

Abstract
MicroRNA-22 (miR-22) was previously reported to elicit cardiac myocyte hypertrophy and had an anti-apoptotic effect on neurons. However, its effects on cardiac myocyte apoptosis and cardiac function during ischemia and reperfusion (I/R) are not clear. In the present study, we demonstrate that pre-administration of miR-22 mimic reduced I/R-induced cardiac dysfunction significantly in a rat model. We found that miR-22 overexpression inhibited cardiac myocyte apoptosis, and reduced cardiac remodeling during I/R. Significant cardiac myocyte apoptosis was also observed in a cardiac myocyte model after hypoxia/reoxygenation (H/R), a representative process of I/R. Further experiments showed that eNOS activity and the following NO production were significantly decreased during I/R and H/R, while such decrease was inhibited by overexpression of miR-22. Mechanistically, overexpression of miR-22 had little effect on the total protein level of eNOS, but restored the level of p-eNOS (Ser1177) which was down-regulated during H/R. Further RT-PCR results demonstrated that Caveolin 3 (Cav3), an upstream negative regulator of eNOS, was upregulated during H/R, resulting in a decrease of p-eNOS. However, such upregulation of Cav3 transcript level was inhibited directly by miR-22 during H/R, leading to a restored p-eNOS level and followed NO production in cardiac myocytes. Together, the present study revealed that miR-22 down-regulated Cav3, leading to restored eNOS activity and NO production, which further inhibited cardiac myocyte apoptosis and promoted cardiac function after I/R. Of clinical interest, the present study may highlight miR-22 as a potential therapeutic agent for reducing I/R induced cardiac injury.
Keywords: microRNA-22, cardiac myocyte, ischemia and reperfusion, caveolin-3, eNO
Introduction
Ischemia and reperfusion (I/R) induced cardiac injury contributes to morbidity and mortality in a wide range of pathologies, including cardiac remodeling, cardiac arrest, cardiac arrhythmias, dilated cardiomyopathy, heart failure, and so forth. The injury is due to numerous pathological processes happening during I/R, for example, intracellular accumulation of H+ and Ca2+ as well as disruption of mitochondrial membrane potential, formation of free radicals or reactive oxygen species (ROS), and subsequent activation of pro-inflammatory pathways [1]. At the cellular level, I/R may lead to cardiac myocyte death in myocardium, which contributes to cardiac dysfunction [2]. Despite significant advances in laboratory researches and clinical trials in the past decade, the underlying mechanisms of I/R induced cardiac injury are not yet fully understood. Further research is indispensable to reduce I/R induced cardiac injury and to improve the cardiac prognosis in clinical practice.
MicroRNAs (miRs) are a class of small (~22 nt) phylogenetically conserved noncoding single-stranded RNA molecules. They downregulate gene expression via degradation or translational inhibition of their target mRNAs through direct binding to the targets with imperfect or perfect complement [3]. There are numerous reports demonstrating that microRNAs are involved in cardiac development and different types of cardiac diseases [3]. For example, miR-1 and miR-499 promote differentiation of cardiac progenitor myocytes into cardiac myocytes while suppress proliferation of the progenitor cells [4]. Cardiac myocyte apoptosis is inhibited by overexpression of miR-1, miR-21, miR-24, or miR-499, and is promoted by overexpression of miR-22 [5-13]. miR-1, miR-21, miR-494, and miR-210 are involved in cardiac inflammation [7,14-16]. Overexpression of miR-133 or miR-208 may result in cardiac hypertrophy and dilated cardiomyopathy [17-20]. However, effects of microRNAs during I/R remain to be further investigated.
Of interest, miR-22 was reported to induce cardiac myocyte hypertrophy and it was a key regulator of stress-induced cardiac hypertrophy and in ventricular remolding [21]. Targeted deletion of miR-22 promoted stress-induced cardiac dilation and contractile dysfunction [22]. However, its effect on cardiac myocytes during I/R is not very clear. Meanwhile, it was reported that overexpression of miR-22 had a neuroprotective effect through reduction in caspase activation [23,24]. Its anti-apoptotic effect on neurons may suggest miR-22 has a similar effect on some other types of cells, including cardiac myocytes. Cardiac myocytes have a pivotal role in cardiac function at the cellular level, and a putative anti-apoptotic effect of miR-22 on cardiac myocytes may propose that it is an important effector during I/R induced cardiac injury.
