

Abstract
Head and neck rhabdomyosarcoma (HNRMS) is exceedingly rare and poorly documented. The difficult diagnosis often causes a poor prognosis and high mortality. Hence, we report 4 cases of HNRMS and their follow-up outcomes, and review the clinicopathological features of this rare tumor. The 4 patients ranged in age from 5 to 29 years. Among them, 3 patients had a good prognosis after combination of radiotherapy and chemotherapy or surgery alone. Another patient survived for only 3 months after diagnosis without therapy. Deeply insight into HNRMS might improve the ability of diagnosis and treatment for this disease.
Keywords: Rhabdomyosarcoma, head and neck, follow-up
Introduction
Rhabdomyosarcoma (RMS) is an aggressive soft tissue sarcoma originating from immature striated skeletal muscle [1]. The most common sites include head and neck, genitourinary system and limb-sruncal [2,3]. Although the head and neck region is the mainly primary site for rhabdomyosarcoma, head and neck rhabdomyosarcoma (HNRMS) is very rare [4]. Head and neck sarcoma represented only 1% of all primary tumors arising within the head and neck region, and accounted for 4-10% of all sarcomas [5]. HNRMS is classified into four major histologic subtypes: embryonal, alveolar, pleomorphic, and spindle cell/sclerosing [6].
Due to nonspecific initial manifestations and histological misinterpretation of microscopic finding, diagnosis of HNRMS is often made late in its course. At present, immunohistochemistry, computed tomography (CT), magnetic resonance image (MRI), positron emission tomography-CT (PET-CT), electron microscopy and molecular genetic studies are major avenues of the diagnosis of HNRMS. Modern modalities of combined surgery, radiotherapy and chemotherapy have significantly improved the survival rate of patients with this tumor [7]. However, patients with HNRMS often have a poor prognosis because of late diagnosis, metastasis, inoperability and local recurrence. Therefore, we collected 4 cases of HNRMS in order to improve our recognition on this tumor and avoid misdiagnosis and mistreatment during clinical practice.
Case collections
Case 1
A 23-year-old male presented with a chief complaint of right ear tinnitus in September 2009. Two months later, electronic nasopharyngoscopy showed a nasopharyngeal mass in right lateral wall (Figure 1), and the first nasopharyngeal biopsy revealed chronic mucosal inflammation. Then a month later, the patient suffered from right eye ptosis and diplopia. Besides, his right eye had only partial motility in upgaze and downgaze, with limited ability of adduction and abduction. The second electronic nasopharyngoscopy revealed a larger right nasopharyngeal mass (Figure 2). Head and neck MRI (Figure 3) showed a large neoplasm (5.3 cm × 3.7 cm × 4.4 cm) in right and posterior walls of nasopharynx with the destruction of skull base bone and right parapharyngeal space. However, there was no evidence of intracranial extension. In addition, enlarged bilateral cervical and submandibular lymph nodes (1 cm × 1 cm × 0.8 cm) were found on MRI.
Figure 1.
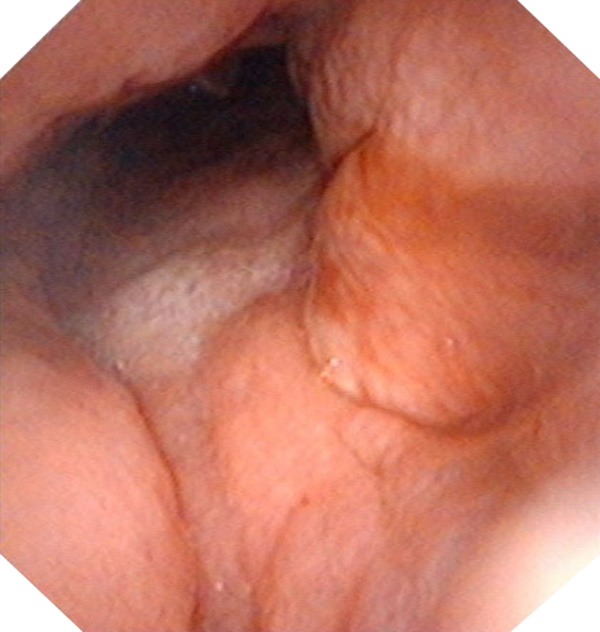
The first electronic nasopharyngoscopy revealed a neoplasm in the right nasopharynx.
Figure 2.
