLTSCR2/PICT-1 (glioma tumor suppressor candidate region 2) promotes PTEN phosphorylation at Ser380.37 ROCK (RhoA-associated, coiled-coil containing kinase) phosphorylates PTEN at Ser229, Thr232, Thr319 and Thr321 in the C2 domain to activate and target PTEN to the plasma membrane.38 Phospho-PTEN typically refers to PTEN characterized by cytoplasmic localization, enhanced stability, lower activity, and a closed structure. Our findings indicate that Ser113 is a new PTEN phosphorylation site that promotes PTEN nuclear translocation. SUMOylation is also a post-translational modification involved in various cellular processes. Bassi C et al. report that SUMOylation of PTEN controlled its nuclear localization. They indicate that unlike wild-type SUMO-PTEN, which can rapidly be excluded from the nucleus, the SUMO-PTENS/T398A mutant cannot exit the nucleus in cells exposed to ionizing radiation.25 In our study, PTEN-WT and PTENS113A were SUMOylated upon treatment with TPT. However, when we treated cells with nuclear-export inhibitor Leptomycin B, our results indicated that the PTENS113A mutant could enter the nucleus but could not be retained there. It suggested that ATM-mediated PTEN (Ser113) phosphorylation was not involved in PTEN SUMOylation. Our findings indicate that Ser113 is a new PTEN phosphorylation site that promotes PTEN nuclear translocation.
As a tumor suppressor, nuclear PTEN may also maintain normal chromosomal structure and function. Based on the nuclear functions of PTEN, we speculate that ATM-mediated PTEN nuclear translocation affects DNA damage signaling. Using γ-H2AFX staining as an indicator of DSBs, we found that TPT-induced DNA damage signaling is perturbed in A549 cells and HeLa cells. Moreover, inhibition of PTEN nuclear translocation increases the number of cells with γ-H2AFX foci staining. Our findings reveal that PTEN nuclear translocation, which is regulated by phosphorylation of PTEN at Ser113, is required to repair DNA damage. This finding is consistent with previous studies. Moreover, DNA break-repair is affected by the cell cycle stage. TPT-stimulated apoptosis has been reported in S-phase A549 cells.28 In this study, we found that PTENS113A mutation had no effect on cell cycle stage and cell apoptosis.
Autophagy is characterized by the sequestration and delivery of bulk cytoplasm and organelles in double or multimembrane autophagic vesicles for subsequent degradation by the cell's own lysosomal system.39 During the formation of autophagosomes, MAP1LC3/LC3 is lipidated. The subsequent LC3-phospholipid conjugate (MAP1LC3-II/LC3-II) is localized to autophagosomes and autolysosomes, and MAP1LC3-II/LC3-II accumulation is used as a marker of autophagy.29 Furthermore, after PTEN nuclear translocation, levels of cytoplasmic PTEN are hypothesized to decrease, leading to PI3K-AKT pathway activation and sequential inhibition of autophagic induction. In contrast, we found that autophagic induction significantly increased in the cells harboring the 3×Flag-PTEN-WT plasmid compared with the 3×Flag-PTENS113A plasmid. This indicated that PTEN nuclear translocation promotes autophagic induction following TPT treatment.
How does PTEN nuclear translocation contribute to autophagic induction? SESN2 is a TP53 target gene that activates the AMPK pathway to induce autophagy. However, TP53 is mutated and nonfunctional in A549 and HeLa cells; thus, TP53 is not involved in the regulation of SESN2 expression in these 2 cell lines. We have previously demonstrated that activated JUN activates SESN2 expression at the transcriptional level through binding to the promoter of SESN2, potentially activating AMPK to induce autophagy through inhibition of MTORC1 (mechanistic target of rapamycin complex 1) in colon cancer cells.31 Here, we found that SESN2 and p-AMPK expression increased and p-RPS6KB expression reduced in the cells with 3×Flag-PTEN-WT following TPT treatment. JUN is a member of the Jun family that consists of JUN, JUNB, and JUND. JUN is a component of the transcription factor activator protein-1 (AP-1).40 In addition, Dunn et al. report that JUN is a principal target of MAPK(8/9)/JNK(1/2). In this study, p-JUN (Ser63), p-AMPK and SESN2 protein levels were relatively increased and p-RPS6KB reduced in the cells harboring 3×Flag-PTEN-WT compared with the cells harboring 3×Flag-PTENS113A after TPT treatment. A relatively high level of MAP1LC3-II/LC3-II was detected in the 3×Flag-PTEN-WT + TPT group in both A549 and HeLa cells. However, p-MAPK(8/9)/JNK(1/2) expression did not vary between the 3×Flag-PTEN-WT + TPT and 3×Flag-PTENS113A + TPT groups (data not show). Therefore, we speculate that PTEN nuclear translocation regulates JUN phosphorylation and stability. The level of JUN phosphorylation at the C-terminal sites Thr239 and Ser243 has been reported to participate in its downregulation.41 Thus, whether nuclear PTEN affects JUN (Ser243) phosphorylation via its phosphatase activity is of interest.