There exists an overproduction of reactive oxygen species (ROS) and a concomitant reduction in NO during I/R [1]. Overproduction of ROS, namely oxidative stress, causes direct damages to cellular DNA, proteins, and lipids in addition to its effects in inflammation [1]. NO can scavenge ROS and reduce detrimental effects of ROS [25-27]. Regulation of ROS and NO balance is important for reduction of cardiac damage during I/R. eNOS is constitutively expressed in cardiac myocytes and enriched in caveolae of cardiac myocytes [28-30]. Caveolins, including caveolin-3 (Cav3), are upstream negative regulators of eNOS [28,29]. Cav3 is the major type of the caveolin family proteins in cardiac myocytes and may form a complex with eNOS, resulting in eNOS inactivity [28-32]. After stimulation, eNOS is released and activated after phosphorylated at serine 1177 (p-eNOS), catalyzing the conversion of L-arginine to L-citrulline and NO [28]. It is found that there exists a putative binding site for miR-22 in the 3’-UTR of Cav3 mRNA predicted by TargetScan 6.0 [22]. Based on its putative binding activity with Cav3 mRNA and anti-apoptotic effect, we hypothesize that miR-22 inhibits Cav3 expression, further promotes eNOS activity and the followed NO production, which may prevent cardiac myocyte apoptosis and reduce I/R induced cardiac injury. In the study, effects of miR-22 on cardiac myocytes and cardiac function are investigated in a rat model of I/R injury as well as in a cell model of hypoxia/reoxygenation (H/R) damage.
Materials and methods
I/R model
Adult male Wistar rats weighing 250-280 g were used in the I/R model in the study. Briefly, an ischemia was simulated by ligation at the left anterior descending coronary artery (LAD). Male Wistar rats were subjected to 30 minutes of myocardial ischemia and 24 hours of reperfusion. 24 rats were randomized into three groups: (1) the sham-operated group (sham group, n=8), the rats underwent sham operation without coronary artery ligation; (2) the I/R group (n=8), where the rats were treated with ischemia for 30 minutes; and (3) the I/R+miR-22 group (n=8); 24 hours before the left anterior descending coronary artery ligation, the rats were given an intramyocardial injection of 2.0 μg of miR-22 mimic.
At the end of 24-hour reperfusion, rats were re-anesthetized and cardiac function was determined by echocardiography in terms of left ventricular end systolic volume (LVESV), left ventricular ejection fraction (%EF), and left ventricular fractional shortening (%FS). After completion of functional determination, the ligature around the left anterior descending coronary artery was retied before the rats were scarified for further examinations on the hearts. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of The Second People’s Hospital of Hefei. The IACUC committee members at The Second People’s Hospital of Hefei approved this study. All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Isolation and primary culture of cardiac myocytes
For primary culture of cardiac myocytes, previous routine protocols were adopted. Briefly, adult male Wistar rats weighing 250-280 g were sacrificed and the heart was quickly removed from the chest and retrogradely aortic perfused at constant pressure (100 cm H2O) at 37°C for 3 minutes with a Ca2+-free PBS solution and 5% taurine (Sigma), gassed with 95% O2-5% CO2. The enzymatic digestion was conducted with collagenase type B (0.5 mg/ml; Life Technologies, USA), collagenase type D (0.5 mg/ml; Life Technologies), and protease type XIV (0.02 mg/ml; Life Technologies). When the heart became swollen and hard after 3 minutes of digestion, 50 μM Ca2+ was added to the enzyme solution. About 7 min later, the heart was cut into chunks, and further digested in a shaker (60-70 rpm) for 10 minutes at 37°C in the same enzyme solution. The supernatant containing the dispersed myocytes was filtered into a sterilized tube and gently centrifuged at 500 rpm for 1 minute before resuspended in PBS solution with 250 μM Ca2+. After pelleted by gravity for 15 minutes, the cardiac myocytes were resuspended in PBS solution with 250 μM Ca2+. The final cell pellet was suspended in PBS with 500 μM Ca2+ for further culture. Meanwhile, the shake-harvest procedure was repeated 3-4 times until all the chunks were digested. Isolated cardiac myocytes were stored in HEPES solution (pH 7.4) consisting of (in mM) 1 CaCl2, 137 NaCl, 5.4 KCl, 15 dextrose, 1.3 MgSO4, 1.2 NaH2PO4, and 20 HEPES.
