

Abstract
Background:
Despite successfully suppressed viremia by treatment, patients with high levels of biomarkers of coagulation/inflammation are at an increased risk of developing non-AIDS defining serious illnesses such as cardiovascular diseases. Thus, there is a relationship between persistent immune activation and coagulation/inflammation, although the mechanisms are poorly understood. Platelets play an important role in this process. Although interactions between platelets and elements of the innate immune system, such as monocytes, are well described, little is known about the interaction between platelets and the adaptive immune system.
Design:
We investigated the interaction of a component of the coagulation system, platelets, and the adaptive immune system T cells.
Methods:
Healthy controls and combination antiretroviral therapy (cART)-treated HIV-infected patients with viral loads of less than 40 copies/ml for more than 15 months were analysed for platelet–T-cell conjugate formation.
Results:
Platelets can form conjugates with T cells and were preferentially seen in CD4+ and CD8+ T-cell subsets with more differentiated phenotypes [memory, memory/effector and terminal effector memory (TEM)]. Compared with healthy controls, these conjugates in patients with HIV infection were more frequent, more often composed of activated platelets (CD42b+CD62P+), and were significantly associated with the D-dimer serum levels.
Conclusion:
These data support a model in which platelet–T-cell conjugates may play a critical role in the fast recruitment of antigen-experienced T cells to the place of injury. This mechanism can contribute in maintaining a state of coagulation/inflammation observed in these patients contributing to the pathology of the disease.
Keywords: coagulation/inflammation and T cells, HIV infection, platelet–T-cell conjugates
Introduction
Immune activation plays an important role in the pathogenesis of HIV infection. Although the introduction of antiretroviral therapy has improved the life expectancy of HIV-infected individuals, an increasing body of data has clearly demonstrated that, despite ‘undetectable’ HIV-RNA plasma levels (generally <50 copies/ml) following initiation of therapy, there remains evidence of persistent immune activation, albeit at a lower level. The evidence of higher levels of immune activation in patients with HIV infection and suppressed viremia has been characterized as increased proliferation of CD4+ and CD8+ T cells, increased expression of T-cell activation markers such as PD1, HLADR+CD38+, and a reduced CD4+/CD8+ ratio [1–5]. The clinical significance of this persistent immune activation is strongly suggested by the increased risk of all-cause mortality associated with elevated levels of soluble markers of inflammation/monocyte activation [interleukin (IL)-6, sCD14] and coagulation (D-dimer, a fibrin degradation product). Patients with higher levels of these biomarkers are at an increased risk of hepatic, metabolic, renal and cardiovascular morbidity despite HIV RNA levels less than 50 copies/ml [6–9]. Taken together, these serious non-AIDS defining illnesses are the leading cause of mortality/morbidity in patients with HIV infection, today. The HIV epidemic presents new challenges that are reflected in an accelerated course of chronic illnesses.
Pathophysiological conditions involving tissue injury by trauma or infection lead to activation of the coagulation cascade in association with an inflammatory response as part of the innate host response to insult. Platelets play a critical role in this process linking haemostasis, coagulation and tissue repair and as an immediate nonspecific host response to pathogens. In some settings, this activation of the coagulation cascade may contribute to disease by perpetuating an inflammatory response [10].
In the context of HIV infection, viral replication and higher levels of immune activation lead to activation of the coagulation cascade resulting in higher levels of certain markers of coagulation/inflammation such as D-dimer, IL-6 and sCD14 [6,7]. Increased tissue factor expression on monocytes and increased proportion of CD8+ T cells expressing the thrombin receptor [protease activated receptor-1 (PAR1)] have been reported in HIV-infected patients [11,12]. CD8+ T-cell activation has been shown to be enhanced by thrombin [12]. These observations suggest an important link between immune activation, coagulation and inflammation during chronic HIV infection.
Recently, several groups have reported that there is an increased platelet activation in patients with HIV infection [13–15]. Platelets play an important role in the early response to pathogens. Upon activation, they accumulate at the site of injury and secrete pro-inflammatory and anti-inflammatory mediators that are prestored in specialized granules. In addition, platelets and leukocytes (neutrophils, monocytes and T cells) can be observed to form conjugates in peripheral blood [16–19]. Increased platelet–monocyte complexes have been observed during inflammatory conditions such as cardiovascular disease, type 1 diabetes, rheumatoid arthritis, systemic lupus erythematosus and end-stage renal disease [20–24]. Platelet–CD4+ T-cell conjugates have been observed in the peripheral blood of patients with rheumatoid arthritis suggesting that platelets maybe involved in regulating T-cell function [18]. There is evidence of an association between platelet activation and hypercoagulability in HIV/simian immunodeficiency virus (SIV) infection. Reports in SIV-infected Pig-tailed Macaques have shown platelet–monocyte complexes formation in this model [25,26]. In patients with HIV infection, platelet activation and platelet microparticles formation (PMPs) have been observed [15].
Platelets and PMP express P-selectin (CD62P) upon activation. Platelets are a major source of P-selectin in circulation (endothelial cells also express CD62P after activation). CD62P is a key selectin that mediates the recruitment of leukocytes into peripheral tissues by facilitating the rolling of leukocytes onto endothelium. This interaction is mediated by P-selectin ligand expressed on lymphocytes (PSGL1). PSGL1 protein is expressed by all T cells; however, the affinity to bind its ligand is determined by the degree of glycosylation [27]. Therefore, platelets bound to inflammatory cells play a crucial role in recruiting them to inflamed tissues. In mice, CD62P+ activated platelets mediate adhesion of lymphocytes to endothelium leading to recruitment of T cells to the place of injury [28].
The interaction between the coagulation system and the immune system is critical for host defense. In HIV infection, ongoing activation of these pathways is associated with increases in serious non-AIDS events by mechanisms that are poorly understood. In the present manuscript, we investigated the relationships between T-cell immune activation, coagulation and inflammation in HIV-infected patients with suppressed viremia to less than 40 copies/ml by cART. We hypothesized that activated T cells can form conjugates with platelets, which may contribute to the increases in coagulation/inflammation observed in these patients.
Materials and methods
Patients and healthy volunteers
Patients and healthy controls were obtained under NIAID Institutional Review Board approved clinical research study protocols in the NIAID/CCMD intramural programme and supplied written informed consent for use of their blood. Samples from healthy controls were obtained from the NIH Blood Bank. HIV-infected patients with suppressed viremia to less than 40 copes/ml were obtained from the NIH Clinical Center (Supplementary data, Table S1, S3 and S4).
