

Abstract
Human intestinal microbiota plays a number of important roles in human health and is also implicated in several gastrointestinal disorders. Though the diversity of human gut microbiota in adults and in young children has been examined, few reports of microbiota composition are available for adolescents. In this work we used Microbiota Array for high-throughput analysis of distal gut microbiota in adolescent children 11-18 years of age. Samples obtained from healthy adults were used for comparison. Adolescent and adult groups could be separated in principal components analysis space based on the relative species abundance of their distal gut microbiota. All samples were dominated by class Clostridia. A core microbiome of 46 species that were detected in all examined samples was established; members of genera Ruminococcus, Faecalibacterium, and Roseburia were well presented among core species. Comparison of intestinal microbiota composition between adolescents and adults revealed a statistically significantly higher abundance of genera Bifidobacterium and Clostridium among adolescent samples. The number of detected species was similar between sample groups indicating that it was relative abundances of the genera and not the presence or absence of a specific genus that differentiated adolescent and adult samples. In summary, contrary to the current belief, this study suggests that the gut microbiome of adolescent children is different from that of adults.
Keywords: adolescent, intestinal microbiota, microbial community, microflora, phylogenetic microarray
INTRODUCTION
The human gastrointestinal tract is an ecosystem rich in microbial species, with an estimated 1013-1014 cells present in the large intestine (Ley, et al., 2006). The sheer numbers of microorganisms and the enclosed space of the intestinal tract create intimate relationships between human host and microbes. This intestinal microbial community is an essential component of human health and disease. The intestinal microbiota is responsible for polysaccharide hydrolysis and fermentation, vitamin production, immune system stimulation, modulation of gut motility, as well as for protection of human host from pathogen invasion (Sekirov, et al., 2010). At the same time, changes in intestinal microbiota are associated with a variety of GI disorders such as irritable bowel syndrome, inflammatory bowel disease, obesity, and gastrointestinal cancer (Turnbaugh, et al., 2006, Frank, et al., 2007, Swidsinski, et al., 2008, Davis & Milner, 2009, Fujimura, et al., 2010).
Though the original studies of intestinal microbial communities were limited by the difficulties of utilizing traditional microbiological techniques for the analysis of this complex community, new methods based on the interrogation of total community DNA and RNA provide means to analyze and compare the presence and abundance of hundreds of microbial species in a single sample (Langendijk, et al., 1995, Suau, et al., 1999, Wang, et al., 2004, Ley, et al., 2005, Palmer, et al., 2006, Biasucci, et al., 2010). A number of projects performed in the last several years focused on sampling the diversity of human microbiota and providing the first large-scale comparative analyses (Eckburg, et al., 2005, Ley, et al., 2005, Bik, et al., 2006, Gill, et al., 2006, Frank, et al., 2007). While several different methods have been utilized for this purpose (Sekirov, et al., 2010), next generation sequencing (NGS) and oligo microarray interrogation of the 16S rRNA gene pool are the primary current approaches that enable high-throughput detection and enumeration of community members. While NGS advantages include new sequence discovery and ability to interrogate any microbial community independent of its source, advantages of phylogenetic oligo microarrays comprise the ability to conduct direct comparisons of each probeset signal among samples making data fully quantitative, ability to interrogate single samples independently, and as of now still lower costs especially when comparing total sample signal recorded by microarray versus total number of reads in NGS per sample. In recent years, several custom microarrays have been designed and successfully utilized for the characterization of complex human-associated microbial communities including PhyloChip and HITChip (Wang, et al., 2004, Brodie, et al., 2006, Palmer, et al., 2006, Rajilic-Stojanovic, et al., 2009), and the Microbiota Array developed in our laboratory (Paliy, et al., 2009).