For primary culture, freshly isolated cardiac myocytes were suspended in minimal essential medium (MEM; Sigma) containing 1.2 mM Ca2+, 2.5% FBS (GIBCO), and 1% PS (pH 7.4). After pelleted by gravity for 15 minutes, the cardiac myocytes were washed 3 more times. The cardiac myocytes were then plated at 0.5-1 × 104 cells/cm2 in MEM containing 2.5% FBS and 1% PS. After 1 hour of culture in a 5% CO2 incubator at 37°C, the medium was changed to FBS-free MEM for further culture. And the medium was changed every 48 hours during culture. To reduce bacterial or viral contamination, all the solutions were filtered (0.2-μm filter) and equilibrated with 95% O2-5% CO2 for at least 20 minutes before use in isolation and culture of cardiac myocytes.
In vitro hypoxia/reoxygenation (H/R) model
To simulate in vivo I/R process, an in vitro H/R model was used in the study. Briefly, cardiac myocytes were firstly perfused in normal Hank’s solution with a gas mixture of 95% O2-5% CO2 at 37°C, pH 7.4. To simulate ischemia, the Hank’s solution was switched to pH 7.4 at 37°C without glucose or calcium (D-Hank’s solution) and then the cells were aerated with a gas mixture of 95% N2-5% CO2. To simulate reperfusion, the cells were again treated with normal Hank’s solution with a gas mixture of 95% O2-5% CO2 at 37°C, pH 7.4.
Echocardiography
Left ventricular function was assessed with two-dimensional M-mode echocardiography in short axis. Images of the left ventricle were obtained using an ultrasound unit Vivid7™ (GE, Milwaukee, WI, USA) equipped with a 13-MHz linear phase arrayed transducer (GEi13L) and an accompanying software for rodent imaging. When the image on the screen was stable for at least 10 seconds, left ventricular end systolic volume (LVESV), left ventricular ejection fraction (%EF) and left ventricular fractional shortening (%FS) were measured. Data represented means ± SD, n=8.
TUNEL assay
Transferase dUTP nick end labeling (TUNEL) apoptotic analysis was performed using an in situ cell death detection kit (Promega, Madison, WI, USA) in accordance to the manufacturer’s instructions. The enzyme TdT was applied to incorporate digoxigenin-conjugated dUTP into the ends of DNA fragments. Cells with clear nuclear labeling were defined as TUNEL-positive cells. Apoptotic cells were calculated as the percentage of TUNEL-positive cells using the following formula: number of TUNEL-positive cell nuclei/(number of TUNEL-positive cell nuclei + the number of total cell nuclei) × 100%.
Caspase-3 activity assay
Caspase-3 activity was evaluated by using a commercialized caspase-3 assay kit (Biovision Inc., USA). The heart was minced and homogenized in lysis buffer (Tris 20 mM, NaCl 50 mM, NaF 50 mM, Na4P2O7 5 mM, C12H22O11 25 mM, DTT 1 mM, Na3VO4 2 mM, and 1% protease inhibitor cocktail, pH 7.4) with a Heidolph DIA900 tissue homogenizer (Heidolph Instruments, Schwabach, Germany). The homogenate was centrifuged (12,000 g at 4°C for 15 minutes) and the supernatant was used to measure caspase-3 activity according to the manufacturer’s instructions. Briefly, an aliquot of protein (10 ml) was incubated with 10 mL of synthetic peptide substrate Ac-DEVD-pNA in a total volume of 100 ml at 37°C for 1 hour before caspase-3 activity detection. The absorbance at 405 nm of the released pNA was monitored in a spectrometer. The relative activity of caspase-3 was monitored by the rate of absorption value.
Lactate dehydrogenase (LDH) assay
After a cell dies, lactate dehydrogenase (LDH) inside the cell is released. LDH is pretty stable, and its level can be used as an indicator to determine cytotoxicity of an agent. Its detection was conducted with the LDH detection activity assay kit (Sigma) at 450 nm according to the manufacturer’s instructions.
Masson’s trichrome staining for collagen fibers
Masson’s trichrome staining was used for detection of collagen fibers in the heart in the study. The rat hearts were formalin-fixed and paraffin-embedded. Masson’s trichrome staining of the heart sections was conducted according to routine protocols. The collagen fibers are stained blue, the nuclei are stained black, and the background is stained red. The percentage of the heart fibrosis area was calculated by the ratio of the blue stained area to the total.