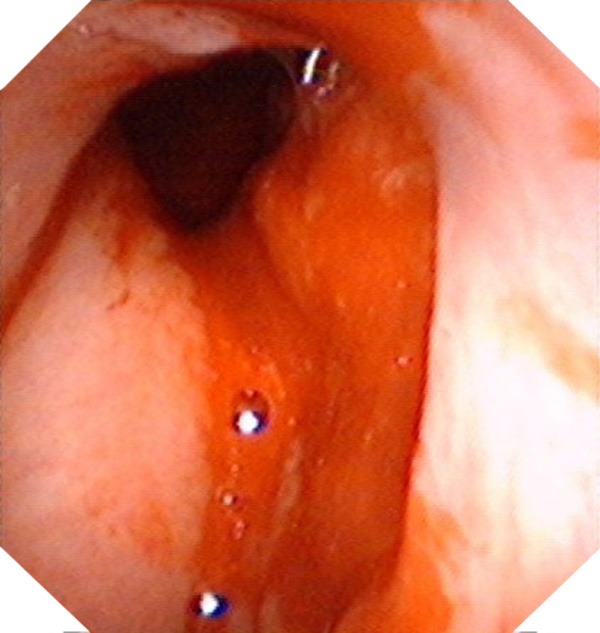
The second electronic nasopharyngoscopy revealed a larger neoplasm in the right nasopharynx than three months before.
Figure 3.
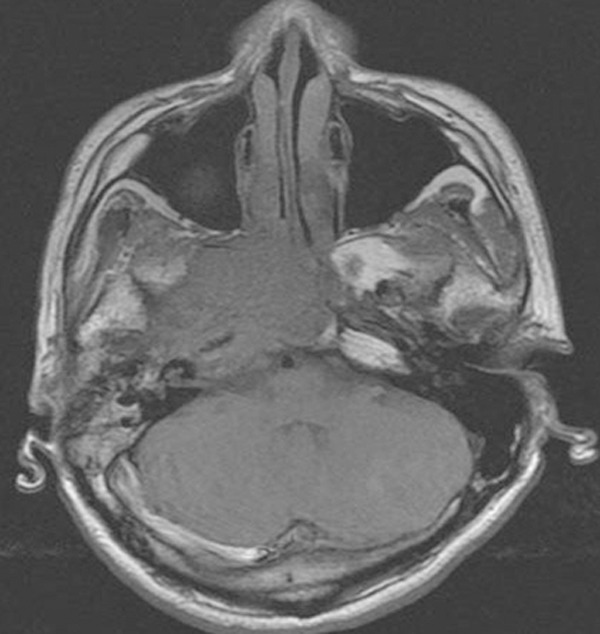
Head and neck MRI showed a large mass (5.3 cm × 3.7 cm × 4.4 cm) in the right nasopharynx extending into skull base and right parapharyngeal space.
The second biopsy (Figure 2) revealed embryonal RMS with an admixture of undifferentiated, small, round or spindle-shaped cells with variable numbers of strap-shaped or tadpole-shaped eosinophilic cells (Figure 4). Immunostaining was positive for MyoD1 (Figure 5). Further investigations were carried out and didn’t detect any distant metastasis. Staging was performed and the tumor was grouped stage III [8,9]. Due to be unfit for operation, it was decided to treat the patient with combination of radiotherapy and chemotherapy. Chemotherapy was started along with radiotherapy. The radiotherapy consisted of a daily dose of 240 cGy and a total dose of 6960 cGy in 29 fractions. Four cycles of chemotherapy with vincristine (1.4 mg/m2), adriamycin (100 mg/m2) and cyclophosphamide (600 mg/m2) every 4 weeks were given with granulocyte colony-stimulatory factor (G-CSF) 5 mcg/kg/d support. The entire course of treatment was completed within 4 months, and the tumor size obviously reduced on MRI (Figure 6). Moreover, the symptoms of ptosis and limited motility in right eye were relieved. After 5.3 years of follow-up, electronic nasopharyngoscopy (Figure 7), head and neck MRI (Figure 8), and other relative examinations revealed neither local recurrence nor distant metastasis.
Figure 4.