While the intestine in a newborn contains no microbes (it is essentially sterile), immediately after birth it becomes colonized by enterobacteria, enterococci, lactobacilli, and bifidobacteria (Hopkins, et al., 2005, Bjorkstrom, et al., 2009, Enck, et al., 2009). The initial microbial gut colonization is dependent upon the mode of infant delivery. While traditional delivery sees the early gut microbiota resembling that of the vaginal canal, infants born by cesarean delivery are exposed to a different bacterial milieu closely related to that of the human skin (Biasucci, et al., 2008, Biasucci, et al., 2010, Dominguez-Bello, et al., 2010). Later, the establishment of microbiota population is dependent on whether the infant is breast or formula fed (Harmsen, et al., 2000). The gut biota of breast milk-fed babies is usually dominated by Bifidobacterium and Lactobacillus, whereas the microbiota of formula-fed babies is more complex and is more similar to that of an adult with increased counts of Clostridia (Hopkins, et al., 2005, Penders, et al., 2005). Gradual changes in microbiota composition occur during early childhood with general reduction in the number of aerobes and facultative anaerobes and an increase in the populations of obligate anaerobic species (Hopkins, et al., 2005). Traditionally, it has been thought that between one and two years of age the human gut microbiota start to resemble that of an adult (Palmer, et al., 2007), which is dominated by species from phyla Firmicutes (predominantly class Clostridia; 50-70% total bacterial numbers), Bacteroidetes (10-30%), Proteobacteria (up to 10%), and Actinobacteria (up to 10%); with 90% believed to be obligate anaerobes (Eckburg, et al., 2005, Lay, et al., 2005, Ley, et al., 2006). Though the intestinal microbiota has been catalogued extensively in adults, and many recent reports were published for children between 0 and 2 years of age (Hopkins, et al., 2005, Palmer, et al., 2007, Bjorkstrom, et al., 2009), few studies were conducted with gut microbiota of older children (Hopkins, et al., 2001, Chernukha, et al., 2005, Enck, et al., 2009, Paliy, et al., 2009). When Hopkins and colleagues analyzed fecal samples of young children between 1-7 years of age (Hopkins, et al., 2001), a less diverse microbiota was evident in child fecal samples, and the numbers of enterobacteria were higher than those in adults. A large scale study by Enck et al. used conventional colony plating to assess numbers of several bacterial genera in children between 0 and 18 years of age (Enck, et al., 2009). Although there were significant shifts in genus abundances during the first 2 years of life, no noticeable changes were evident in children between 2-18 years old including stable levels of Bifidobacterium and Lactobacillus (see (Harmsen, et al., 2000) for a description of limitations of colony plating estimates). A pilot study carried out in our laboratory revealed that adult and pre-adolescent child samples could be easily separated by principal components analysis indicating that gut communities were different between adults and older children (Paliy, et al., 2009).
Based on the results of these initial experiments, we hypothesized that older children might harbor different intestinal microbial communities than what has been observed either in adults or younger children. To that end we utilized Microbiota Array to quantitatively compare fecal microbiota communities of healthy children of pre- and adolescent age group to that of healthy adults.
MATERIALS AND METHODS
Fecal sample collection and processing
Healthy children of pre- and adolescent age (n=22; age range: 11-18 years, average - 12.6 years; BMI – 19.4 ± 3.7; 10 males, 12 females) and adults (n=10, age range: 22-61 years, average – 34.3 years; BMI – 22.0 ± 2.7; 4 males, 6 females) were recruited at Dayton Children’s Medical Center and Wright State University. All volunteers consumed standard western diet. The study was approved by Wright State University IRB committee. All volunteers had no gastrointestinal symptoms and did not consume antibiotics or probiotics for three months prior to sample collection. Fresh fecal samples were collected, immediately homogenized, and frozen as described (Paliy, et al., 2009, Rigsbee, et al., 2011).
Isolation of genomic DNA and hybridization to Microbiota Array
Total genomic DNA (gDNA) was isolated from 150 mg of fecal samples using ZR Fecal DNA Isolation kit (Zymo Research Corporation) according to manufacturer’s protocol. Genomic DNA was amplified with phylogenetically conserved primers targeting full length 16S rRNA gene as described (Paliy, et al., 2009, Rigsbee, et al., 2011). PCR reaction conditions were 250 ng starting DNA, 25 cycles of PCR amplification, total reaction volume – 50 μl. At least 4 separate PCR reactions were pooled together, fragmented, and then hybridized to Microbiota Array. The pooling of 4 separate PCR reactions was sufficient to eliminate PCR drift (Rigsbee, et al., 2011). Microarray hybridization, washing, and scanning were carried out as described previously (Paliy, et al., 2009, Rigsbee, et al., 2011). Two replicate arrays were run for each fecal sample, each utilizing independently amplified 16S rDNA pools. The replicate arrays produced highly correlated signal values for most probesets – an average Pearson correlation between replicates was 0.94.
Microarray data analysis
Raw microarray data was analyzed through a previously developed analysis pipeline as described (Rigsbee, et al., 2011). Data adjustments and signal summations were carried out in Microsoft Excel template file that accounted for signal intensity, probeset presence call, cross-hybridization reduction, and 16S rRNA gene copy number (Rigsbee, et al., 2011). To obtain average relative abundances of phylogenetic groups among all samples within each sample type, weighted mean around median was calculated in order to reduce the effect of outliers on the mean estimate. The weighted mean was calculated as (Wilcox, 2003)
where θ̂ is the weighted mean value for a given phylogenetic group, xi is the relative abundance of phylogenetic group in sample i, and wi is the corresponding weight. Weights were calculated generally following bi-weight approach of robust statistics as
where m is the median of distribution and τ̂ is the median absolute deviation τ̂ = median{|xi−m|}. This bi-weight calculation insured that values close to the median had weights of 1, and the value contribution to the weighted mean was reduced as the distance from the median increased. No weight was lower than 0.5.