In conclusion, the results presented here provide evidence that ATM regulates PTEN phosphorylation at Ser113, as a result of exposure to DNA-damaging agents, and promotes PTEN nuclear translocation, which further promotes autophagy through activation of the p-JUN-SESN2-AMPK pathway in tumor cells. We conclude that autophagy requires PTEN nuclear translocation that is regulated by PTEN (Ser113) phosphorylation in response to DNA-damaging agent treatment in cancer cells.
Materials and Methods
Cell culture
The human cervical cancer (HeLa) and lung adenocarcinoma (A549) cell lines were obtained from American Type Culture Collection (CCL-2 and CRM-CCL-185, respectively) and have been stored in liquid nitrogen at our lab. Both cell lines were cultivated in DMEM and RMPI 1640 medium, respectively, supplemented with 10% fetal bovine serum, penicillin (100 μg/mL) and streptomycin (100 μg/mL) in a 5% CO2 humidified atmosphere at 37°C. All experiments were performed in these cells between passage 10 and 20 at the confluency of 70%.
Drugs and reagents
Topotecan (TPT; 004) was purchased from GlaxoSmithKline. KU55933 (587871–26–9) was purchased from Millipore. RPMI-1640 medium (31800–022), DMEM medium (12800–017), fetal bovine serum, the anti-flag antibody, anti-TUBA4A (t6199) and ATP (A1582) were obtained from Sigma Aldrich. Anti-SQSTM1/p62 (8025), phospho-H2AFX (γ-H2AFX; 9718), anti-PTEN (9559), anti-ATM (2873), phospho-ATM (5883), anti-HIST1H3A/histone H3 (9715), anti-RPS6KB/p70S6K (2708), phospho-RPS6KB/p70S6K (9234) and anti-normal mouse IgG (7076) were purchased from Cell Signaling Technology. The anti-MAP1LC3/LC3 antibody (NB100–2220) was obtained from Novus Biologicals. Anti-phospho-PTEN antibody was prepared by the Abgent, Inc.. Lipofectamine 2000 (11668–027), goat anti-rabbit Alexa 488 (997773) and opti-MEM medium (22600050) were purchased from the InvitrogenTM. Purified ATM protein (active) (14–933) was purchased from Millipore, and purified PTEN recombinant protein (TP710023) was obtained from Origene. ATM siRNA, JUN siRNA and AMPK siRNA were obtained from GenePharma Co, Ltd. The 3×Flag-PTEN-WT plasmid and the 3×Flag-PTENS113A plasmid were prepared by Fulen Gene Company. Leptomycin B was purchased from Beyotime Institute of Biotechnology (S1726).
Immunoblotting analysis
A549 and HeLa cells were harvested and lysed in 100 μL of lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton X-100 (Sigma-Aldrich, 234729), 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/mL leupeptin) after treatment with TPT for the indicated times. Lysates were centrifuged at 13,400 g for 15 min at 4°C. The supernatant fraction was then collected. Protein concentrations were determined using the Pierce BCA protein assay kit (Thermo, 1862634). Whole cell lysates were prepared by adding 6×SDS-PAGE buffer (Serva, 151–21–3) and incubation at 100°C for 10 min for protein denaturation. Equivalent aliquots of protein samples (20 to 40 μg) were loaded and electrophoresed on SDS-PAGE gels and then transferred to polyvinylidene fluoride/PVDF membranes. The membranes were first blocked with 5% nonfat dry milk for 60 min at room temperature and then incubated with primary antibodies overnight at 4°C. Next, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology, sc-2004 and sc-2005) for 1 h at room temperature. After 3 washes with TBST (10 mM Tris, 150 mM NaCl, pH 8.0, 0.1% Tween 20 [MP Biomedicals, 103168]) for 5 min, the proteins were visualized with the ECL chemiluminescence reagent and exposure to radiograph film.