Platelet–T-cell conjugates analysis
Platelet–T-cell conjugates, recombinant CD62P-Fc binding and thrombin stimulations were analysed by flow cytometry. T-cell conjugates were imaged by Amnis and scanning electron microscopy as described at Supplementary Materials and Methods.
Statistical analysis method
Described in supplementary Material and Methods, statistically significant differences were considered to be those whose P value was less than 0.05.
Results
CD4+ and CD8+ T-cell subsets with memory phenotypes show high affinity binding to CD62P
Selectins are involved in the trafficking of T lymphocytes, and the interaction of selectins with their ligands mediates rolling of lymphocytes on the endothelium. In circulation, platelets are the main source of CD62P, a selectin expressed upon activation. T cells express PSGL1 protein; however, the binding affinity for its ligand is determined by the degree of glycosylation of PSGL1 [27]. To directly analyse the contribution of CD62P to platelet–T-cell interactions, we performed a functional staining using recombinant CD62P-Fc fusion protein. CD4+ and CD8+ T-cell subsets from healthy controls (n = 13) and HIV-infected patients with suppressed viremia to less than 40 copies/ml (n = 18, Supplementary Table S1) showed similar protein expression levels of PSGL1 (Figure S1). In contrast, recombinant P selectin bound with higher affinity to CD4+ and CD8+ T-cell subsets with memory phenotypes (Fig. 1b, c). This interaction was ion dependent, as incubation in the presence of EDTA inhibited CD62P-Fc binding (Fig. 1c). In addition, a higher binding for CD62P was observed on CD4+ and CD8+ T cells expressing HLADR+CD38+, although the proportion of these cells in treated HIV-infected patients was low (Figure S1, Supplementary Table S1). These data suggest that CD4+ and CD8+ T cells with memory phenotypes express a high affinity ligand for CD62P and potentially can bind to CD62P expressing platelets.
Fig. 1.
Preferential binding of recombinant CD62P-Fc to CD4+ and CD8+ T-cell subsets with memory, memory/effector and TEM phenotypes.
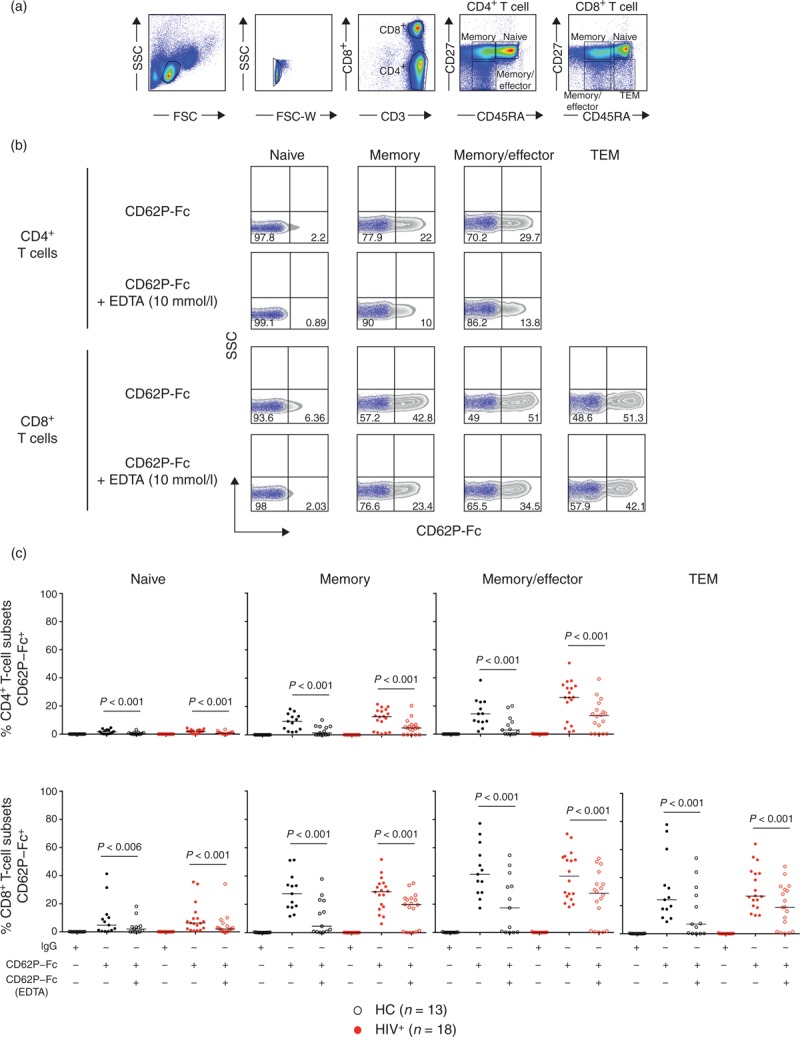
(a) Flow cytometry gating strategy for T-cell subsets: Naive (CD45RA+CD27+), memory (CD45RA-CD27+), memory/effector (CD45RA-CD27-), TEM (CD45RA+CD27-). (b) Purified T cells were incubated with IgG control or CD62P-Fc in the absence or presence of 10 mmol/l EDTA. Representative CD62P-Fc binding zebra plots (gray) overlaid with the media control (blue dot plot) of an HIV-infected patient. (c) The graph shows the proportion of CD4+ and CD8+ T-cell subsets CD62P-Fc+ in the presence and absence of EDTA from healthy controls (n = 13, black symbols) and HIV-infected patients (n = 18, red symbols). Paired Wilcoxon test was used for comparisons between binding IgG control, CD62P-Fc and CD62P-Fc in the presence of EDTA; P < 0.05 was considered significant.
Presence of activated platelet–T-cell conjugates in the peripheral blood of HIV-infected patients
We next looked ex vivo for the presence of platelet–CD4+ T and CD8+ T-cell conjugates in whole blood from 20 patients with chronic HIV infection and successfully suppressed viremia to less than 40 copies/ml for more than 15 months and 23 healthy controls (HC1) (Supplementary Table S1). To examine only those platelets bound to T cells, we used a gating strategy to exclude the free/unbound platelets using a specific platelet receptor glycoprotein Ib+ (CD42b+), Fig. 2a. CD4+ and CD8+ T-cell subsets were identified using cell surface markers CD45RA and CD27 (Fig. 2b) and the frequency of platelet–T-cell conjugates were determined by those T-cell subsets expressing the platelet receptor CD42b+.
Fig. 2.
Platelet–T-cell conjugates in peripheral blood mononuclear cells (PBMCs) of HIV-infected patients and healthy controls.