NO and eNOS activity assay
NO concentration and eNOS activity were measured with detection kits (Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instructions. NO concentration was determined by measuring the concentration of nitrite, a stable metabolite of NO in vitro. Briefly, rat serum or whole cell lystate supernatant was added into the reaction buffer and incubated for 10 minutes in the dark at room temperature. The resultant chromophore was determined at 540 nm using a spectrometer (SpectraMax 190, Molecular Device, USA). The nitrite concentrations in the samples were calculated according to the freshly prepared nitrite standard curve made from sodium nitrite with Krebs buffer. For eNOS activity assay, 100 μl serum or cell lysate supernatant was added into the reaction buffer. 30 minutes after incubation at 37°C, the reaction was stopped by adding a termination buffer. Formazan was the reaction product of NBT/PMS with NADPH in the presence of NO, and its level was measured at 530 nm. Results were normalized against the mean value of control and expressed as fold changes. The experiments were repeated for at least 3 independently.
Flow cytometry for cell apoptosis assay Annexin V-FITC/PI double marking
FITC-conjugated Annexin V and propidium iodide (PI) (Annexin-V-FLUOS, Boehringer Mannheim, Mannheim, Germany) were used to identify cardiac myocyte apoptosis with a flow cytometer following the manufacturer’s instructions. The result was analyzed using CELLQuest software.
Western blot
Western blot were performed according to routine protocols. Primary antibodies used were listed as below: eNOS, 1:1000 (BD Biosciences, San Jose, CA, USA); p-eNOS, 1:1000 (BD Biosciences); GAPDH, 1:2000 (Sigma). This experiment was repeated for at least three times independently.
Real-time reverse transcription polymerase chain reaction
Cav3 mRNA level was detected with RT-PCR. Total RNA was extracted from cardiac myocytes after H/R for reverse transcription using TRIzol reagent (Invitrogen, New York, NY, USA). cDNA was synthesized using a PrimeScript 1st Strand cDNA synthesis kit (TaKaRa Biotechnology, Dalian, China). Further, RT-PCR was performed in a single tube using SYBR Premix kit (TaKaRa Biotechnology) with an ABI 7300 real-time PCR machine (Life Technologies, USA).
Primers used in the experiment were as follows:
Cav3, 5’-CGG AGTGTTACTACTGGCTTCTC-3’ (forward) and 5’-GTGCGCACAAGCT GCATGGAAAC-3’ (reverse); GAPDH, 5’-GAAGGTGAAGGTCGGAGTC-3’ (forward) and 5’-GAGATGGTGATGGGATTTC-3’ (reverse).
The relative Cav3 mRNA level was calculated by normalizing the quantified cDNA transcript level to the GAPDH transcript level. Each value represents the average of at least 3 independent experiments.
Statistical analysis
Values are reported as mean ± SD (standard deviation) unless indicated otherwise. The 2-tailed Mann-Whitney U test was used for comparing 2 means (Prism, GraphPad). Values of P<0.05 were considered statistically significant.
Results
In vivo differences in left ventricular (LV) M-mode echocardiograms in short axis of three groups
Previous studies showed that miR-22 was involved in cardiac myocyte hypertrophy and left ventricular remodeling [21,22] and its overexpression inhibited neuron apoptosis [23,24]. However, its effect on cardiac myocytes and cardiac function during I/R is not yet studied. To examine its effects during I/R, a rat model was employed in the study. 24 rats were randomly and evenly divided into 3 groups: the sham group, the I/R group, and the I/R+miR-22 group. MiR-22 mimic was pre-administrated in the rat hearts in the I/R+miR-22 group 24 hours before experiments. After 30 minutes of ischemia and 24 hours of reperfusion, M-mode echocardiographic assessment was applied to detect the function of the left ventricle in all the 3 groups of rats based on left ventricular end systolic volume (LVESV), left ventricular ejection fraction (%EF), and left ventricular fractional shortening (%FS).
Compared to the sham group, the LVESV value was significantly higher in the I/R group (Figure 1B), and the %EF and %FS were significantly lower (Figure 1C and 1D) (all P<0.05). It indicated that the left ventricular function was impaired after I/R, as previously reported. However, with pre-administration of miR-22 mimic, the impaired function in the left ventricle was restored markedly, as evidenced by increased %EF and %FS and decreased LVESV, compared to the I/R group (Figure 1). In short, pre-administration of miR-22 mimic promoted the left ventricular function and reduced I/R induced cardiac injury significantly during I/R. Of note, there was no significant difference observed in the left ventricular function in all the 3 groups right before experiments.