Immunofluorescence
A549 and HeLa cells were seeded in 24-well plates containing NUNC Thermanox™ coverslips. Upon treatment, cells were fixed with a 4% paraformaldehyde fixative solution at room temperature for 20 min and washed 3 times in 1×PBS (0.8% NaCl, 0.02% KCl, 0.144% Na2HPO4, 0.024% KH2PO4, pH7.4) for 5 min each. Cells were then permeabilized with 0.25% Octoxynol 9/Triton X-100 (Sigma-Aldrich, 9002–93–1) in PBS at 4°C for 10 min and blocked with 4% BSA (MP Biomedicals, 0218054991) blocking solution at 37°C for 30 min. The cells were incubated with the primary antibody (1:300) overnight at 4°C. Cells were washed thrice in PBST for 5 min and incubated with the fluorescent secondary antibody at room temperature for 1 h. DAPI was added for 5 min to visualize nuclei. After using an antifluorescence quenching reagent, the coverslips were stored at 4°C. Cells were observed using a laser scanning confocal microscope (Olympus, FV-1000).
shRNA and siRNA transfection
The target sequence for ATM or JUN or AMPK siRNA was respectively 5′-AAGGTGAA GATATATTCCTCC-3′,5′-UGCCUACCAUCUCAUAAUAtt-3′, 5′-AGAUGG AAACGACCUUCUATT-3′. The hairpin RNA (shRNA) sequence targeting PTEN was 5′-GATCTTGACCAATGGCTAAGT-3′. The control siRNA sequence was 5′-UUC UCC GAA CGU GUC ACG UTT-3′ and synthesized by GenChem Co. HeLa and A549 cells were seeded into 6-well plates the day before transfection. When the cells reached 40% to 60% confluence, transfection was performed using serum-free Opti-MEM media (Gibco, 31985–070), lipofectamine 2000 (Invitrogen, 11668–019) and siRNA according to the manufacturer's recommendations. The medium was replaced with fresh growth medium containing 10% fetal bovine serum 6 h after transfection, and cells were incubated for an additional 24 h for subsequent experiments.
In vitro phosphorylation assay
This assay was carried out using commercially available active ATM kinase (1 μg) and PTEN recombinant protein (1 μg). Reaction was initiated by adding ATP and carried out at 30 °C in 25 μL kinase buffer [20 mM Tris·HCl (pH 7.5), 2 mM EDTA, 10 mM MgCl2, 1 mM DTT] for 30 min. The reaction was terminated by adding 12.5 μL 3x loading buffer and incubating at 100°C for 10 min.
Cell cycle analysis
Cell cycle analysis was carried out by flow cytometry. Briefly, A549 cells were seeded into 6-well culture plates, treated as indicated, collected, fixed, stained with propidium iodide (1 μg/mL) and flow cytometric analysis on a FACScan with a 488 nm Argon laser (Backman Coulter, FC500). Quantitation was performed by scoring cells for sub-G0/G1 DNA content using the SigmaPlot software.
In vitro SUMOylation
HeLa cells were transiently transfected with 10 μg of the HA-SUMO1 and 10 μg 3×Flag-PTEN-WT or 3×Flag-PTENS113A plasmid. Equal amounts of protein sample were incubated with a flag monoclonal antibody for 2 h at 4°C. The antibody-protein complex was mixed with protein A Sepharose 4 fast flow solution for 16 h at 4°C. After centrifugation, the pellet fraction was washed 4 times and resuspended in loading buffer. The suspension was boiled for 10 min at 100°C, centrifuged and analyzed by SDS–PAGE and western blotting with anti-HA antibody.