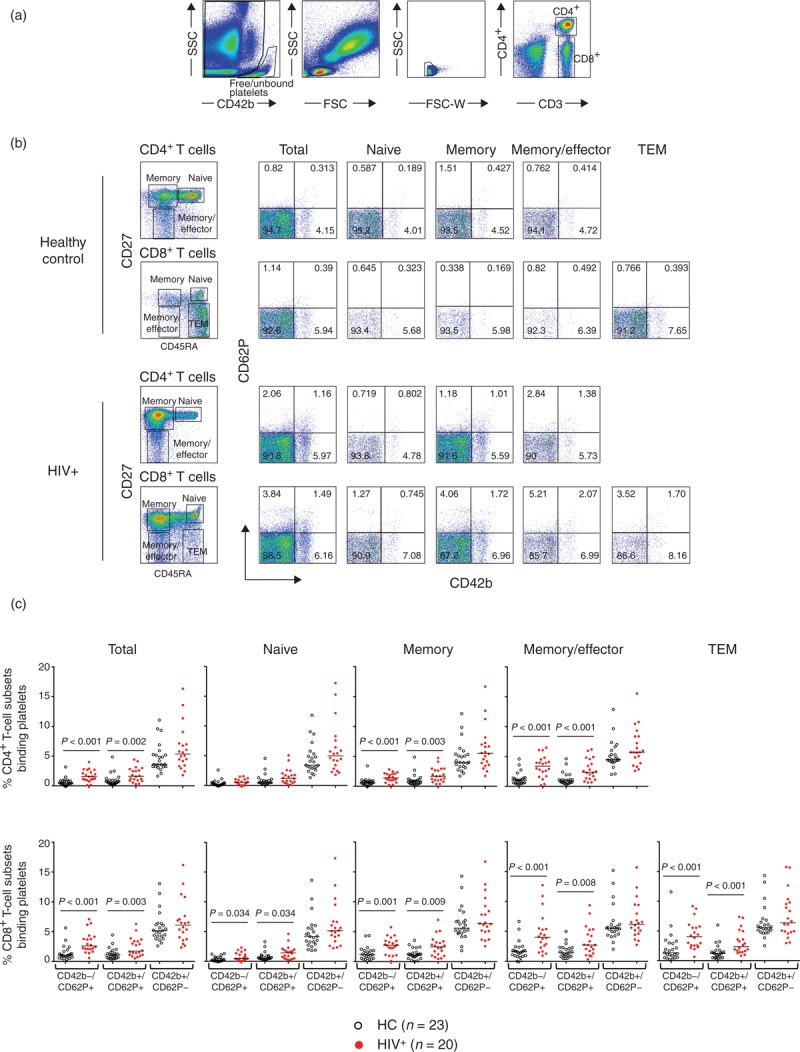
(a) Whole blood from HIV-infected patients (n = 20) and healthy controls (HC1, n = 23) were stained to analyse platelet–T-cell conjugates by flow cytometry. (b) CD4+ and CD8+ T-cell subsets were defined as above. Frequency of platelet–CD4+ and platelet–CD8+ T cell conjugates were determined by those T-cell subsets expressing the platelet markers CD42b and CD62P. (c) The graph depicts the proportion of platelet–CD4+ and platelet–CD8+ T-cell subset conjugates in healthy control (black symbol) and HIV-infected patient (red symbol) groups. Mann–Whitney test was used for the group comparisons: P < 0.05 was considered significant.
The median proportion of CD4+ T cells that had bound platelets was 7.3% [interquartile range (IQR) 4.5–10] in the HIV-infected patients compared with 4.0% (IQR 3.8–6.3) in healthy controls. The CD8+ T cells from HIV-infected patients showed 8.2% (IQR 5.5–11.6) compared with 6.3% (IQR 4.8–7.4) in healthy controls. A significantly higher proportion of bound platelets were observed in CD4+ and CD8+ T-cell subsets with more differentiated phenotypes than their naive counterparts (P < 0.001) (Fig. 2b).
We next investigated the activation status of the bound platelets by analysing the surface expression of P-selectin (CD62P). CD62P is stored in the alpha-granules of platelets under steady-state conditions and is expressed on their surface or released as a soluble form (sCD62P) upon platelet activation. HIV-infected patients showed a 2.5 and 1.6 higher proportions of activated platelets (CD42b+CD62P+) on CD4+ and CD8+ total T cells, respectively, when compared with the platelets bound to T cells from healthy controls (Fig. 2c). In addition, CD4+ and CD8+ T cells from HIV-infected patients showed a significantly increased proportion of T cells expressing CD42b-CD62P+ likely corresponding to bound sCD62P (Fig. 2c), when compared with healthy controls (P < 0.001). In contrast, similar proportions of CD42b+CD62P- bound CD4+ and CD8+ T-cell subsets were observed in both groups (Fig. 2c). Other subsets of leukocytes in the blood of HIV-infected patients showed an increased proportion of bound activated platelets to CD3-negative lymphocytes. In cells with higher forward side scatter (FSC) and side scatter (SSC) (likely monocytes), there was a trend that did not reach statistical significance (Supplementary Figure S2). In addition, free/unbound-circulating platelets of HIV-infected patients showed a significantly increased proportion (median 16%, IQR 12–21) of activated phenotype (CD42b+CD62P+) when compared with healthy controls (median 11%, IQR 8–14). In addition, the proportion of activated circulating platelets (free/unbound) in HIV-infected patients, but not in healthy controls, was positively associated with the proportion of activated bound platelets on CD4+ or CD8+ T cells (R = 0.66, P = 0.004 and R = 0.65, P = 0.005 respectively), suggesting that activated platelets are prone to form T-cell conjugates (Supplementary Figure S3A).