Figure 1.
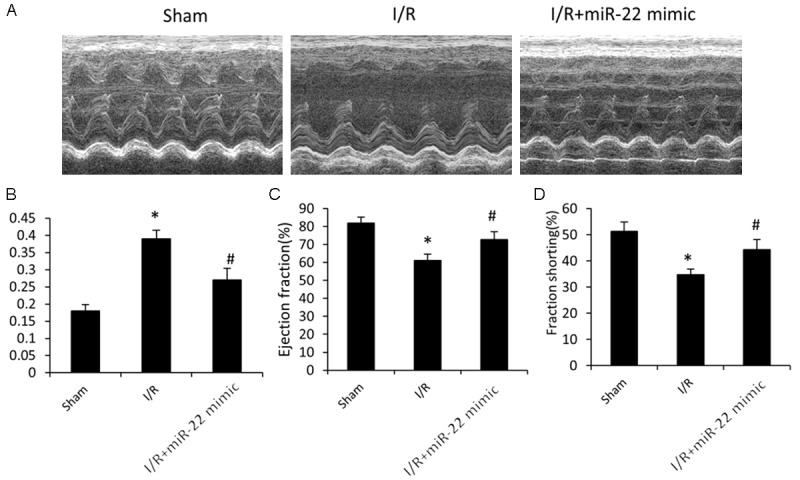
The effect of microRNA-22 on I/R induced cardiac dysfunction. A. Representative left ventricular (LV) M-mode echocardiograms in short axis. B. Left ventricular end systolic volume (LVESV). C. Left ventricular ejection fraction (%EF). D. Left ventricular fractional shortening (%FS). Data represent means ± SD, n=8. *P≤0.05 vs. sham, #P<0.05 vs. I/R.
The effect of miR-22 on I/R induced cardiac myocytes injury in vivo
After I/R, the rat hearts were excised for further examination of the effect of miR-22 on cardiac myocytes. TUNEL analysis on heart sections revealed that a large amount of cardiac myocytes died during I/R in the I/R group, compared to the sham group (Figure 2A). However, such cell death was reduced significantly in the I/R+miR-22 group with pre-administration of miR-22 mimic. Meanwhile, the caspase-3 activity was found to be increased significantly after I/R (Figure 2B), suggesting that cardiac myocytes underwent significant apoptosis during I/R. And such apoptosis was prevented markedly by miR-22 overexpression in the I/R+miR-22 group (Figure 2B).
Figure 2.
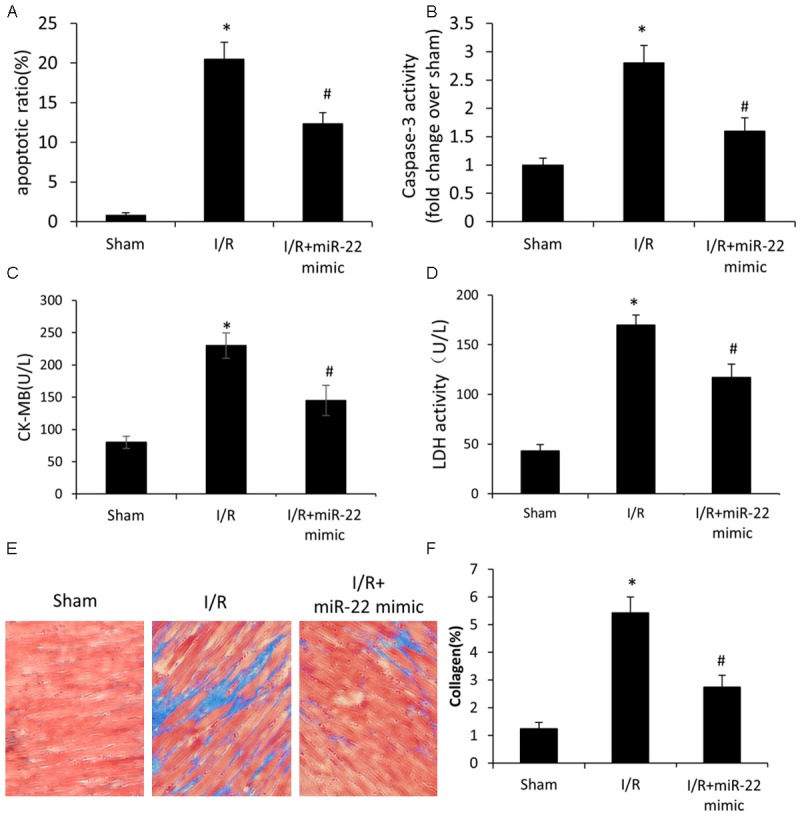
The effect of microRNA-22 on I/R induced cardiac myocytes injury in vivo. A. TUNEL analysis of cardiac myocytes apoptosis. B. Caspase-3 activity in myocardial tissues. C. ELISA analysis of the level of CK-MB in the serum. D. ELISA analysis of the activity of LDH in the serum. E. Representative histological images with Masson’s trichrome staining of heart sections. F. Quantification of collagen accumulation areas for Masson’s trichrome staining. Data represent means ± SD, n=8. *P≤0.05 vs. sham, #P<0.05 vs. I/R.