Plasmids and transfection
The vector pcDNA3.1 and YFP-LC3 plasmids were kindly gifted by Dr. Rong Deng, Dr. Dandan Li and Dr. Xiaoqi Wu, Cancer Center, Sun Yat-sen University, Guangzhou, China. The 3×Flag-PTEN-WT and 3×Flag-PTENS113A, 3×Flag-PTEN-WT (shPTEN-resistant) and 3×Flag-PTENS113A (shPTEN-resistant) plasmids were produced by the Gene Copoeia Company. HA-SUMO1 plasmid was purchased from Addgene (plasmid 21154). All mutations were verified by DNA sequencing. Transfection was performed as previously described.42,43
Cytoplasmic and nuclear protein extraction
Cytosolic and nuclear fractions were prepared using a nuclear-cytosolic protein isolation kit (Beyotime Institute of Biotechnology). HeLa and A549 cells were seeded in 10 cm plates. After treatment with 3 μg/mL TPT for 24 h, cells were collected and washed with 1 mL of PBS and then centrifuged in 4°C at 90 g for 3 min. The resultant supernatant fraction was discarded, and the sediment was resuspended in reagent A with fresh PMSF. Samples were vortexed vigorously for 5 sec and placed on ice for 15 min before adding reagent B. The samples were centrifuged at 13,400 g for 3 min at 4°C. The supernatant fraction containing the cytosolic proteins was collected and used immediately or stored at −80°C. The sediment containing the nuclear proteins was resuspended in nuclear protein extraction reagent with PMSF. The samples were vortexed at 13,400 g for 30 sec, placed on ice for 2 min, and vortexed for 30 sec. The last 2 steps were repeated for 30 min, and the samples were centrifuged at 13,400 g for 10 min at 4°C. The supernatant fractions containing the nuclear proteins was used immediately or stored at −80°C. The concentrations of the cytosolic and nuclear proteins were measured using the bicinchoninic acid/BCA method (Thermo, 1862634).
Coimmunoprecipitation
A549 and HeLa cells were transiently transfected with the 3×Flag-PTEN-WT plasmid. Cells were lysed with RIPA lysis buffer, and the protein concentration for each group was measured using the bicinchoninic acid method. Equal amounts of protein sample were incubated with a flag monoclonal antibody for 2 h at 4°C. The antibody-protein complex was mixed with Protein A/G Agarose (Roche, 11243233001) for 16 h at 4°C. After centrifugation, the pellet fraction was washed 4 times and resuspended in loading buffer. The suspension was boiled for 10 min at 100°C and centrifuged, and the supernatant fraction was used for western blotting to identify the coprecipitated proteins.
Statistical analysis
All assays were performed in triplicate. Data are expressed as the mean ± standard deviation (SD). Statistical analyses were performed with the 2-tailed Student t test for comparison of 2 groups by SPSS 13.0 software. P < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by grants from the National Basic Research Program of China (2012CB967004; 2011CB504300), the National Natural Science Foundation of China (81272199;81272895) and the Natural Science Foundation of Guangdong (S2011020002759).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997; 275:1943-7; PMID:9072974; http://dx.doi.org/ 10.1126/science.275.5308.1943 [DOI] [PubMed] [Google Scholar]
- 2. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 1998; 273:13375-8; PMID:9593664; http://dx.doi.org/ 10.1074/jbc.273.22.13375 [DOI] [PubMed] [Google Scholar]
- 3. Parsons R. Human cancer, PTEN and the PI-3 kinase pathway. Semin Cell Dev Biol 2004; 15:171-6; PMID:15209376; http://dx.doi.org/ 10.1016/j.semcdb.2003.12.021 [DOI] [PubMed] [Google Scholar]
- 4. Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 2000; 275:992-8; PMID:10625637; http://dx.doi.org/ 10.1074/jbc.275.2.992 [DOI] [PubMed] [Google Scholar]
- 5. Weng L, Brown J, Eng C. PTEN induces apoptosis and cell cycle arrest through phosphoinositol-3-kinase/Akt-dependent and -independent pathways. Hum Mol Genet 2001; 10:237-42; PMID:11159942; http://dx.doi.org/ 10.1093/hmg/10.3.237 [DOI] [PubMed] [Google Scholar]
