

Abstract
In addition to supporting cell survival in response to starvation or stress, autophagy promotes basal protein and organelle turnover. Compared to our understanding of stress-induced autophagy, little is known about how basal autophagy is regulated and how its activity is coordinated with other cellular processes. We recently identified a novel interaction between the ATG12–ATG3 conjugate and the ESCRT-associated protein PDCD6IP/Alix that promotes basal autophagy and endolysosomal trafficking. Moreover, ATG12–ATG3 is required for diverse PDCD6IP-mediated functions including late endosome distribution, exosome secretion, and viral budding. Our results highlight the importance of late endosomes for basal autophagic flux and reveal distinct roles for the core autophagy proteins ATG12 and ATG3 in controlling late endosome function.
Keywords: ATG3, ATG12, basal autophagy, exosome, late endosome, PDCD6IP/Alix, viral budding
Two ubiquitin-like conjugations systems are required for autophagosome formation. The first pathway attaches the ubiquitin-like molecule ATG12 to ATG5, while the second conjugates MAP1LC3/LC3 with the lipid phosphatidylethanolamine. Our lab previously identified ATG3, the E2-like conjugating enzyme that mediates LC3 lipidation, as an alternative ATG12 conjugation target. Although ATG12 and ATG3 each mediate essential steps in the early autophagy pathway, cells lacking ATG12–ATG3 have normal LC3 lipidation and starvation-induced autophagy. However, in full-nutrient conditions, cells lacking ATG12–ATG3 exhibit increased numbers of autophagosomes, indicative of either increased autophagosome formation or decreased turnover. Hence, we sought to clarify how ATG12–ATG3 regulates autophagic trafficking in basal, nutrient-rich conditions and to dissect the mechanistic basis for this phenotype.
To interrogate the functions of ATG12–ATG3 in basal autophagy, we reconstituted autophagy-deficient atg3−/− fibroblasts with wild-type ATG3 or a lysine to arginine mutant (ATG3KR) that is capable of LC3 lipidation but unable to form the ATG12–ATG3 conjugate. Using the tandem mCherry-GFP-LC3 reporter, we observed no differences in autophagosome maturation between nutrient-starved ATG3 and ATG3KR cells. However, in full-media conditions, ATG3KR cells have significantly reduced autolysosome formation, indicating a specific role for ATG12–ATG3 in promoting basal autophagosome maturation. In addition, ATG3KR cells exhibit increased basal accumulation of the autophagy substrates, SQSTM1/p62 and NBR1, further corroborating the requirement for the ATG12–ATG3 conjugate in basal autophagy. Notably, we previously demonstrated that cells lacking ATG12–ATG3 display increased mitochondrial mass and fragmentation, which arose from defects in mitophagy and mitochondrial fusion. We speculate that these disruptions in mitochondrial homeostasis in ATG3KR cells are at least partly due to defective basal autophagy and mitophagy.
Efficient autophagy depends on both lysosome and endosome function. Autophagosomes can fuse with late endosomes rather than directly with the lysosome; this hybrid compartment then fuses with the lysosome where its contents are degraded. Electron microscopy and immunostaining for endosomal and lysosomal markers revealed that ATG3KR cells accumulate enlarged and perinuclear late endosomes, suggesting that defective basal autophagy in cells lacking ATG12–ATG3 is due to impaired late endosome function. Indeed, we corroborated that ATG3KR cells exhibit impaired endolysosomal trafficking, with a specific block at the late endosome. Moreover, this role for ATG12–ATG3 in endolysosomal trafficking is distinct from the canonical role of either ATG protein in autophagosome formation. To further define how ATG12–ATG3 promotes late endosome function we performed mass spectrometry and identified the ESCRT-accessory protein PDCD6IP as a binding partner of ATG12–ATG3. PDCD6IP associates with lysobisphosphatidic acid at the late endosome membrane and subunits of the ESCRT machinery to promote membrane abscission events such as intralumenal vesicle formation, exosome biogenesis, and viral budding. PDCD6IP also controls the spatial distribution of late endosomes, likely via interactions with the actin-remodeling protein CTTN (cortactin).