Clinically, CK-MB (creatine kinase-muscle/brain type of subunit) in the serum is a marker of myocardial infarction or severe heart damage. In the study, the level of CK-MB increased significantly during I/R (Figure 2C), and its increase was reduced in the rats pre-administrated with miR-22 mimic, which indicated that the I/R induced cardiac damage was ameliorated significantly by overexpression of miR-22. Similarly, the LDH level in the serum, a marker of cell death, increased significantly during I/R, and the increase was reduced in the I/R+miR-22 group, which suggested that cell death was inhibited by miR-22 overexpression.
Masson’s trichrome staining of heart sections showed no obvious fibrosis in the sham group. However, there was increased fibrosis observed in the I/R group (Figure 2E). The increased fibrosis was due to I/R induced cardiac injury, which was in line with previously published studies. Compared to the I/R group, late fibrotic replacement of dead cardiac myocytes was reduced in the I/R+miR-22 group (Figure 2E). Quantification of collagen accumulation areas for Masson’s trichrome staining confirmed that pre-administration of miR-22 mimic reduced cell death and cardiac remodeling significantly during I/R (Figure 2F).
Together, our results demonstrated that overexpression of miR-22 significantly reduced cardiac myocyte apoptosis during I/R.
Effect of miR-22 on H/R induced cardiac myocyte injury in vitro
We further employed an in vitro cardiac myocyte model of H/R damage. The effect of miR-22 on cardiac myocyte apoptosis during H/R was examined with flow cytometry. During apoptosis, phosphatidylserine (PS) translocation in the cell membrane is assumed to be an early feature of apoptosis, and Annexin V has the ability to bind to the translocated PS on the cell membrane during the early apoptotic stage. Propidium iodide (PI) can bind to DNA in the middle and late stage of apoptosis when cell membrane and nucleus membrane are permeable. Therefore, FITC-conjugated Annexin V and PI were used to identify apoptotic cells during H/R in the study. Experimental results showed that cardiac myocyte apoptosis was induced after H/R (Figure 3A) and such induction was significant (Figure 3B). Overexpression of miR-22 showed little effect on cardiac myocyte apoptosis under normal condition (without H/R), compared to the control (Figure 3A and 3B). However, the induced apoptosis after H/R was suppressed by overexpression of miR-22 significantly (Figure 3A and 3B). Together, overexpression of miR-22 suppressed cardiac myocyte apoptosis during H/R. The inhibitory effect on cardiac myocyte apoptosis of miR-22 was further confirmed by LDH assay (Figure 3C) and caspase-3 activity assay (Figure 3D), where overexpression of miR-22 inhibited the increased LDH level and caspase-3 activity (Figure 3D) induced by H/R.
Figure 3.