We next analysed the association between activated platelet–CD4+ and platelet–CD8+ T-cell conjugates with the serum levels of D-dimer (Supplementary Figure S3B). D-dimer serum levels correlated with the frequency of activated platelet–CD4+ (R = 0.56, P = 0.015) and platelet–CD8+ T-cell conjugates (R = 0.52, P = 0.023) (Supplementary Figure S3B and Table S2). This association was not observed in healthy controls (group HC1, Table S2). Because aspirin's mode of action targets platelet function, when three HIV patients under aspirin treatment were removed from the analysis, the association between activated platelet–CD4+ (R = 0.68, P = 0.007) and platelet–CD8+ T-cell conjugates (R = 0.62, P = 0.013) with serum levels of D-dimer remained significant. In addition, D-dimer serum levels were directly associated with the proportion of free/unbound activated (CD42b+CD62P+) platelets (R = 0.51, P = 0.026) and similarly, as above, when patients taking aspirin were excluded from the analysis, the association remained significant (R = 0.68, P = 0.007). As platelet activation could induce release of sCD62P, we measured serum levels of sCD62P. We found that the proportion of CD62P+ CD4+ T cells was positively associated with its serum levels (R = 0.54, P = 0.031), in contrast, this was not observed in CD8+ T cells. Such associations were not observed in the healthy controls (data not shown). We next examined a second cohort of healthy controls (HC2, Supplementary Table S1, and Figure S4); of note, this group had higher serum levels of D-dimer than HC1. In this group, D-dimer serum levels were not associated with the proportion of activated platelets (free/unbound or those forming conjugates, supplementary Table S2). These data suggest that chronic HIV infection may contribute to maintaining higher levels of platelet activation promoting formation of conjugates with T cells. In addition, no association was observed with T-cell activation (HLADR+CD38+), CD4+ and CD8+ cell counts with either CD42b+CD62P+ platelets both unbound/free or forming conjugates in both healthy controls and HIV-infected patients (Supplementary Table S2).
These data suggest that, in vivo, patients with HIV infection have an increased proportion of activated circulating platelets and an increased proportion of activated platelet–T-cell conjugates that are positively associated with serum levels of D-dimer.
Thrombin-activated platelets showed increased binding ability to memory phenotype CD4+ and CD8+ T-cell subsets
Tissue damage induced by trauma or infection leads to a cascade of events including the generation of thrombin and activation of platelets. CD62P expression upon platelet activation bridges the endothelium and circulating leukocytes through its interaction with CD62P-ligand in both cells. Thus, platelets play a critical role in the recruitment of cells to sites of injury.
To address whether upon activation platelets can form conjugates with T cells, we analysed the ability of platelets to bind T cells after in-vitro thrombin stimulation in healthy controls (n = 8) and HIV-infected patients (n = 8) presented in Figs. 3 and 4. PBMCs isolated by Ficoll gradient already contain considerable numbers of free/unbound platelets (Fig. 3a), therefore we stimulated freshly isolated PBMCs with 50 U/ml of thrombin for 5 and 15 min and measured the expression of CD62P and CD42b on CD4+ (Fig. 3) and on CD8+ (Fig. 4) T-cell subsets. As thrombin stimulation induces downregulation of CD42b on platelets, we gated on the total CD62P to account for platelets CD42blow[29]. Thrombin stimulation significantly increased the proportion of CD62P+ T cells. CD4+ T-cell memory and memory/effector phenotypes exhibited high levels of CD62P+ indicating preferential binding of activated platelets to these subsets (Fig. 3b). Similarly, CD8+ T cells showed an increase in CD62P+ cells following thrombin stimulation. This was most notable in those subsets with a more differentiated phenotype (Fig. 4). Interestingly, CD4+ and CD8+ T cells subsets from HIV-infected patients tended to show lower expression of CD62P+ than healthy controls after thrombin stimulation. This could reflect a higher in-vivo activation state of platelets in HIV-infected patients. These results suggest that thrombin-induced platelet activation enhanced platelet–CD4+ and platelet–CD8+ T-cell conjugate formation particularly with memory, memory/effector and TEM phenotype subsets. This mechanism may represent an early step in the recruitment of antigen specific T cells to the site of injury.
Fig. 3.
Increased platelet–T-cell conjugates formation after thrombin stimulation in CD4+ T-cell subsets.
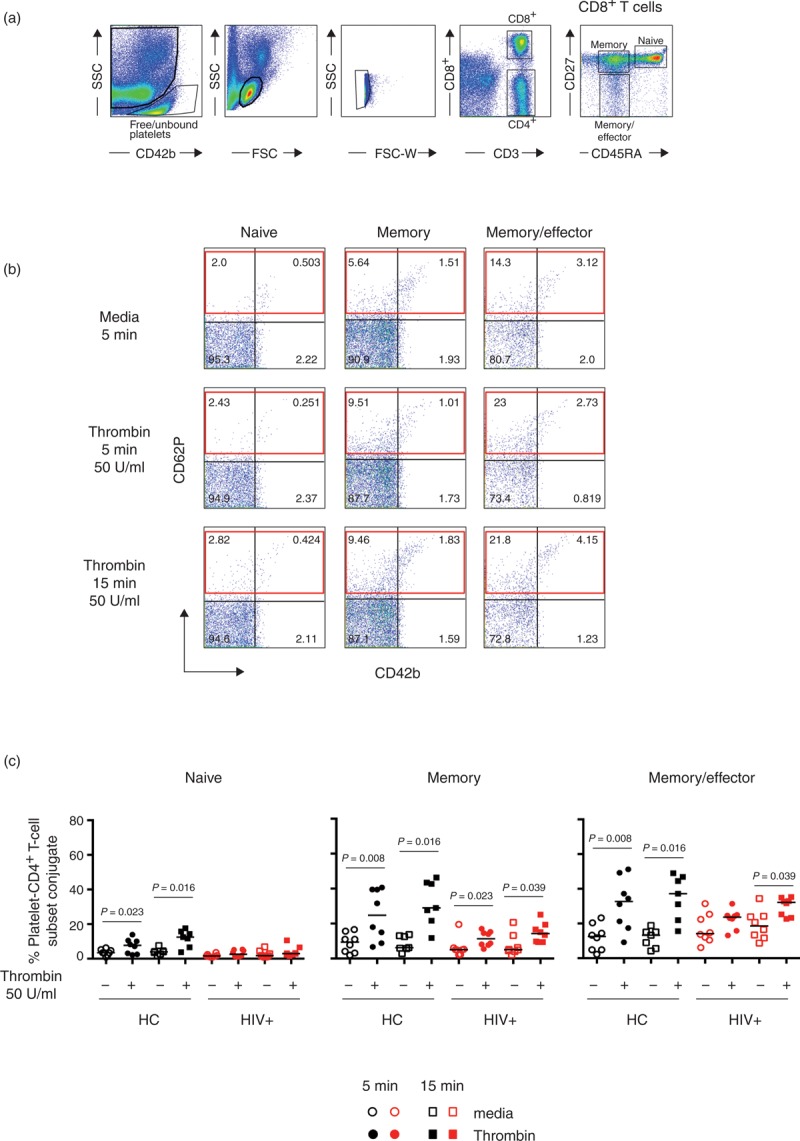
PBMCs isolated from healthy controls (n = 8) or HIV-infected patients (n = 8) were stimulated at 37°C for 5 (circle symbols) or 15 min (square symbols) in the presence or absence of thrombin (50 U/ml). (a) PBMC gating strategy for platelet–T-cell conjugate formation. (b) Representative example of the effects of thrombin stimulation on PBMCs from an HIV-infected patient. The red rectangle represents the proportion of CD4+ T cells binding platelets after thrombin activation. (c) Proportion of CD4+ T-cell subsets binding platelets after thrombin stimulation in healthy controls (solid black symbols) and HIV-infected patients (solid red symbols). Nonparametric Wilcoxon paired test was used for comparisons between unstimulated and thrombin; P < 0.05 was considered significant.