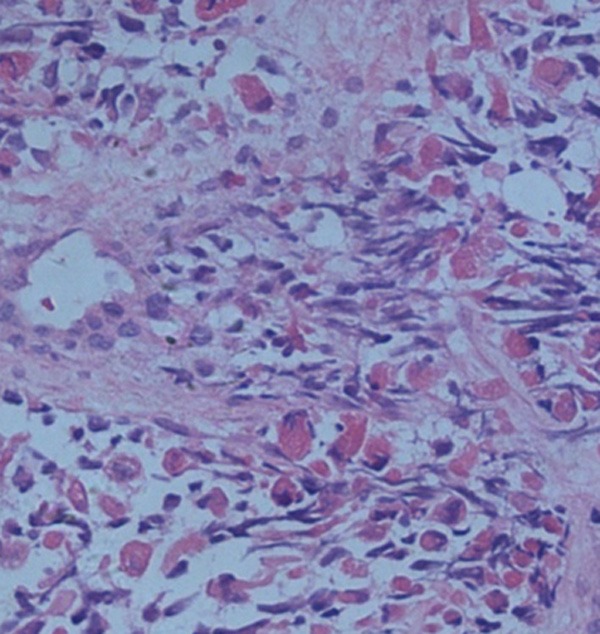
Histopathological staining showed blue round cells arranged around the vessel (hematoxylin and eosin stain, × 100).
Figure 5.
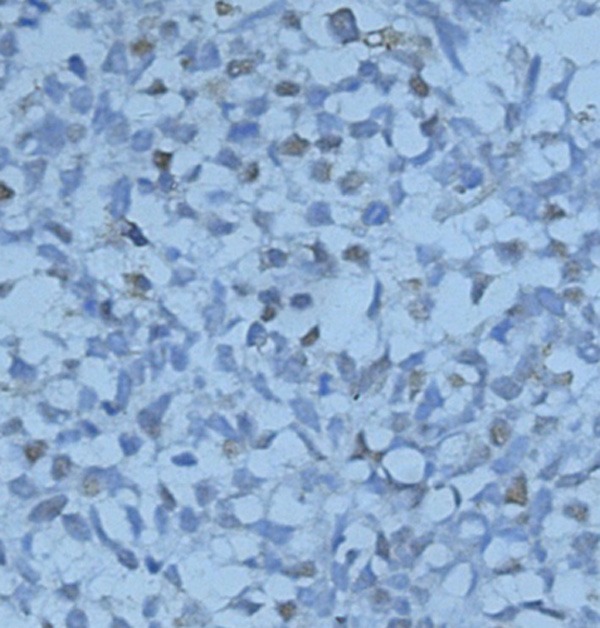
Immunohistochemical staining was positive for MyoD1 which was compatible with a diagnosis of embryonal RMS (× 100).
Figure 6.
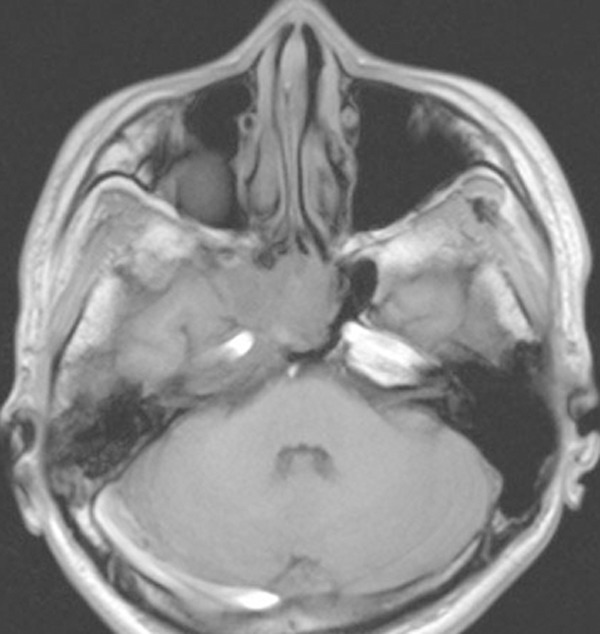
Head and neck MRI confirmed significant decreasing in the tumor size 4 months later after therapy.
Figure 7.
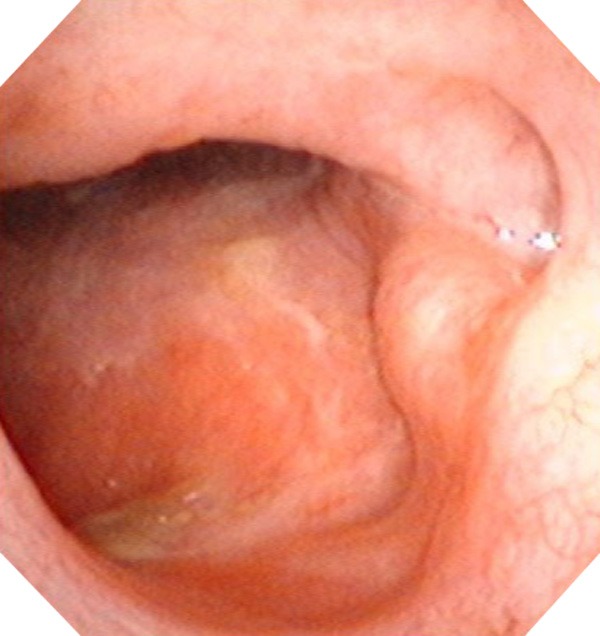
The electronic nasopharyngoscopy showed no neoplasm in the right nasopharynx 5.3 years later after therapy.