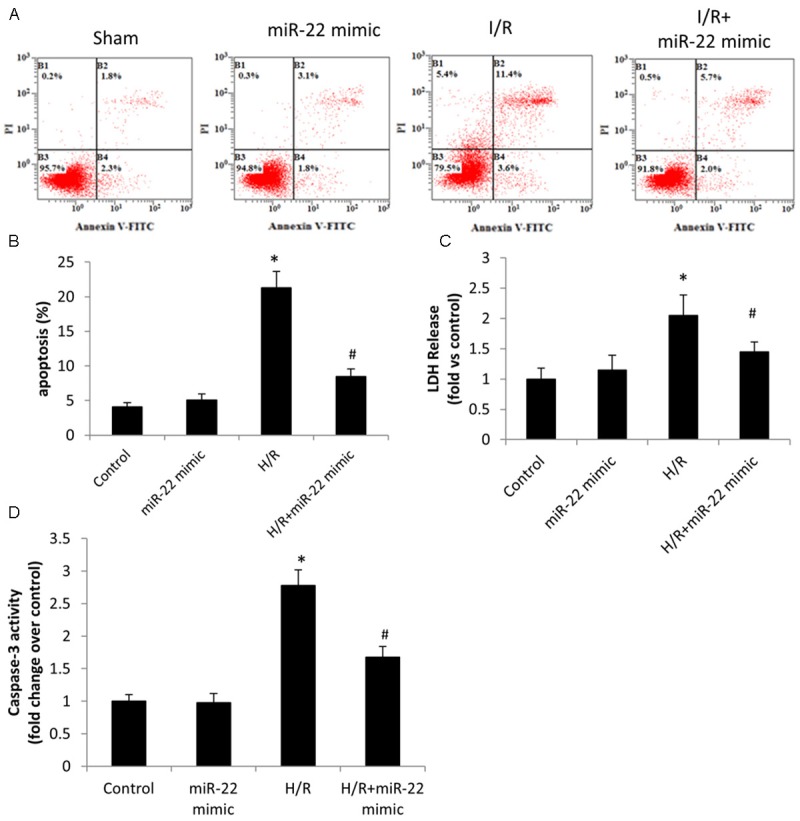
The effect of microRNA-22 on H/R induced cardiac myocytes injury in vitro. A. The cells were stained with propidium iodide and annexin V for flow cytometry and analyzed for apoptosis using CELLQuest software. B. The analysis of cell apoptosis in each groups. C. The level of LDH release. D. Caspase-3 activity in cardiac myocytes. Data were presented as mean ± S.E.M. *P≤0.05 vs. control, #P<0.05 vs. H/R.
Effect of miR-22 on I/R induced NO production and eNOS activity
Oxidative stress plays an important role in I/R induced cardiac injury. A severe oxidative stress is detrimental, for example it may induce inflammation and elicit cell apoptosis. Serum eNOS activity and NO level in rats were examined after I/R. eNOS activity decreased during I/R in the I/R group, compared to the sham group (Figure 4B). And such decrease was restored significantly by overexpression of miR-22 in the I/R+miR22 group. Meanwhile, NO production exhibited the same phenomenon with or without overexpression of miR-22 during I/R (Figure 4A). It was not surprising since eNOS catalyzed the conversion of L-arginine to L-citrulline and NO. In short, overexpression of miR-22 restored eNOS activity and the followed NO production which was reduced during I/R.
Figure 4.
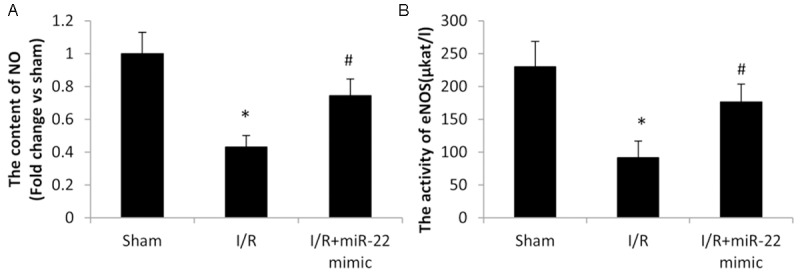
The effect of microRNA-22 on I/R induced NO production and eNOS activity. A. Serum NO was detected by commercial NO Detection Kit. B. Serum eNOS was detected by commercial eNOS Detection Kit. Data represent means ± SD, n=8. *P≤0.05 vs. sham, #P<0.05 vs. I/R.
Cav3/eNOS pathway accounts for the regulatory mechanism for the role of miR-22 in cardiac myocyte
To further investigate the underlying mechanism for the cardiac-protective effect of miR-22, the in vitro cardiac myocyte model of H/R was further used. It was shown that NO production decreased significantly in cardiac myocytes after H/R (Figure 5A) and such decrease was suppressed by overexpression of miR-22, whereas miR-22 overexpression showed little effect on NO production under normal condition (without H/R) (Figure 5A). Western blot analysis further showed that the eNOS protein level did not change noticeably after H/R, and its level was not affected by overexpression of miR-22 either (Figure 5B). However, the p-eNOS level decreased dramatically after H/R and such dramatic decrease was restored significantly by transfection of miR-22 mimic. p-eNOS is active, catalyzing the conversion of L-arginine to L-citrulline and NO, while dephosphorylated eNOS is inactive. Together, the p-eNOS level decreased significantly after H/R, which resulted in decreased eNOS activity and followed NO production (Figure 5A and 5B). However, such decrease in p-eNOS and followed NO production was restored significantly by overexpression of miR-22 (Figure 5B).