We confirmed that PDCD6IP specifically interacts with the ATG12–ATG3 conjugate at endogenous protein levels. Overexpression studies indicated that this interaction is mediated by ATG12, as unconjugated ATG12 but not ATG3 co-immunoprecipitates with PDCD6IP upon exogenous expression of both proteins. PDCD6IP itself contains 3 interaction domains: the N-terminal Bro1 domain binds to ESCRT-III CHMP4 subunits, the V domain recognizes YPXnL motifs found in retroviral Gag proteins and the exosomal cargo SDCBP/syntenin, and the C-terminal proline rich domain (PRD) interacts with the ESCRT-I subunit TSG101. Intramolecular interaction of the PRD with the Bro1 and V domains maintains PDCD6IP in an inhibitory conformation that blocks access to its YPXnL-binding site (Fig. 1A). Whereas CHMP4 family members bind the Bro1 domain independent of PRD displacement, YPXnL-containing proteins such as SDCBP and Gag require PRD displacement for binding to the V domain. Mutational analysis demonstrated that ATG12 interacts with the Bro1 and V domains of PDCD6IP and that both domains are required for binding to PDCD6IP mutants possessing the auto-inhibitory PRD. Notably, while ATG12–ATG3 conjugation does not affect the interaction of CHMP4B with PDCD6IP, co-immunoprecipitation of MLV-Gag with PDCD6IP is significantly enhanced by ATG12–ATG3 conjugation. Together, these results support a model in which ATG12–ATG3 binding to PDCD6IP displaces the PRD from the Bro1 and V domains, thereby promoting an open PDCD6IP conformation and enhancing its binding to YPXnL-containing effectors (Fig. 1B).
Figure 1.
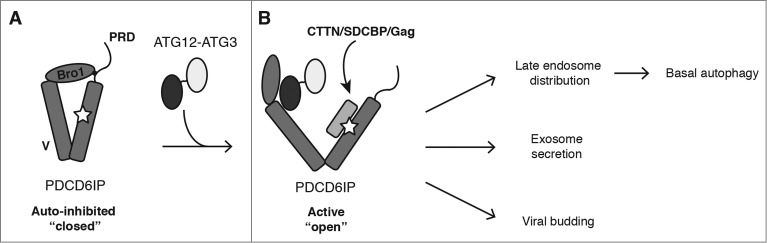
Model for ATG12–ATG3 function in PDCD6IP/Alix activation and basal autophagy. (A) Intramolecular interaction of the PRD with the Bro1 and V domains maintains PDCD6IP in an inhibitory conformation that blocks access to its YPXnL-binding site (star). (B) ATG12–ATG3 binding to PDCD6IP displaces the PRD from the Bro1 and V domains, leading to its open conformation. The accessible V domain enables recruitment of partner proteins, including CTTN (cortactin), SDCBP/syntenin and viral Gag, thereby supporting diverse PDCD6IP functions including late endosome distribution, exosome release, and viral budding. We propose basal autophagy to be dependent on the fusion of autophagosomes with late endosomes. Thus, cells lacking either PDCD6IP or ATG12–ATG3 accumulate abnormal perinuclear late endosomes that are unable to fuse with lysosomes, thereby abrogating autolysosome formation.
Based on this model, we postulated that ATG12–ATG3 promotes PDCD6IP activities requiring PRD displacement for PDCD6IP activation. PDCD6IP interacts with CTTN to control late endosome distribution, with the exosomal scaffold protein SDCBP to promote exosome biogenesis, and with viral Gag proteins to facilitate viral budding. All 3 of these PDCD6IP-mediated processes depend on accessibility of and binding to its V domain (Fig. 1B). Accordingly, cells lacking ATG12–ATG3 phenocopy the principal defects observed upon PDCD6IP depletion, including accumulation of perinuclear late endosomes, and impairments in exosome release and viral budding. Conversely, PDCD6IP depletion reduces basal autophagic flux but does not affect starvation-induced autophagy, similar to ATG12–ATG3 loss. We speculate that basal autophagy is uniquely dependent upon fusion of autophagosomes with late endosomes for efficient autophagic flux. In contrast, because starvation activates transcriptional pathways that lead to coordinated lysosome biogenesis and induction of autophagy genes, starvation-induced autophagy may occur primarily through direct autophagosome-lysosome fusion. Hence, ATG12–ATG3 and PDCD6IP promote basal autophagic flux, yet both are dispensable for starvation-induced autophagy. Our results provide new insight into the differential regulation of basal versus starvation-induced autophagy and how this process is coordinated with other homeostatic pathways, including endosomal trafficking and secretion.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the NIH (CA126792, CA188404) and a Howard Hughes Medical Institute Physician-Scientist Early Career Award to JD and an NSF Graduate Research Fellowship (DGE-1144247) to LM.