Fig. 4.
Increased platelet–T-cell conjugates after thrombin stimulation in CD8+ T-cell subsets.
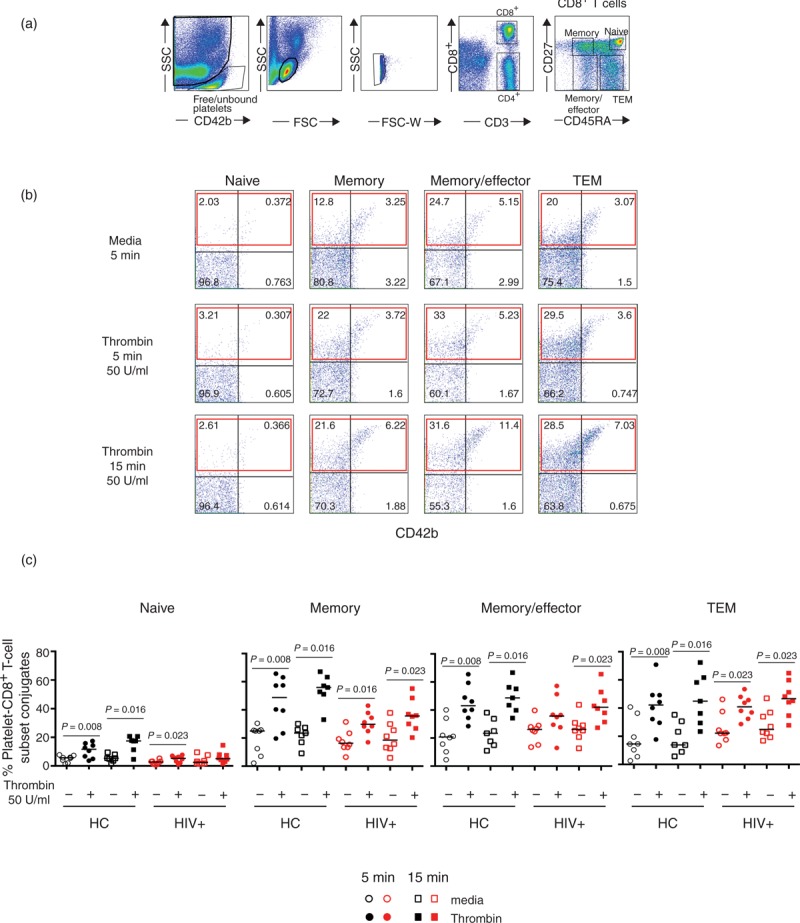
PBMCs isolated from healthy controls (n = 8) or HIV-infected patients (n = 8) were stimulated at 37°C for 5 (circle symbols) or 15 min (square symbols) in the presence or absence of thrombin (50 U/ml). (a) PBMC gating strategy for platelet–T-cell conjugate formation. (b) Representative example of the effects of thrombin stimulation on PBMCs from an HIV-infected patient. The red rectangle represents the proportion of CD8+ T cells binding platelets after thrombin activation. (c) Proportion of CD8+ T-cell subsets binding platelets after thrombin stimulation in healthy controls (solid black symbols) and HIV-infected patients (solid red symbols). Nonparametric Wilcoxon paired test was used for comparisons between unstimulated and thrombin; P < 0.05 was considered significant.
In-vitro thrombin stimulation enhances formation of platelet–CD4+ and platelet–CD8+ T-cell conjugates
To directly visualize whether thrombin activation of platelets leads to an increase in platelet binding to T lymphocytes, we used an image stream technology that combined high-resolution microscopy in flow (Fig. 5 and Supplementary Figure S5). PBMCs from healthy controls (n = 10) and HIV infected patients (n = 5) were cultured in vitro with media or thrombin. After 15 min stimulation, there was an increase in the binding of CD42b+CD62P+ platelets to both CD4+ and CD8+ T cells (Supplementary Figure S5A, dot plot pink gate). Overlaid images of 10 representative platelet–T-cell conjugates stimulated with thrombin showed that platelets bound to T cells undergo a visible change in shape and upregulation of CD62P upon activation (Fig. 5a and Supplementary Figure S5). An increased proportion of activated platelets–CD4+ and CD8+ T cell conjugates formation was observed in healthy controls (P = 0.01). A similar trend was observed in the HIV patients (Fig. 5b). The fraction of CD42b+CD62P- bound platelets to T cells observed in the PBMCs cultured in media (Supplementary Figure S5A, orange gate) was significantly reduced in both CD4+ (P = 0.004) and CD8+ T cells (P = 0.002) after thrombin stimulation in healthy controls but did not reach significance in HIV-infected patients (data not shown). These observations suggest that the platelets bound to lymphocytes in steady-state conditions are able to respond to thrombin stimulation.
Fig. 5.
Thrombin enhances formation of platelet–CD4+ and platelet–CD8+ T-cell conjugates.