Standard error of the weighted mean (SEθ̂) was calculated as (Wilcox, 2003)
Statistical tests of significance of observed differences were carried out in SPSS v16 (SPSS, Inc.) and in CARMAweb (Rainer, et al., 2006) using moderated T test and rank correlations. Non-parametric rank tests were used for correlation analyses because the datasets were not found to be normally distributed. Principal components analysis was performed in Matlab (The Mathworks, Inc.).
Phylogenetic analyses were carried out on the Fast UniFrac web server (Hamady, et al., 2010). Phylogenetic trees were constructed in Bosque (Ramirez-Flandes & Ulloa, 2008). The results of the weighted and unweighted UniFrac analyses were displayed through principal coordinates analysis as described (Hamady, et al., 2010).
Quantitative real-time PCR
Quantitative real-time PCR (qPCR) was carried out on ABI Prism 7000 Sequence Detection System using PERFECTa SYBR Green qPCR mastermix (Quanta Biosciences, Inc.) essentially as described (Paliy, et al., 2009) including mathematical calculations taking into account unequal amplification efficiency for different primer pairs. Group-specific primers were designed as described (Rigsbee, et al., 2011).
RESULTS
Profiling of distal gut microbiome of healthy adolescent children and adults
The goal of this study was to assess whether the intestinal microbiota of teenage children resembles that of adults, or if any specific differences can be detected. Fecal samples representing members of human distal gut microbiota (Gill, et al., 2006) were collected from 22 healthy pre- and adolescent children (designated kHLT for “healthy kids”). A smaller group of healthy adult volunteers was used for comparison (designated aHLT). Microbial populations in the fecal samples were analyzed by hybridizing 16S rDNA-enriched genomic DNA to the Microbiota Array, which provided both detection and quantitative measurements of 775 different species of human gut microbiota. The number of detected species varied appreciably from sample to sample, with a minimum of 209 and 276 species detected in adolescent and adult samples, respectively, and a maximum of 407 and 385 species, respectively. Though we detected slightly higher number of species in adult samples on average, the difference was not statistically significant (306 and 330 species for kHLT and aHLT groups, respectively).
Principal components analysis (PCA) of the species abundance data set was employed to test if we could separate samples that belonged to different groups. As is shown in Figure 1, PCA achieved relatively good separation between adolescent and adult groups. Among adolescent samples, two significant outliers were observed. Examination of the bacteria detected in those samples revealed that both harbored an unusually high abundance of members of genus Holdemania (class Mollicutes; 5.1% and 6.0% relative abundance in outlier samples; weighted mean for adolescent group was 1.0%). The main separation between kHLT and aHLT samples was observed in the principal component 1 (PC1) dimension. The PC1 values for each sample correlated negatively with the age of the corresponding participant, the negative correlation was highly significant (Spearman rank correlation Rs=-0.55, p<0.01). This can be interpreted to imply that participant’s age was one of the main factors differentiating different fecal samples in the PCA space. We have also employed both unweighted as well as weighted UniFrac analyses (Hamady, et al., 2010) to assess if similar separation could also be achieved when phylogenetic information about detected sequences was taken into account. Both types of UniFrac analyses did not produce a clear separation of kHLT and aHLT samples in the principal coordinates analysis (PCoA) plots (data not shown). The observed lack of concordance between PCA and PCoA results might indicate that the differences between kHLT and aHLT samples are caused by bacterial species and genera spread out among different phylogenetic groups rather than all clustered within one family, order, or class (see below).
Figure 1. Results of the principal components analysis of the microarray dataset.
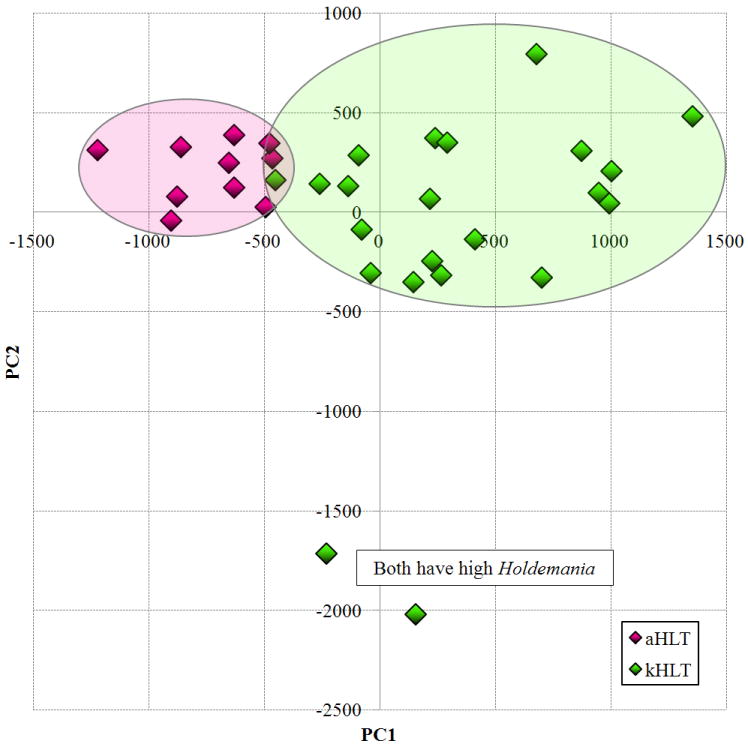
Principal component 1 values for each sample are plotted on the X axis and principal component 2 values are plotted on the Y axis. aHLT – healthy adult samples (n=10); kHLT – healthy adolescent samples (n=22).