Figure 8.
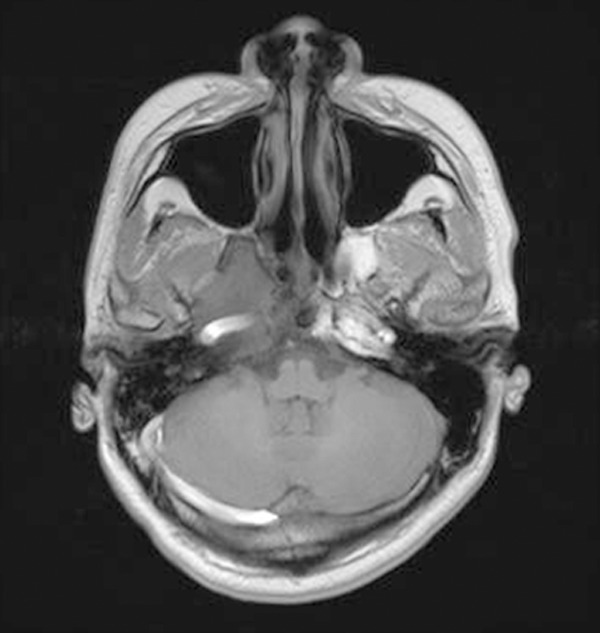
Head and neck MRI exhibited stable tumor control 5.3 years later after therapy.
Case 2
A 25-year-old man referred to our department in November 2012 with symptoms of left nasal congestion, yellow discharge and blurred vision. MRI showed a solid mass centered in the left parapharyngeal space, left ethmoid sinus, left superior and middle meatus. Biopsy was performed and histopathologic examination showed the typical features of embryonal RMS. Immunostaining was positive for Desmin and CD56. After diagnosed, the patient gave up therapy because of poor economic condition. Unfortunately, the man lost his young life 3 months later.
Case 3
A previously well, 5-year-old girl presented in October 2007 with a painless swelling in the right cheek for one year. Physical examination found a round, firm mass measuring approximately 1 cm × 1 cm × 0.8 cm in her right cheek. Palpation of the neck was negative for any cervical lymphadenopathy. Imaging studies indicated no regional or distant spread. Shortly afterward, she underwent surgical resection. Postoperative pathological results revealed embryonal RMS. Immunostaining was positive for Myogenin (Myf4), Calponin, CD10, Desmin, Actin and Vimentin. The patient is now in persistent complete remission 7.2 years after resection without evidence of metastasis or recurrence.
Case 4
A 29-year-old female exhibited a painless right facial swelling during one year with rapid evolution, who was admitted to our hospital in April 2002. CT found a mass measuring approximately 10 cm × 9 cm × 8 cm in the right cheek without evidence of metastasis. The following workup revealed no local or distant spread. The tumor was removed completely and the surgical margins were tumor-free. Due to a huge skin defect after removing the mass, we repaired widespread traumatic soft tissue defects on her face with free latissimus dorsi muscle-skin flap. The preoperative and postoperative information showed in serial photographs (Figures 9, 10, 11 and 12). Postoperative pathological results revealed embryonal RMS with positive immunostaining for Desmin and MyoD1. During her 12.8-year follow-up visit, there was no obvious evidence of recurrence or metastasis.
Figure 9.
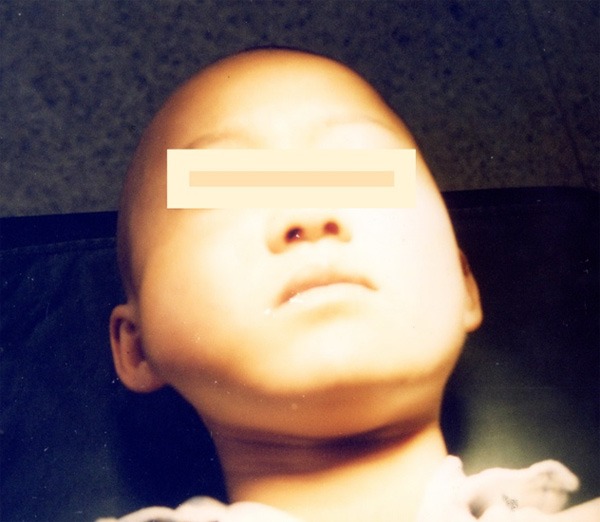
Diffuse swelling on the right cheek.