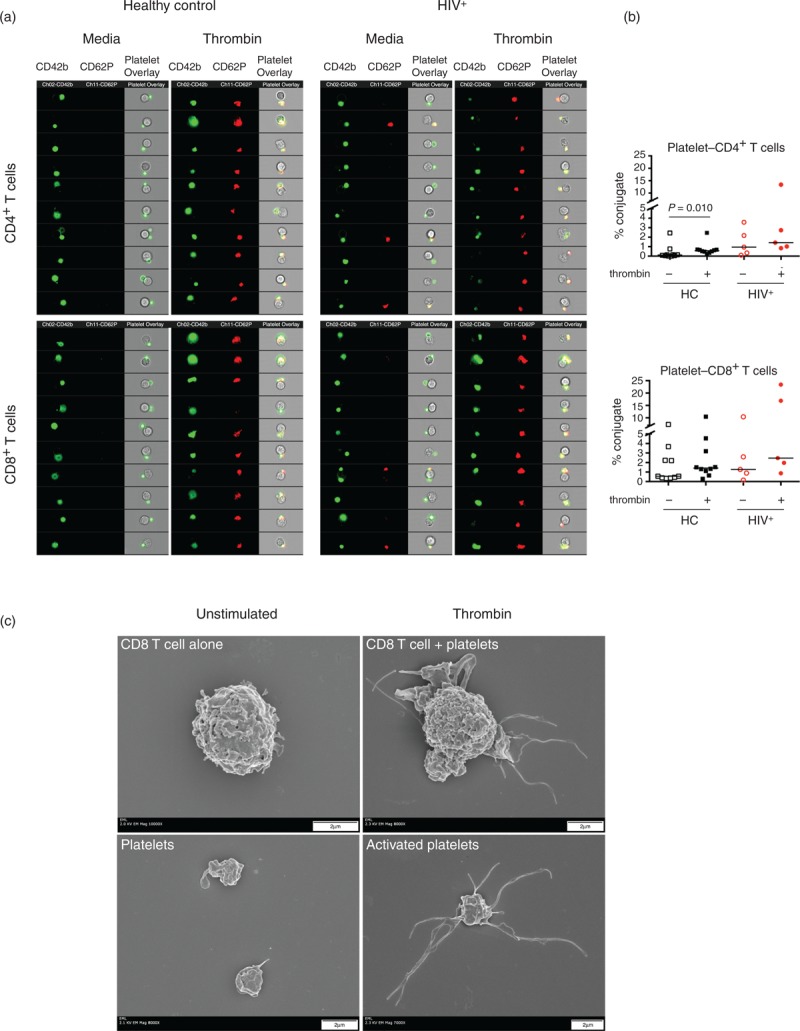
PBMCs from healthy controls (n = 10) and HIV-infected patients (n = 5) were stimulated in the presence or absence of thrombin (50 U/ml) for 15 min. Cells were stained with CD3, CD4, CD42b and CD62P mAbs and fixed prior to acquisition in ImageStream (Amnis). (a) Representative analysis of samples from a healthy control and HIV-infected patient. Samples were pregated in CD4+ and CD8+ T cells as described in Supplementary Figure S5. Overlay of the brightfield morphology, CD62P and CD42b channels in 10 representative platelet–T-cell conjugates. (b) Percentage of platelet–CD4+ and platelet–CD8+ T-cell conjugates before and after thrombin stimulation. (c) Sorted memory (CD45RA-CD27+) CD8+ T cells were cocultured at 1 : 1 ratio of autologous platelets and activated during 15 min with 50 U/ml of thrombin. Scanning electron microscope images of CD8+ memory T cells and platelets unstimulated (left) and stimulated (right).
We next tested whether sorted memory CD8+ T cells cocultured with platelets (1 : 1 ratio) were able to form conjugates and the role of thrombin activation in this process using a direct visualization by scanning electron microscopy (Fig. 5c). We found that thrombin-activated platelets underwent a drastic change in shape (right lower panel, Fig. 5c). In addition, several activated platelets were able to bind an individual memory CD8+ T cells (right upper panel, Fig. 5c).
These data suggest that, in vivo, thrombin-induced platelet activation enhances the recruitment of memory T cells to the place of injury and thus platelets act as ‘lymphocyte chaperone’. These results support a mechanism in which in-vivo circulating platelet–lymphocyte conjugates can facilitate migration of memory CD8+ T cells to the place of tissue insult.
Discussion
Patients with HIV infection, despite virologic suppression, are at an increased risk of developing serious non-AIDS defining illnesses. The risk of developing these complications is associated with elevated serum biomarkers of coagulation/inflammation such as D-dimer and IL-6 [6,30,31]. The mechanisms by which HIV infection maintains higher levels of coagulation/inflammation are not totally understood. In the present manuscript, we investigated the interaction between a component of the coagulation system, platelets and the adaptive immune system, T cells. We found that platelets can form conjugates with T cells in peripheral blood from healthy controls and HIV-infected patients. Platelets bound preferentially to those subsets of CD4+ and CD8+ T cells that had a more differentiated phenotype (memory, memory/effector and TEM). In contrast to healthy controls, HIV-infected patients showed an increase in the proportion of T cells that formed conjugates with activated platelets.
Platelets play a role in very early steps of response to an injury by trauma or infection. Their high numbers in circulation compared with that of circulating leukocytes and their ability to release mediators (pro-inflammatory and anti-inflammatory) stored in specialized secretory granules suggest that platelets are critical players in the early phase of the host response [32,33]. Platelets are known to be involved in functions beyond haemostasis and have been identified as regulators of both innate and adaptive immune systems [34–36]. In the immune system, one of these functions is to promote trafficking of immune cells into injured tissues by upregulation of CD62P leading to enhanced rolling of leukocytes along vascular endothelium. Thus, activated platelets mediate the interaction between the endothelium and circulating immune cells and can participate in the pathology of the disease during chronic inflammatory conditions such as atherosclerosis, sepsis and rheumatoid arthritis [37–39]. Interestingly, circulating platelet-monocytes conjugates have been found to be a clinical predictor of myocardial infarction [21,40]. During HIV/SIV infection, circulating platelet-monocyte conjugates have been observed. Platelet-monocyte complexes have been suggested to play a role in the thrombocytopenia observed during acute SIV infection in Pig-tailed Macaques [25].
Upon activation, platelets can shed PMP, which are small fragments (size 0.1–1 μm) of plasma membrane. PMP express receptors and cytoplasmic contents, which have been suggested to function as a transcellular delivery system [41,42]. An increased proportion of activated platelets and PMP have been observed in some patients during HIV infection [15]. In addition, circulating platelet-monocyte complexes in patients with HIV infection have been proposed to play a role in neuroinflammation [13,43]. Platelet–monocyte complexes may mediate adherence to the brain microvascular endothelial cells promoting extravasation of platelet-monocytes complexes in the central nervous system (CNS) of HIV-infected patients with associated encephalitis [43]. Our data support a model in which platelets can also mediate the trafficking of cells of the adaptive immune system. In the context of HIV infection, the increased proportion of activated platelets–T-cell conjugate suggest that these cells are more prone to traffic into inflamed tissues harbouring HIV replication. In the present study, activation of platelets by thrombin led to enhanced formation of platelet–T-cell conjugates and this interaction was mediated at least in part by CD62P. Interestingly, HIV-infected patients tended to show lower upregulation of CD62P+ than healthy controls after thrombin stimulation; this could reflect the in-vivo activation status of HIV-infected patients [14]. It is possible that these activated platelets–T-cell conjugates represent a fraction of more easily recruited cells from the adaptive immune system into peripheral tissues. Although all T-cell subsets express CD62P ligand (PSGL1), the binding affinity of this interaction is determined by the proper glycosylation of PSGL1, which is mediated by glycosytransferases and tyrosine sulphotransferases. The expression of these enzymes is inducible and regulated during T-cell activation [27,44,45]. Accordantly, we found that those subsets with memory, memory/effector and TEM phenotype had higher affinity for recombinant CD62P-Fc than naive T cells, suggesting that these subsets may express the enzymes involved in the glycosylation of PSGL1. In addition, CD4+ and CD8+ T-cell expressing the activation markers HLADR+CD38+ showed higher affinity for CD62P binding. The higher level of immune activation (HLADR+CD38+) in CD8+ T and CD4+ T cells during chronic HIV infection may enhance the formation of these conjugates and in doing so maintain a state of inflammation by increasing T-cell trafficking through inflamed tissues. It is still to be determined whether this pathway regulates in-situ T-cell function, as has been suggested in patients with rheumatoid arthritis, leading to inefficient viral elimination and perpetuating inflammation [18]. In addition, one could consider that recruitment of memory CD4+ T cells to places of ‘ongoing viral replication’ might perpetuate continuous inflammation and facilitate infection of CD4+ T cells. Therefore, it is important to better understand the ways in which platelets can modulate T-cell function.