- 6. Whang YE, Wu X, Suzuki H, Reiter RE, Tran C, Vessella RL, Said JW, Isaacs WB, Sawyers CL. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci U S A 1998; 95:5246-50; PMID:9560261; http://dx.doi.org/ 10.1073/pnas.95.9.5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perren A, Weng LP, Boag AH, Ziebold U, Thakore K, Dahia PL, Komminoth P, Lees JA, Mulligan LM, Mutter GL, et al. Immunohistochemical evidence of loss of PTEN expression in primary ductal adenocarcinomas of the breast. Am J Pathol 1999; 155:1253-60; PMID:10514407; http://dx.doi.org/ 10.1016/S0002-9440(10)65227-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gimm O, Attie-Bitach T, Lees JA, Vekemans M, Eng C. Expression of the PTEN tumour suppressor protein during human development. Hum Mol Genet 2000; 9:1633-9; PMID:10861290; http://dx.doi.org/ 10.1093/hmg/9.11.1633 [DOI] [PubMed] [Google Scholar]
- 9. Gimm O, Perren A, Weng LP, Marsh DJ, Yeh JJ, Ziebold U, Gil E, Hinze R, Delbridge L, Lees JA, et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am J Pathol 2000; 156:1693-700; PMID:10793080; http://dx.doi.org/ 10.1016/S0002-9440(10)65040-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Perren A, Komminoth P, Saremaslani P, Matter C, Feurer S, Lees JA, Heitz PU, Eng C. Mutation and expression analyses reveal differential subcellular compartmentalization of PTEN in endocrine pancreatic tumors compared to normal islet cells. Am J Pathol 2000; 157:1097-103; PMID:11021813; http://dx.doi.org/ 10.1016/S0002-9440(10)64624-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Whiteman DC, Zhou XP, Cummings MC, Pavey S, Hayward NK, Eng C. Nuclear PTEN expression and clinicopathologic features in a population-based series of primary cutaneous melanoma. Int J Cancer 2002; 99:63-7; PMID:11948493; http://dx.doi.org/ 10.1002/ijc.10294 [DOI] [PubMed] [Google Scholar]
- 12. Lobo GP, Waite KA, Planchon SM, Romigh T, Nassif NT, Eng C. Germline and somatic cancer-associated mutations in the ATP-binding motifs of PTEN influence its subcellular localization and tumor suppressive function. Hum Mol Genet 2009; 18:2851-62; PMID:19457929; http://dx.doi.org/ 10.1093/hmg/ddp220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu F, Wagner S, Campbell RB, Nickerson JA, Schiffer CA, Ross AH. PTEN enters the nucleus by diffusion. J Cell Biochem 2005; 96:221-34; PMID:16088943; http://dx.doi.org/ 10.1002/jcb.20525 [DOI] [PubMed] [Google Scholar]
- 14. Gil A, Andres-Pons A, Fernandez E, Valiente M, Torres J, Cervera J, Pulido R. Nuclear localization of PTEN by a Ran-dependent mechanism enhances apoptosis: Involvement of an N-terminal nuclear localization domain and multiple nuclear exclusion motifs. Mol Biol Cell 2006; 17:4002-13; PMID:16807353; http://dx.doi.org/ 10.1091/mbc.E06-05-0380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chung JH, Ginn-Pease ME, Eng C. Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) has nuclear localization signal-like sequences for nuclear import mediated by major vault protein. Cancer Res 2005; 65:4108-16; PMID:15899801; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-0124 [DOI] [PubMed] [Google Scholar]
- 16. Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 2007; 128:141-56; PMID:17218261; http://dx.doi.org/ 10.1016/j.cell.2006.11.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 2007; 128:129-39; PMID:17218260; http://dx.doi.org/ 10.1016/j.cell.2006.11.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ortega-Molina A, Serrano M. PTEN in cancer, metabolism, and aging. Trends Endocrinol Metab 2013; 24:184-9; PMID:23245767; http://dx.doi.org/ 10.1016/j.tem.2012.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chung JH, Ostrowski MC, Romigh T, Minaguchi T, Waite KA, Eng C. The ERK1/2 pathway modulates nuclear PTEN-mediated cell cycle arrest by cyclin D1 transcriptional regulation. Hum Mol Genet 2006; 15:2553-9; PMID:16849370; http://dx.doi.org/ 10.1093/hmg/ddl177 [DOI] [PubMed] [Google Scholar]
- 20. Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007; 128:157-70; PMID:17218262; http://dx.doi.org/ 10.1016/j.cell.2006.11.042 [DOI] [PubMed] [Google Scholar]
- 21. Puc J, Parsons R. PTEN loss inhibits CHK1 to cause double stranded-DNA breaks in cells. Cell Cycle 2005; 4:927-9; PMID:15970699; http://dx.doi.org/ 10.4161/cc.4.7.1795 [DOI] [PubMed] [Google Scholar]
- 22. Li AG, Piluso LG, Cai X, Wei G, Sellers WR, Liu X. Mechanistic insights into maintenance of high p53 acetylation by PTEN. Mol Cell 2006; 23:575-87; PMID:16916644; http://dx.doi.org/ 10.1016/j.molcel.2006.06.028 [DOI] [PubMed] [Google Scholar]