Distribution of class and genera abundances among samples
Figure 2 displays a distribution of relative abundances of different bacterial classes among samples and groups. As expected, Clostridia was by far the most abundant class in all 32 samples profiled, with an average of 72.2% and 74.4% of total sample abundance among kHLT and aHLT samples, respectively. Actinobacteria (10.3% and 6.9%), Bacteroidetes (8.1% and 8.7%), and Bacilli (4.4% and 3.6%) were also well represented. Different classes of Proteobacteria combined to account for 2.4% and 2.2% total abundance in the kHLT and aHLT samples, respectively. For most classes, similar average numbers of detected species and relative class abundance were observed between kHLT and aHLT groups with two exceptions. Members of Actinobacteria were more abundant among adolescent samples, whereas adult samples contained more Betaproteobacteria. However, neither of these differences was statistically significant due to considerable variability of each class’ abundance from sample to sample within each group. Indeed, as can be surmised from Figure 2, a general trend of higher sample-to-sample class abundance variability that corresponded to lower average class abundance was evident in our experiments. This trend was found to be statistically significant as assessed by both the Spearman rank and Kendall’s tau non-parametric tests (Rs=-0.71, p=0.01; τ=-0.54, p=0.01). One possible explanation of this trend is that abundant classes are indispensable for the normal functioning of human intestinal microbiota in most people, whereas low abundance classes can vary from person to person based on a particular diet or environment.
Figure 2. Distribution of microbial species detection and abundance among adolescent and adult samples.

The main figure displays a group scatter plot with the X axis listing different bacterial classes and the Y axis representing relative combined abundance of the members of each class in individual samples. Note that the Y axis is shown in linear scale separated into 3 segments. aHLT – healthy adult samples (triangle; n=10); kHLT – healthy adolescent samples (diamond; n=22). Horizontal bars represent weighted means of the abundance of each class among all samples of a particular group. Three sets of values below the scatter plot show (i) the number of samples in which members of each class were detected (“samples”); (ii) an average number of detected species of each class per sample (“avg det”); and (iii) an average combined relative abundance (weighted mean) of class members in each sample (“avg abnd”).
The distribution of relative abundances among different bacterial genera is shown in Figure 3. Similar to the spread of class abundances, the fecal microbiota was dominated by relatively few genera. The top 12 abundant genera contributed between 72% and 76% of total microbial abundance in both kHLT and aHLT samples. Genus Ruminococcus was by far the most abundant with about 21-22% of total abundance in all samples. This genus also contained the largest number of detected species (73 species per sample on average). Among the dozen most abundant genera nine belonged to class Clostridia (see Figure 3); the remaining three genera were Streptococcus, Bifidobacterium, and Bacteroides. Relative abundance values for these twelve genera were generally similar between adolescent and adult sample groups for all genera but Clostridium and Bifidobacterium, which both displayed a considerably higher level in kHLT samples. In addition, statistically significant differences between kHLT and aHLT sample groups were observed for several low abundance genera as we describe below.
Figure 3. Distribution of genus relative abundances among samples.

Different experiments are plotted as columns; 115 analysed genera are plotted as rows. Relative abundances of each genus are plotted using a gradient scale as shown in the legend. aHLT – healthy adult samples; kHLT – healthy adolescent samples. Vertical line separates aHLT and kHLT samples. The 12 most abundant genera are shown on the right side; numbers represent relative average abundance of each genus (weighted mean) in aHLT and kHLT samples, respectively. Genus assignments to four most abundant phyla are shown on the left side of the image. Sample kHLT07 lacking members of Bacteroidetes is highlighted.