Figure 10.
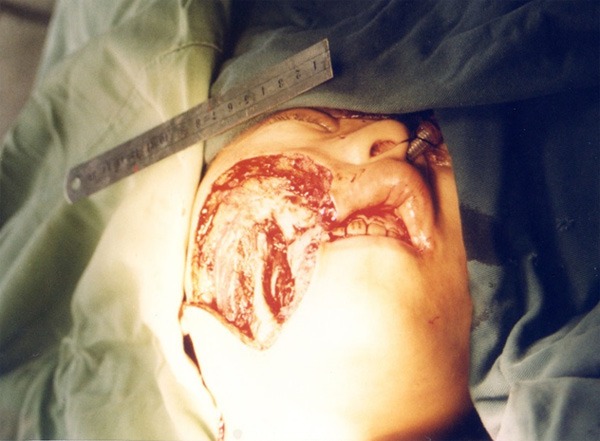
Huge skin defect after removing the mass.
Figure 11.
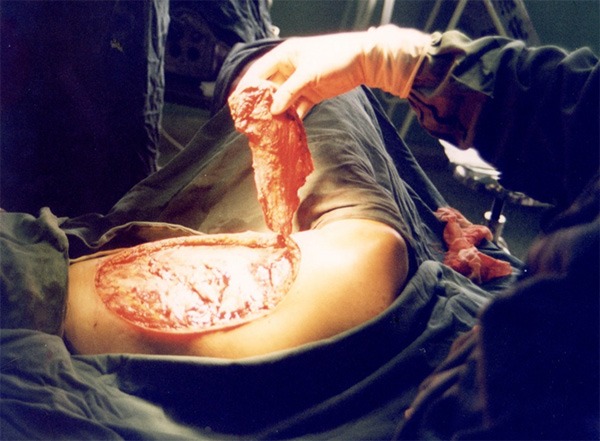
Repairing widespread traumatic soft tissue defects with free latissimus dorsi muscle-skin flap.
Figure 12.
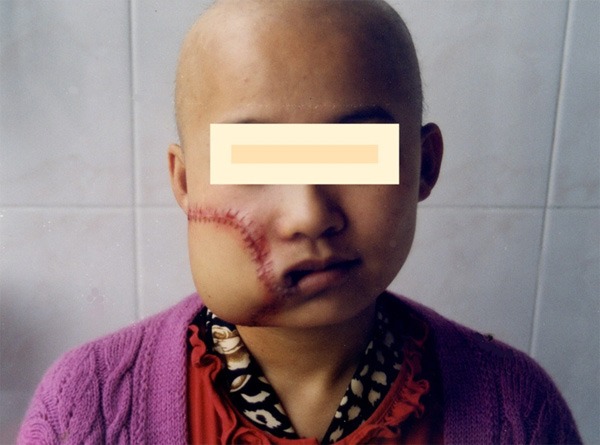
Ten days after operation.
Discussion
RMS is the most frequent soft tissue malignancy in children and adolescents but infrequent in adults. Although these tumors can arise almost anywhere, the most common locations for these tumors to develop are in the structures of the head and neck (nearly 40% of all cases), the genitourinary tract (20%), the extremities (20%) and other locations (20%) [2,10]. Usually, pediatric RMS occurs with a predilection in the head and neck sites, whereas adult RMS occurs predominantly in the extremities. The annual incidence of RMS is 4.5 cases per million children/adolescents in the United States. Between 1975 and 2005, the incidence of embryonal RMS was stable, whereas a significant increase in the incidence of alveolar RMS was observed with an annual percentage change of 4.20% [11,12]. The incidence of HNRMS was increased significantly with an annual percentage change of 1.16% [13]. It is largely thought that RMS arises because of regulatory disruption of skeletal muscle progenitor cell growth and differentiation [3]. More and more studies have suggested the distinct genetic factors were involved in the development and tumor progression of RMS, such as loss of heterozygosity, specific chromosomal translocations, abnormal gene alterations [3,14,15]. Although the overwhelming majority of cases of RMS occur sporadically, between 10-33% of children who develop RMS are thought to have an underlying genetic risk factor [16].