The present findings propose a link between an arm of the adaptive immune system (T cells) and the coagulation system (platelets). This interface may play an important role in host defense and in the chronic inflammatory diseases associated with HIV infection. Studies designed to block platelet activation may provide insights about the in-vivo mechanisms maintaining higher levels of coagulation/inflammation in these patients.
Acknowledgements
We would like to thank the patients of the NIAID HIV-Clinic and the healthy controls of the NIH Blood bank for their participation in this study.
Leidos Biomedical Research Inc has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the U.S. Government. This research was supported [in part] by the National Institute of Allergy and Infectious Disease.
S.A.G. performed the experiments, analysed and interpreted data and wrote the manuscript. M.S., R.B.H., D.S., A.H., K.N., A.R., S.A. and S.P. performed the experiments. T.I. critically reviewed the manuscript. A.O. helped with patient recruitment. J.Q. performed statistical analysis of the data. H.C.L. interpreted the data and critically reviewed the article. M.C. contributed to design of the study and analysed/interpreted the data and wrote the manuscript.
Conflicts of interest
There are no conflicts of interest.
Supplementary Material
References
- 1.Catalfamo M, Di Mascio M, Hu Z, Srinivasula S, Thaker V, Adelsberger J, et al. HIV infection-associated immune activation occurs by two distinct pathways that differentially affect CD4 and CD8 T cells. Proc Natl Acad Sci U S A 2008; 105:19851–19856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Catalfamo M, Wilhelm C, Tcheung L, Proschan M, Friesen T, Park JH, et al. CD4 and CD8 T cell immune activation during chronic HIV infection: roles of homeostasis, HIV, type I IFN, and IL-7. J Immunol 2011; 186:2106–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee SA, Sinclair E, Jain V, Huang Y, Epling L, Van Natta M, et al. Low proportions of CD28- CD8+ T cells expressing CD57 can be reversed by early ART initiation and predict mortality in treated HIV infection. J Infect Dis 2014; 210:374–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serrano-Villar S, Sainz T, Lee SA, Hunt PW, Sinclair E, Shacklett BL, et al. HIV-infected individuals with low CD4/CD8 ratio despite effective antiretroviral therapy exhibit altered T cell subsets, heightened CD8+ T cell activation, and increased risk of non-AIDS morbidity and mortality. PLoS Pathog 2014; 10:e1004078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Z, Cumberland WG, Hultin LE, Prince HE, Detels R, Giorgi JV. Elevated CD38 antigen expression on CD8+ T cells is a stronger marker for the risk of chronic HIV disease progression to AIDS and death in the Multicenter AIDS Cohort Study than CD4+ cell count, soluble immune activation markers, or combinations of HLA-DR and CD38 expression. J Acquir Immune Defic Syndr Hum Retrovirol 1997; 16:83–92. [DOI] [PubMed] [Google Scholar]
- 6.Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med 2008; 5:e203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker JV, Neuhaus J, Duprez D, Kuller LH, Tracy R, Belloso WH, et al. Changes in inflammatory and coagulation biomarkers: a randomized comparison of immediate versus deferred antiretroviral therapy in patients with HIV infection. J Acquir Immune Defic Syndr 2011; 56:36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deeks SG, Lewin SR, Havlir DV. The end of AIDS: HIV infection as a chronic disease. Lancet 2013; 382:1525–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duprez DA, Neuhaus J, Kuller LH, Tracy R, Belloso W, De Wit S, et al. Inflammation, coagulation and cardiovascular disease in HIV-infected individuals. PLoS One 2012; 7:e44454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levi M, van der Poll T, Buller HR. Bidirectional relation between inflammation and coagulation. Circulation 2004; 109:2698–2704. [DOI] [PubMed] [Google Scholar]
- 11.Funderburg NT, Mayne E, Sieg SF, Asaad R, Jiang W, Kalinowska M, et al. Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation. Blood 2010; 115:161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hurley A, Smith M, Karpova T, Hasley RB, Belkina N, Shaw S, et al. Enhanced effector function of CD8(+) T cells from healthy controls and HIV-infected patients occurs through thrombin activation of protease-activated receptor 1. J Infect Dis 2013; 207:638–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh MV, Davidson DC, Kiebala M, Maggirwar SB. Detection of circulating platelet-monocyte complexes in persons infected with human immunodeficiency virus type-1. J Virol Methods 2012; 181:170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holme PA, Muller F, Solum NO, Brosstad F, Froland SS, Aukrust P. Enhanced activation of platelets with abnormal release of RANTES in human immunodeficiency virus type 1 infection. FASEB J 1998; 12:79–89. [DOI] [PubMed] [Google Scholar]
- 15.Mayne E, Funderburg NT, Sieg SF, Asaad R, Kalinowska M, Rodriguez B, et al. Increased platelet and microparticle activation in HIV infection: upregulation of P-selectin and tissue factor expression. J Acquir Immune Defic Syndr 2012; 59:340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rinder HM, Bonan JL, Rinder CS, Ault KA, Smith BR. Dynamics of leukocyte-platelet adhesion in whole blood. Blood 1991; 78:1730–1737. [PubMed] [Google Scholar]
- 17.de Bruijne-Admiraal LG, Modderman PW, Von dem Borne AE, Sonnenberg A. P-selectin mediates Ca(2+)-dependent adhesion of activated platelets to many different types of leukocytes: detection by flow cytometry. Blood 1992; 80:134–142. [PubMed] [Google Scholar]