Plotting genus-level relative abundances for all samples together revealed several specific cases of unusual genus abundance distribution. For example, Figure 3 highlights fecal sample kHLT07 that lacked members of class Bacteroidetes almost entirely (total abundance less than 0.01%). This sample was taken from a healthy 13 year old female adolescent with no indication of gastrointestinal disorders or any other unusual gut physiology. No other genus was found to be uniquely increased in its abundance in this sample to take the place of Bacteroidetes in the community. Rather, the members of several other genera such as Haemophilus, Anaerostipes, Veillonella, Holdemania, and Streptococcus were disproportionately more abundant in this sample compared to their average abundance among other samples.
On the other hand, the detection of microbial species was very similar between sample groups at all levels from genus to phylum. Comparison of average species detection between kHLT and aHLT groups at the genus level produced a highly statistically significant correlation (Spearman Rs=0.87, p<0.001; Kendall τ=0.95, p<0.001) indicating that it was not the presence or absence of particular species or genera that differentiated kHLT and aHLT samples, but rather the relative abundances of the species within each sample group.
Core microbiome
Microbiota Array contains probesets for 775 different intestinal bacterial species. Among these, we were able to detect 647 in at least 1 sample out of combined 32 samples. Most of these species (543) belonged to the “shared” category of those common to multiple but not all samples. A total of 58 species were uniquely present in only a single sample. We have also identified a “core” of 46 species shared among all analyzed fecal samples. This core microbiome was dominated by genus Ruminococcus represented by 27 species; Faecalibacterium and Roseburia contributed 4 core species each. In fact, 44 out of 46 core species belonged to class Clostridia (other two were Streptococcus species from class Bacilli), again showing the dominance of this class in the fecal microbiota samples examined. Two species of genus Bacteroides, B. ovatus and B. thetaiotaomicron, were present in 31 out of 32 analyzed samples with the lone exception being sample kHLT07 that lacked all members of Bacteroidetes class (see Figure 3). Core species factored considerably in the overall microbiota composition – on average they contributed 24.8% to the total microbiota abundance (range: 17.5% - 34.4%) even though they only constituted 1/7 fraction of detected species in each sample on average.
Differences in genus relative abundance among groups
Because Microbiota Array provides direct quantitative measurements of 16S rDNA amounts and thus directly estimates the abundance of each of the 775 microbiota species, we were able to quantitatively compare genus abundances among samples examined in this study. Among 115 different genera that microarray can detect, 15 showed substantial differences in their abundance levels between kHLT and aHLT groups (Table 1). The most prominent difference detected was a statistically significantly higher abundance of Bifidobacterium members among adolescent samples (9.0% and 5.4% relative abundance in kHLT and aHLT samples, respectively; p=0.05). This difference was due to an increase in the relative abundance of genus members rather than their presence or absence – the number of Bifidobacterium species detected per sample was practically identical for both groups (6.5). Specifically, both B. longum and B. catenulatum species were more abundant among adolescents (2.9% and 1.2% average abundance in adolescents, respectively; 0.9% and 0.7% in adults, respectively), whereas B. adolescentis had similar abundance in kHLT and aHLT samples (2.0% versus 1.8% average abundance). Other notable differences included higher abundance of Clostridium and lower abundance of Prevotella and Sutterella in adolescent samples. Consistent with previous reports (Iizuka, et al., 2004), Clostridium difficile (opportunistic human pathogen causing diarrhea) was detected in most samples at a low abundance level (kHLT samples: 18 out of 22 positive, 0.007% average abundance; aHLT samples: 4 out of 10 positive, 0.003% average abundance). Note that because of significant variability in the relative genus abundance from sample to sample not all differences were statistically significant as shown in Table 1. Members of genus Escherichia were detected significantly more often in adolescent than in adult samples (15 out of 22 had detectable levels in kHLT samples; 2 out of 10 – in aHLT samples), though average abundance level was too low in both groups for us to assess relative abundance difference with confidence.
Table 1.
Genera present at different relative abundances in samples from adults and adolescent children.