At present, histopathologic test and immunohistochemical staining are the most reliable method of diagnosis for RMS. In immunohistochemical tests, the RMS tumor cells can express Desmin, Myogenin, CD56, muscle-specific actin, Myoglobin, Vimentin and MyoD1, and so on. Differential diagnosis includes Ewing’s sarcoma, neuroblastoma, primitive neuroectodermal tumor, malignant lymphoma, nasopharyngeal carcinoma, desmoplastic small round cell tumor, or chordocarcinoma, etc. Keeping the differential diagnosis of a head and neck mass and specific clinicopathology characteristic in mind may lead us to avoid the misdiagnosis and mistreatment of RMS patients.
Moreover, genetic assessment currently appears to be a more meaningful endeavor for clinical management than histologic classification. Categorization of future therapeutic trials will depend on genetic testing for selecting high-risk patients [17-19].
In addition, better definition of the stage of RMS, as important as the deeply mastering of treatment strategy might bring us an expectation to prolong the survival time of patients with the tumor. Staging definitions were determined using the Intergroup RMS Study (IRS) TNM classification for pretreatment clinical assessment of disease, and IRS group was assigned at diagnosis according to the extent of residual tumor after initial surgery [8,9].
RMS can spread locally, regionally, or distantly. The commonest site of metastatic spread was the lungs, followed by the bone marrow, lymph node, and bones. All patients with alveolar subtype had metastasis at time of diagnosis while only 31% of embryonal subtype had metastasis [20]. At least 15% of children and adolescents with RMS present with distant metastases [21,22]. Based on HNRMS in children, metastatic disease at diagnosis (33% of all cases) occurred in the bone marrow (11%), cerebrospinal fluid (6%), peritoneal fluid (6%), lung (4%), parietal pleura (2%), pleural fluid (2%) and pericardial fluid (2%) [10]. For HNRMS in adults, the rate of cervical lymph node involvement was 28% [7].
In order to evaluation the prognostic factors of RMS, the combination of stage, group, site, size, age, histologic subtype, and the presence or absence of regional nodes or distant metastases is used to stratify patient into one of four ‘risk-groups’ (Low A, Low B, Intermediate, High) [17,23]. These risk groups provide important information about the potential curability of the tumor with treatments of lesser or greater intensity.
The treatment of patients with RMS is multi-disciplinary and multimodal. New avenues into biologically-based treatments are being gained. Highly specific small molecule tyrosine kinase inhibitors targeted against the insulin-like growth factor-1 receptor tyrosine kinase have been synthesized and shown to inhibit tumor xenograft growth, both alone and in combination with cytotoxic chemotherapy [24]. Because of the presence of the unique, tumor-cell specific ‘translocation’ gene in cases of alveolar RMS, the potential exists to utilize immune-based therapies to recognize and kill cells that contain this abnormal gene. New therapeutic strategies based on targeted immunotherapy are therefore much in demand. The epidermal growth factor receptor has all the characteristics of an ideal target [25]. Recently, clinical report showed successful treatment of metastatic RMS with radiochemotherapy and allogeneic hematopoietic stem cell transplantation. Allogeneic hematopoietic stem cell transplantation for RMS should be a novel promising therapeutic means method in future [26].
A multicenter study showed that the overall survival (OS) rate of RMS was 56.9% ± 8.4 and the failure free survival rate was 68.3% ± 7.6 [20]. In patients with localized disease, overall 5-year survival rates have improved to more than 80% with the combined use of surgery, radiotherapy, and chemotherapy. However, in patients with metastatic disease, 5-year event-free survival rate less than 30% [20,27]. In children and adolescents with RMS, the 5-year OS rate was 64.5% and the 10-year OS was 61.8% for an entire cohort [28].
For HNRMS, the OS rate was 28.7% in 5 and 10 years, and p53 expression may be related with poor prognosis [29], while relative 5-year survival was statistically unchanged during the study period at 62.8% ± 2.3 [13]. For pediatric patients with HNRMS, total survival rate was 86.5%, complete remission rate was 67.6%, partial remission rate was 18.9%, and recurrence rate was 18.9% [30]. However, the estimated 5-year OS rate was 36% for HNRMS in adults [7].
Acknowledgements
This work was supported by the Science and Technology Project of Xiamen Science and Technology Bureau (No. 3502Z20134019) and the Natural Science Fund of Fujian Province (No. 2013D003), China.
Disclosure of conflict of interest
None.