- 23. Chung JH, Eng C. Nuclear-cytoplasmic partitioning of phosphatase and tensin homologue deleted on hromosome 10 (PTEN) differentially regulates the cell cycle and apoptosis. Cancer Res 2005; 65:8096-100; PMID:16166282; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-1888 [DOI] [PubMed] [Google Scholar]
- 24. Chang CJ, Mulholland DJ, Valamehr B, Mosessian S, Sellers WR, Wu H. PTEN nuclear localization is regulated by oxidative stress and mediates p53-dependent tumor suppression. Mol Cell Biol 2008; 28:3281-9; PMID:18332125; http://dx.doi.org/ 10.1128/MCB.00310-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B, Stambolic V. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 2013; 341:395-9; PMID:23888040; http://dx.doi.org/ 10.1126/science.1236188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 2000; 10:886-95; PMID:10959836; http://dx.doi.org/ 10.1016/S0960-9822(00)00610-2 [DOI] [PubMed] [Google Scholar]
- 27. Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 2001; 276:42462-7; PMID:11571274; http://dx.doi.org/ 10.1074/jbc.C100466200 [DOI] [PubMed] [Google Scholar]
- 28. Tanaka T, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants. Cell Cycle 2006; 5:1940-5; PMID:16940754; http://dx.doi.org/ 10.4161/cc.5.17.3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005; 1:84-91; PMID:16874052; http://dx.doi.org/ 10.4161/auto.1.2.1697 [DOI] [PubMed] [Google Scholar]
- 30. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/ 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:17580304; http://dx.doi.org/ 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- 32. Gagliardi S, Rees M, Farina C. Chemistry and structure activity relationships of bafilomycin A1, a potent and selective inhibitor of the vacuolar H+-ATPase. Curr Med Chem 1999; 6:1197-212; PMID:10519916 [PubMed] [Google Scholar]
- 33. Zhang XY, Wu XQ, Deng R, Sun T, Feng GK, Zhu XF. Upregulation of sestrin 2 expression via JNK pathway activation contributes to autophagy induction in cancer cells. Cell Signal 2013; 25:150-8; PMID:22982090; http://dx.doi.org/ 10.1016/j.cellsig.2012.09.004 [DOI] [PubMed] [Google Scholar]
- 34. Tanaka T, Huang X, Halicka HD, Zhao H, Traganos F, Albino AP, Dai W, Darzynkiewicz Z. Cytometry of ATM activation and histone H2AX phosphorylation to estimate extent of DNA damage induced by exogenous agents. Cytometry A 2007; 71:648-61; PMID:17622968; http://dx.doi.org/ 10.1002/cyto.a.20426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem 2001; 276:993-8; PMID:11035045; http://dx.doi.org/ 10.1074/jbc.M009134200 [DOI] [PubMed] [Google Scholar]
- 36. Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T. Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J Biol Chem 2005; 280:35195-202; PMID:16107342; http://dx.doi.org/ 10.1074/jbc.M503045200 [DOI] [PubMed] [Google Scholar]
- 37. Okahara F, Itoh K, Nakagawara A, Murakami M, Kanaho Y, Maehama T. Critical role of PICT-1, a tumor suppressor candidate, in phosphatidylinositol 3,4,5-trisphosphate signals and tumorigenic transformation. Mol Biol Cell 2006; 17:4888-95; PMID:16971513; http://dx.doi.org/ 10.1091/mbc.E06-04-0301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, Tang L, Hla T, Zeng R, Li L, et al. Regulation of PTEN by Rho small GTPases. Nat Cell Biol 2005; 7:399-404; PMID:15793569; http://dx.doi.org/ 10.1038/ncb1236 [DOI] [PubMed] [Google Scholar]
- 39. Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004; 23:2891-906; PMID:15077152; http://dx.doi.org/ 10.1038/sj.onc.1207521 [DOI] [PubMed] [Google Scholar]
- 40. Jochum W, Passegue E, Wagner EF. AP-1 in mouse development and tumorigenesis. Oncogene 2001; 20:2401-12; PMID:11402336; http://dx.doi.org/ 10.1038/sj.onc.1204389 [DOI] [PubMed] [Google Scholar]
- 41. Morton S, Davis RJ, McLaren A, Cohen P. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J 2003; 22:3876-86; PMID:12881422; http://dx.doi.org/ 10.1093/emboj/cdg388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Deng R, Tang J, Ma JG, Chen SP, Xia LP, Zhou WJ, Li DD, Feng GK, Zeng YX, Zhu XF. PKB/Akt promotes DSB repair in cancer cells through upregulating Mre11 expression following ionizing radiation. Oncogene 2011; 30:944-55; PMID:20956948; http://dx.doi.org/ 10.1038/onc.2010.467 [DOI] [PubMed] [Google Scholar]
- 43. Li DD, Wang LL, Deng R, Tang J, Shen Y, Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 2009; 28:886-98; PMID:19060920; http://dx.doi.org/ 10.1038/onc.2008.441 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.