| Genus | Corresponding class | Adult samples
|
Child samples
|
Log ratio‡ | P value** | ||
|---|---|---|---|---|---|---|---|
| No* | % † | No* | % † | ||||
| Oxalobacter | βProteobacteria | 2 | 0.2±<0.1 | 2 | <0.1±<0.1 | 4.9 | 0.13 |
| Sutterella | βProteobacteria | 5 | 0.8±0.2 | 5 | 0.2±<0.1 | 2.2 | 0.02 |
| Limnobacter | βProteobacteria | 10 | 0.2±0.1 | 20 | 0.4±0.1 | -1.1 | 0.14 |
| Enterobacter | γProteobacteria | 6 | <0.1±<0.1 | 5 | <0.1±<0.1 | 2.4 | 0.04 |
| Clostridium | Clostridia | 10 | 1.8±0.3 | 22 | 2.8±0.2 | -0.6 | 0.01 |
| Parasporobacterium | Clostridia | 9 | 0.1±<0.1 | 15 | <0.1±<0.1 | 2.7 | <0.01 |
| Butyrivibrio | Clostridia | 8 | 0.1±0.1 | 6 | <0.1±<0.1 | 4.9 | 0.01 |
| Peptococcus | Clostridia | 4 | 0.3±0.1 | 6 | <0.1±<0.1 | 2.9 | 0.02 |
| Hespellia | Clostridia | 6 | 0.2±<0.1 | 4 | <0.1±<0.1 | 3.9 | <0.01 |
| Turicibacter | Clostridia | 9 | 0.2±0.1 | 22 | 0.4±0.1 | -1.3 | 0.05 |
| Slackia | Actinobacteria | 6 | 0.1±<0.1 | 2 | <0.1±<0.1 | 3.4 | 0.01 |
| Bifidobacterium | Actinobacteria | 9 | 5.4±1.3 | 22 | 9.0±0.8 | -0.7 | 0.05 |
| Prevotella | Bacteroidetes | 5 | 0.7±0.3 | 12 | 0.2±0.1 | 1.6 | 0.18 |
| Anaerophaga | Bacteroidetes | 8 | 0.3±0.1 | 12 | 0.1±<0.1 | 1.1 | 0.24 |
| Victivallis | Lentisphaerae | 4 | 0.2±0.1 | 7 | 0.1±0.1 | 1.6 | 0.15 |
Number of samples in which members of this genus were detected out of 10 adult samples and 22 adolescent samples.
Percent contribution of species in each bacterial genus to the total hybridization signal measured by Microbiota Array. Data is shown as weighted mean ± weighted standard error.
Logarithm with base 2 of the ratio of relative abundances of adult and adolescent samples.
Statistical significance of observed differences between sample groups as measured by the moderated T- test. P values less than or equal to 0.05 are underlined.
Validation of array results with qPCR
We have used quantitative real time PCR to validate the findings revealed by the Microbiota Array. Primers developed specifically to amplify 16S rDNA from members of three separate genera were used in qPCR tests (Rigsbee, et al., 2011). As can be assessed in Table 2, qPCR results matched microarray data well, with only a slight deviation observed for Faecalibacterium genus abundance estimate of adult sample aHLT05.
Table 2.
Validation of microarray results with quantitative PCR
| Bacterial group * | Adult sample aHLT02 †
|
Adult sample aHLT05 †
|
||
|---|---|---|---|---|
| qPCR ‡ | Array ‡ | qPCR ‡ | Array ‡ | |
| Faecalibacterium | 11.7%±5.0% | 11.0%±0.3% | 4.9%±3.5% | 9.1%±0.2% |
| Bifidobacterium | 5.7%±2.2% | 6.4%±0.3% | 0.3%±0.2% | 0.1%±<0.1% |
| Collinsella | 1.0%±0.3% | 0.7%±0.1% | 0.1%±0.1% | 0.3%±<0.1% |
Primers were developed to cover most of the 16S rDNA sequences in the RDP database belonging to each group (Rigsbee, et al., 2011).
Numbers represent relative abundances of genomic DNA of each genus in the total sample; no 16S rRNA gene copy adjustment was applied to the normalized values.
Data is shown as arithmetic mean ± standard error.
DISCUSSION
The present study is the first to assess distal gut microbiota of adolescent children with a high-throughput technology. Though it is generally assumed that older children house gut microbial populations similar to those of adults, our results indicate that such gut communities are sufficiently different (see Figure 1) in the population examined. Few distinctions were found when relative abundances were compared at the class level; however, many differences were observed at the genus level between adolescent and adult fecal microbiota. Most strikingly, we found that relative combined abundance of members of genus Bifidobacterium was statistically significantly higher in adolescent children (close to 2-fold difference). This high prevalence of Bifidobacterium is well known for children of young age, in fact bifidobacteria are some of the first species to colonize newborn gut (Bjorkstrom, et al., 2009, Enck, et al., 2009). However, an increased prevalence of this genus was not recognized previously among pre- and adolescent age groups. Importantly, the level of bifidobacteria correlated negatively with age in our sample set (Spearman rank Rs=-0.29, p=0.03; Kendall’s tau τ=-0.36, p=0.05). Thus, our data is more consistent with the model where levels of bifidobacteria in children decrease gradually between 2 and 18 years of life until reaching stable levels in the early adulthood, rather than with the model that assumes that levels of these bacteria drop quickly after weaning.