References
- 1.Goosens V, Van den Berghe I, De Clercq C, Casselman J. Radiation-induced mandibular adult spindle cell rhabdomyosarcoma. Int J Oral Maxillofac Surg. 2008;37:395–7. doi: 10.1016/j.ijom.2007.09.173. [DOI] [PubMed] [Google Scholar]
- 2.Paulino AC, Okcu MF. Rhabdomyosarcoma. Curr Probl Cancer. 2008;32:7–34. doi: 10.1016/j.currproblcancer.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Dasgupta R, Rodeberg DA. Update on rhabdomyosarcoma. Semin Pediatr Surg. 2012;21:68–78. doi: 10.1053/j.sempedsurg.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Zhu J, Zhang J, Tang G, Hu S, Zhou G, Liu Y, Dai L, Wang Z. Computed tomography and magnetic resonance imaging observations of rhabdomyosarcoma in the head and neck. Oncol Lett. 2014;8:155–60. doi: 10.3892/ol.2014.2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barosa J, Ribeiro J, Afonso L, Fernandes J, Monteiro E. Head and neck sarcoma: analysis of 29 cases. Eur Ann Otorhinolaryngol Head Neck Dis. 2014;131:83–6. doi: 10.1016/j.anorl.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 6.Hisaoka M, Nishio J. Elastofibroma. In: Fletcher CD, Bridge JA, Hogendoorn PC, Mertens F, editors. WHO Classification of Tumours of Soft Tissue and Bone. 4th edition. Lyon: IARC Press; 2013. pp. 53–54. [Google Scholar]
- 7.Wu Y, Li C, Zhong Y, Guo W, Ren G. Head and neck rhabdomyosarcoma in adults. J Craniofac Surg. 2014;25:922–5. doi: 10.1097/SCS.0000000000000704. [DOI] [PubMed] [Google Scholar]
- 8.Raney RB, Maurer HM, Anderson JR, Andrassy RJ, Donaldson SS, Qualman SJ, Wharam MD, Wiener ES, Crist WM. The Intergroup Rhabdomyosarcoma Study Group (IRSG): major lessons from the IRS-I through IRS-IV studies as background for the current IRS-V treatment protocols. Sarcoma. 2001;5:9–15. doi: 10.1080/13577140120048890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lawrence W Jr, Anderson JR, Gehan EA, Maurer H. Pretreatment TNM staging of childhood rhabdomyosarcoma: a report of the Intergroup Rhabdomyosarcoma Study Group. Children’s Cancer Study Group. Pediatric Oncology Group. Cancer. 1997;80:1165–70. [PubMed] [Google Scholar]
- 10.Hicks J, Flaitz C. Rhabdomyosarcoma of the head and neck in children. Oral Oncol. 2002;38:450–9. doi: 10.1016/s1368-8375(01)00105-1. [DOI] [PubMed] [Google Scholar]
- 11.Ognjanovic S, Linabery AM, Charbonneau B, Ross JA. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975-2005. Cancer. 2009;115:4218–26. doi: 10.1002/cncr.24465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Egas-Bejar D, Huh WW. Rhabdomyosarcoma in adolescent and young adult patients: current perspectives. Adolesc Health Med Ther. 2014;5:115–25. doi: 10.2147/AHMT.S44582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turner JH, Richmon JD. Head and neck rhabdomyosarcoma: a critical analysis of population-based incidence and survival data. Otolaryngol Head Neck Surg. 2011;145:967–73. doi: 10.1177/0194599811417063. [DOI] [PubMed] [Google Scholar]
- 14.Rezvani G, Lui JC, Barnes KM, Baron J. A set of imprinted genes required for normal body growth also promotes growth of rhabdomyosarcoma cells. Pediatr Res. 2012;71:32–8. doi: 10.1038/pr.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Megiorni F, Cialfi S, McDowell HP, Felsani A, Camero S, Guffanti A, Pizer B, Clerico A, De Grazia A, Pizzuti A, Moles A, Dominici C. Deep Sequencing the microRNA profile in rhabdomyosarcoma reveals down-regulation of miR-378 family members. BMC Cancer. 2014;14:880. doi: 10.1186/1471-2407-14-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartley AL, Birch JM, Blair V, Kelsey AM, Harris M, Jones PH. Patterns of cancer in the families of children with soft tissue sarcoma. Cancer. 1993;72:923–30. doi: 10.1002/1097-0142(19930801)72:3<923::aid-cncr2820720343>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 17.Parham DM, Barr FG. Classification of rhabdomyosarcoma and its molecular basis. Adv Anat Pathol. 2013;20:387–97. doi: 10.1097/PAP.0b013e3182a92d0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rudzinski ER. Histology and fusion status in rhabdomyosarcoma. Am Soc Clin Oncol Educ Book. 2013:425–8. doi: 10.14694/EdBook_AM.2013.33.425. [DOI] [PubMed] [Google Scholar]