- 18.Zamora C, Canto E, Nieto JC, Ortiz MA, Diaz-Torne C, Diaz-Lopez C, et al. Functional consequences of platelet binding to T lymphocytes in inflammation. J Leukoc Biol 2013; 94:521–529. [DOI] [PubMed] [Google Scholar]
- 19.Li N, Ji Q, Hjemdahl P. Platelet-lymphocyte conjugation differs between lymphocyte subpopulations. J Thromb Haemost 2006; 4:874–881. [DOI] [PubMed] [Google Scholar]
- 20.Sarma J, Laan CA, Alam S, Jha A, Fox KA, Dransfield I. Increased platelet binding to circulating monocytes in acute coronary syndromes. Circulation 2002; 105:2166–2171. [DOI] [PubMed] [Google Scholar]
- 21.Michelson AD, Barnard MR, Krueger LA, Valeri CR, Furman MI. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation 2001; 104:1533–1537. [DOI] [PubMed] [Google Scholar]
- 22.Harding SA, Sommerfield AJ, Sarma J, Twomey PJ, Newby DE, Frier BM, et al. Increased CD40 ligand and platelet-monocyte aggregates in patients with type 1 diabetes mellitus. Atherosclerosis 2004; 176:321–325. [DOI] [PubMed] [Google Scholar]
- 23.Joseph JE, Harrison P, Mackie IJ, Isenberg DA, Machin SJ. Increased circulating platelet-leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br J Haematol 2001; 115:451–459. [DOI] [PubMed] [Google Scholar]
- 24.Ashman N, Macey MG, Fan SL, Azam U, Yaqoob MM. Increased platelet-monocyte aggregates and cardiovascular disease in end-stage renal failure patients. Nephrol Dial Transplant 2003; 18:2088–2096. [DOI] [PubMed] [Google Scholar]
- 25.Metcalf Pate KA, Lyons CE, Dorsey JL, Shirk EN, Queen SE, Adams RJ, et al. Platelet activation and platelet-monocyte aggregate formation contribute to decreased platelet count during acute simian immunodeficiency virus infection in pig-tailed macaques. J Infect Dis 2013; 208:874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandrea I, Cornell E, Wilson C, Ribeiro RM, Ma D, Kristoff J, et al. Coagulation biomarkers predict disease progression in SIV-infected nonhuman primates. Blood 2012; 120:1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ley K, Kansas GS. Selectins in T-cell recruitment to nonlymphoid tissues and sites of inflammation. Nat Rev Immunol 2004; 4:325–335. [DOI] [PubMed] [Google Scholar]
- 28.Diacovo TG, Catalina MD, Siegelman MH, von Andrian UH. Circulating activated platelets reconstitute lymphocyte homing and immunity in L-selectin-deficient mice. J Exp Med 1998; 187:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michelson AD, Ellis PA, Barnard MR, Matic GB, Viles AF, Kestin AS. Downregulation of the platelet surface glycoprotein Ib-IX complex in whole blood stimulated by thrombin, adenosine diphosphate, or an in vivo wound. Blood 1991; 77:770–779. [PubMed] [Google Scholar]
- 30.Lundgren JD, Baxter J, Deeks SG, Lane HC. Biomarkers in HIV disease. Curr Opin HIV AIDS 2010; 5:459–462. [DOI] [PubMed] [Google Scholar]
- 31.Baker JV, Neuhaus J, Duprez D, Kuller LH, Tracy R, Belloso WH, et al. Changes in inflammatory and coagulation biomarkers: a randomized comparison of immediate versus deferred antiretroviral therapy in patients with HIV infection. J Acquir Immune Defic Syndr 2011; 56:36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coppinger JA, O’Connor R, Wynne K, Flanagan M, Sullivan M, Maguire PB, et al. Moderation of the platelet releasate response by aspirin. Blood 2007; 109:4786–4792. [DOI] [PubMed] [Google Scholar]
- 33.Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol 2011; 11:264–274. [DOI] [PubMed] [Google Scholar]
- 34.Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998; 391:591–594. [DOI] [PubMed] [Google Scholar]
- 35.Iannacone M, Sitia G, Isogawa M, Marchese P, Castro MG, Lowenstein PR, et al. Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat Med 2005; 11:1167–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shiraki R, Inoue N, Kawasaki S, Takei A, Kadotani M, Ohnishi Y, et al. Expression of Toll-like receptors on human platelets. Thromb Res 2004; 113:379–385. [DOI] [PubMed] [Google Scholar]
- 37.Burger PC, Wagner DD. Platelet P-selectin facilitates atherosclerotic lesion development. Blood 2003; 101:2661–2666. [DOI] [PubMed] [Google Scholar]
- 38.Wagner DD, Burger PC. Platelets in inflammation and thrombosis. Arterioscler Thromb Vasc Biol 2003; 23:2131–2137. [DOI] [PubMed] [Google Scholar]
- 39.Knijff-Dutmer EA, Koerts J, Nieuwland R, Kalsbeek-Batenburg EM, van de Laar MA. Elevated levels of platelet microparticles are associated with disease activity in rheumatoid arthritis. Arthritis Rheum 2002; 46:1498–1503. [DOI] [PubMed] [Google Scholar]
- 40.Furman MI, Barnard MR, Krueger LA, Fox ML, Shilale EA, Lessard DM, et al. Circulating monocyte-platelet aggregates are an early marker of acute myocardial infarction. J Am Coll Cardiol 2001; 38:1002–1006. [DOI] [PubMed] [Google Scholar]
- 41.George JN, Thoi LL, McManus LM, Reimann TA. Isolation of human platelet membrane microparticles from plasma and serum. Blood 1982; 60:834–840. [PubMed] [Google Scholar]
- 42.George JN, Pickett EB, Heinz R. Platelet membrane microparticles in blood bank fresh frozen plasma and cryoprecipitate. Blood 1986; 68:307–309. [PubMed] [Google Scholar]
- 43.Singh MV, Davidson DC, Jackson JW, Singh VB, Silva J, Ramirez SH, et al. Characterization of platelet-monocyte complexes in HIV-1-infected individuals: possible role in HIV-associated neuroinflammation. J Immunol 2014; 192:4674–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nolz JC, Harty JT. IL-15 regulates memory CD8+ T cell O-glycan synthesis and affects trafficking. J Clin Invest 2014; 124:1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moore KL, Thompson LF. P-selectin (CD62) binds to subpopulations of human memory T lymphocytes and natural killer cells. Biochem Biophys Res Commun 1992; 186:173–181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.