Considering distal gut microbiota of healthy adolescent children and adults on a global level, microbial populations were dominated by class Clostridia in all cases. In particular, Ruminococcus was the most prevalent genus in all samples examined, which is consistent with an important role of the members of this genus as the primary carbohydrate degraders in the human gut (Flint, et al., 2008). Most of the highly abundant genera and classes showed relatively consistent abundances among samples, whereas levels of low abundance genera and classes varied widely from sample to sample (see Figures 2 and 3).
We have identified a core set of 46 bacterial species that we detected in every sample examined; as expected this core set was dominated by members of Clostridia and genus Ruminococcus in particular, which is in general agreement with another study that was focused on core microbiome identification (Tap, et al., 2009). It is tempting to speculate that Ruminococcus serves as the “irreplaceable” primary degrader of complex carbohydrates in the ecological food chain in the gut, which would explain its very consistent level of high abundance in all samples. At the same time, functions of the secondary and tertiary degraders can be performed (interchangeably) by many different bacterial species and genera, leading to a wider spread of their abundances among different samples (Flint, et al., 2008, Karasov & Carey, 2009). Future mechanistic studies using artificial gut fermentor models and engineered microbiota populations in gnotobiotic mice will be required to shed light on this hypothesis.
Supplementary Material
{kind=link}
Acknowledgments
Our thanks to Vijay Shankar for valuable comments on the manuscript and to the members of the WSU Center for Genomics Research for access to the facility. This work was supported by the National Institutes of Health grants AT003423 and HD065575 to OP.
Footnotes
Supplementary material. Additional figures, tables, and data are available as supplementary material at http://www.wright.edu/~oleg.paliy/Papers/MB_kHLT/MB_kHLT.html.
References
- Biasucci G, Benenati B, Morelli L, Bessi E, Boehm G. Cesarean delivery may affect the early biodiversity of intestinal bacteria. J Nutr. 2008;138:1796S–1800S. doi: 10.1093/jn/138.9.1796S. [DOI] [PubMed] [Google Scholar]
- Biasucci G, Rubini M, Riboni S, Morelli L, Bessi E, Retetangos C. Mode of delivery affects the bacterial community in the newborn gut. Early Hum Dev. 2010;86:13–15. doi: 10.1016/j.earlhumdev.2010.01.004. [DOI] [PubMed] [Google Scholar]
- Bik EM, Eckburg PB, Gill SR, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A. 2006;103:732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkstrom MV, Hall L, Soderlund S, Hakansson EG, Hakansson S, Domellof M. Intestinal flora in very low-birth weight infants. Acta Paediatr. 2009;98:1762–1767. doi: 10.1111/j.1651-2227.2009.01471.x. [DOI] [PubMed] [Google Scholar]
- Brodie EL, Desantis TZ, Joyner DC, et al. Application of a high-density oligonucleotide microarray approach to study bacterial population dynamics during uranium reduction and reoxidation. Appl Environ Microbiol. 2006;72:6288–6298. doi: 10.1128/AEM.00246-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernukha M, Alekseeva GV, Avetisian LR, Kuznetsova OV, Tolovskaia KR, Shaginian IA. Microbiological aspects of enteric dysbacteriosis in patients of different age groups, visiting outpatient clinics of Moscow. Zh Mikrobiol Epidemiol Immunobiol. 2005;4:69–73. [PubMed] [Google Scholar]
- Davis CD, Milner JA. Gastrointestinal microflora, food components and colon cancer prevention. J Nutr Biochem. 2009;20:743–752. doi: 10.1016/j.jnutbio.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107:11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enck P, Zimmermann K, Rusch K, Schwiertz A, Klosterhalfen S, Frick JS. The effects of maturation on the colonic microflora in infancy and childhood. Gastroenterol Res Pract. 2009;2009:752401. doi: 10.1155/2009/752401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint HJ, Bayer EA, Rincon MT, Lamed R, White BA. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Microbiol. 2008;6:121–131. doi: 10.1038/nrmicro1817. [DOI] [PubMed] [Google Scholar]
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura KE, Slusher NA, Cabana MD, Lynch SV. Role of the gut microbiota in defining human health. Expert Rev Anti Infect Ther. 2010;8:435–454. doi: 10.1586/eri.10.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SR, Pop M, Deboy RT, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamady M, Lozupone C, Knight R. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. Isme J. 2010;4:17–27. doi: 10.1038/ismej.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmsen HJ, Wildeboer-Veloo AC, Raangs GC, Wagendorp AA, Klijn N, Bindels JG, Welling GW. Analysis of intestinal flora development in breast-fed and formula-fed infants by using molecular identification and detection methods. J Pediatr Gastroenterol Nutr. 2000;30:61–67. doi: 10.1097/00005176-200001000-00019. [DOI] [PubMed] [Google Scholar]