- 19.Rekhi B, Singhvi T. Histopathological, immunohistochemical and molecular cytogenetic analysis of 21 spindle cell/sclerosing rhabdomyosarcomas. APMIS. 2014;122:1144–52. doi: 10.1111/apm.12272. [DOI] [PubMed] [Google Scholar]
- 20.Badr MA, Al-Tonbary YA, Mansour AK, Hassan TH, Beshir MR, Darwish A, El-Ashry RA. Epidemiological characteristics and survival studies of rhabdomyosarcoma in East egypt: a five-year multicenter study. ISRN Oncol. 2012;2012:674523. doi: 10.5402/2012/674523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oberlin O, Rey A, Lyden E, Bisogno G, Stevens MC, Meyer WH, Carli M, Anderson JR. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J. Clin. Oncol. 2008;26:2384–9. doi: 10.1200/JCO.2007.14.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiss AR, Lyden ER, Anderson JR, Hawkins DS, Spunt SL, Walterhouse DO, Wolden SL, Parham DM, Rodeberg DA, Kao SC, Womer RB. Histologic and clinical characteristics can guide staging evaluations for children and adolescents with rhabdomyosarcoma: a report from the Children’s Oncology Group Soft Tissue Sarcoma Committee. J. Clin. Oncol. 2013;31:3226–32. doi: 10.1200/JCO.2012.44.6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hosoi H, Teramukai S, Matsumoto Y, Tsuchiya K, Iehara T, Hara J, Mitsui T, Kaneko M, Hatae Y, Hayashi Y, Mabuchi O, Adachi N, Morikawa Y, Nishimura S, Kumagai M, Takamatsu H, Sawada T, Sugimoto T. A review of 331 rhabdomyosarcoma cases in patients treated between 1991 and 2002 in Japan. Int J Clin Oncol. 2007;12:137–45. doi: 10.1007/s10147-006-0638-6. [DOI] [PubMed] [Google Scholar]
- 24.Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Shringarpure R, Akiyama M, Hideshima T, Chauhan D, Joseph M, Libermann TA, García-Echeverría C, Pearson MA, Hofmann F, Anderson KC, Kung AL. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell. 2004;5:221–30. doi: 10.1016/s1535-6108(04)00050-9. [DOI] [PubMed] [Google Scholar]
- 25.Niesen J, Brehm H, Stein C, Berges N, Pardo A, Fischer R, Ten Haaf A, Gattenlöhner S, Tur MK, Barth S. In vitro effects and ex vivo binding of an EGFR-specific immunotoxin on rhabdomyosarcoma cells. J Cancer Res Clin Oncol. 2014;141:1049–61. doi: 10.1007/s00432-014-1884-z. [DOI] [PubMed] [Google Scholar]
- 26.Yamazaki F, Osumi T, Shigematsu N, Morioka H, Shimada H. Successful treatment of metastatic rhabdomyosarcoma with radiochemotherapy and allogeneic hematopoietic stem cell transplantation. Jpn J Clin Oncol. 2015;45:225–8. doi: 10.1093/jjco/hyu189. [DOI] [PubMed] [Google Scholar]
- 27.McDowell HP, Foot AB, Ellershaw C, Machin D, Giraud C, Bergeron C. Outcomes in paediatric metastatic rhabdomyosarcoma: results of The International Society of Paediatric Oncology (SIOP) study MMT-98. Eur J Cancer. 2010;46:1588–95. doi: 10.1016/j.ejca.2010.02.051. [DOI] [PubMed] [Google Scholar]
- 28.Yang L, Takimoto T, Fujimoto J. Prognostic model for predicting overall survival in children and adolescents with rhabdomyosarcoma. BMC Cancer. 2014;14:654. doi: 10.1186/1471-2407-14-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andrade CR, Takahama Junior A, Nishimoto IN, Kowalski LP, Lopes MA. Rhabdomyosarcoma of the head and neck: a clinicopathological and immunohistochemical analysis of 29 cases. Braz Dent J. 2010;21:68–73. doi: 10.1590/s0103-64402010000100011. [DOI] [PubMed] [Google Scholar]
- 30.Zhang WL, Zhang Y, Huang DS, Guo F, Han T, Hong L, Hu HM, Zhi T. Clinical character of pediatric head and neck rhabdomysarcomas: a 7-year retrospective study. Asian Pac J Cancer Prev. 2013;14:4089–93. doi: 10.7314/apjcp.2013.14.7.4089. [DOI] [PubMed] [Google Scholar]