- Hopkins MJ, Sharp R, Macfarlane GT. Age and disease related changes in intestinal bacterial populations assessed by cell culture, 16S rRNA abundance, and community cellular fatty acid profiles. Gut. 2001;48:198–205. doi: 10.1136/gut.48.2.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins MJ, Macfarlane GT, Furrie E, Fite A, Macfarlane S. Characterisation of intestinal bacteria in infant stools using real-time PCR and northern hybridisation analyses. FEMS Microbiol Ecol. 2005;54:77–85. doi: 10.1016/j.femsec.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Iizuka M, Konno S, Itou H, et al. Novel evidence suggesting Clostridium difficile is present in human gut microbiota more frequently than previously suspected. Microbiol Immunol. 2004;48:889–892. doi: 10.1111/j.1348-0421.2004.tb03607.x. [DOI] [PubMed] [Google Scholar]
- Karasov WH, Carey HV. Metabolic Teamwork between Gut Microbes and Hosts. Microbe. 2009;4:323–328. [Google Scholar]
- Langendijk PS, Schut F, Jansen GJ, Raangs GC, Kamphuis GR, Wilkinson MH, Welling GW. Quantitative fluorescence in situ hybridization of Bifidobacterium spp. with genus-specific 16S rRNA-targeted probes and its application in fecal samples. Appl Environ Microbiol. 1995;61:3069–3075. doi: 10.1128/aem.61.8.3069-3075.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lay C, Rigottier-Gois L, Holmstrom K, et al. Colonic microbiota signatures across five northern European countries. Appl Environ Microbiol. 2005;71:4153–4155. doi: 10.1128/AEM.71.7.4153-4155.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliy O, Kenche H, Abernathy F, Michail S. High-throughput quantitative analysis of the human intestinal microbiota with a phylogenetic microarray. Appl Environ Microbiol. 2009;75:3572–3579. doi: 10.1128/AEM.02764-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C, Bik EM, Digiulio DB, Relman DA, Brown PO. Development of the Human Infant Intestinal Microbiota. PLoS Biol. 2007;5:e177. doi: 10.1371/journal.pbio.0050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C, Bik EM, Eisen MB, et al. Rapid quantitative profiling of complex microbial populations. Nucleic Acids Res. 2006;34:e5. doi: 10.1093/nar/gnj007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penders J, Vink C, Driessen C, London N, Thijs C, Stobberingh EE. Quantification of Bifidobacterium spp., Escherichia coli and Clostridium difficile in faecal samples of breast-fed and formula-fed infants by real-time PCR. FEMS Microbiol Lett. 2005;243:141–147. doi: 10.1016/j.femsle.2004.11.052. [DOI] [PubMed] [Google Scholar]
- Rainer J, Sanchez-Cabo F, Stocker G, Sturn A, Trajanoski Z. CARMAweb: comprehensive R- and bioconductor-based web service for microarray data analysis. Nucleic Acids Res. 2006;34:W498–503. doi: 10.1093/nar/gkl038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajilic-Stojanovic M, Heilig HG, Molenaar D, Kajander K, Surakka A, Smidt H, de Vos WM. Development and application of the human intestinal tract chip, a phylogenetic microarray: analysis of universally conserved phylotypes in the abundant microbiota of young and elderly adults. Environ Microbiol. 2009;11:1736–1751. doi: 10.1111/j.1462-2920.2009.01900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Flandes S, Ulloa O. Bosque: integrated phylogenetic analysis software. Bioinformatics. 2008;24:2539–2541. doi: 10.1093/bioinformatics/btn466. [DOI] [PubMed] [Google Scholar]
- Rigsbee L, Agans R, Foy BD, Paliy O. Optimizing the analysis of human intestinal microbiota with phylogenetic microarray. FEMS Microbiol Ecol. 2011;75:332–342. doi: 10.1111/j.1574-6941.2010.01009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90:859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, Dore J. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol. 1999;65:4799–4807. doi: 10.1128/aem.65.11.4799-4807.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swidsinski A, Loening-Baucke V, Vaneechoutte M, Doerffel Y. Active Crohn’s disease and ulcerative colitis can be specifically diagnosed and monitored based on the biostructure of the fecal flora. Inflamm Bowel Dis. 2008;14:147–161. doi: 10.1002/ibd.20330. [DOI] [PubMed] [Google Scholar]
- Tap J, Mondot S, Levenez F, et al. Towards the human intestinal microbiota phylogenetic core. Environ Microbiol. 2009;11:2574–2584. doi: 10.1111/j.1462-2920.2009.01982.x. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Wang RF, Beggs ML, Erickson BD, Cerniglia CE. DNA microarray analysis of predominant human intestinal bacteria in fecal samples. Mol Cell Probes. 2004;18:223–234. doi: 10.1016/j.mcp.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Wilcox RR. Applying Contemporary Statistical Techniques. Academic Press; New York: 2003. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.