Figure 5.
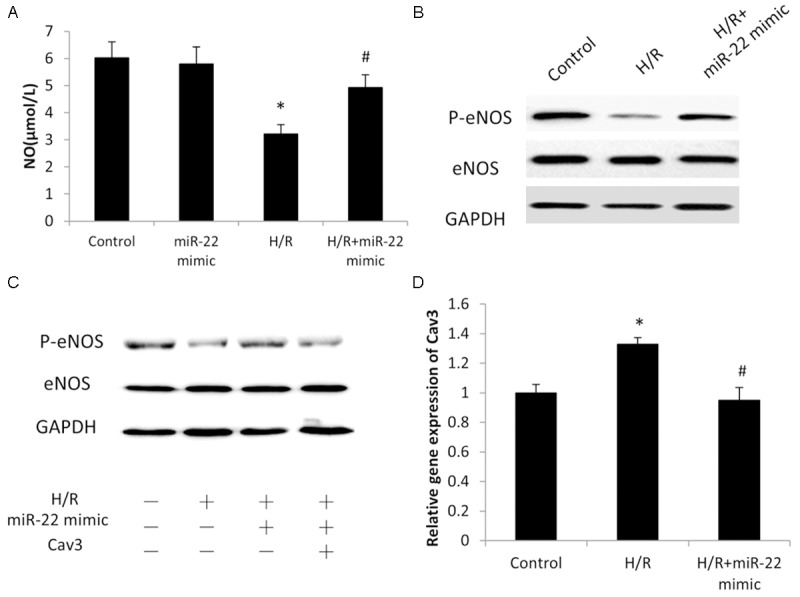
The Cav3/eNOS pathway is the regulatory mechanism for the role of miRNA-22 in cardiac myocytes. A. NO measurement by commercial NO Detection Kit in cardiac myocytes. B, C. Western blot analysis showing the expression of p-eNOS. D. Cav3 gene expression was evaluated by quantitative polymerase chain reaction analysis. Data represent means ± SD, *P≤0.05 vs. Control, #P<0.05 vs. H/R.
Cav3 is an upstream negative regulator of eNOS, which is the major type of caveolin family proteins in cardiac myocytes [28,29]. p-eNOS was upregulated by transfection of miR-22 mimic during H/R (Figure 5C, lane 3). However, such upregulation was markedly reduced by overexpression of Cav3 (Figure 5C), indicating it was involved in the upregulation of p-eNOS induced by miR-22 during H/R. Further RT-PCR experiments showed that the mRNA level of Cav3 increased significantly during H/R while it was suppressed by transfection of miR-22 mimic (Figure 5D). Taken together, miR-22 downregulated Cav3 transcript level, promoted p-eNOS activation and followed NO production during H/R.
Discussion
MiR-22 was originally reported as a tumor-suppressive miRNA which induced cellular senescence in cancer cell lines [33,34]. Recently, a cell model showed that its overexpression significantly induced cardiac myocyte hypertrophy via the upregulation of PTEN, while its reduction attenuated either PE- or Ang II-induced hypertrophy [35]. A transgenic mice model with enhanced levels of miR-22 in adult cardiac myocytes showed that it promoted heart failure through coordinate suppression of PPAR/ERR-nuclear hormone receptor transcription [36]. In another research, miR-22 was found to induce cardiomyocyte hypertrophy and it was a key regulator of stress-induced cardiac hypertrophy and ventricular remodeling [21]. Targeted deletion of miR-22 in myocardium promoted stress-induced cardiac dilation and contractile dysfunction [22]. Meanwhile, it was revealed that its overexpression reduced apoptosis of neurons by inhibiting caspase-3 activity [23], and a cerebral I/R model confirmed its anti-apoptotic effect on neurons after I/R [24]. However, its effect on cardiac myocytes and cardiac function during I/R remains to be addressed.
In this study, we first examined effects of miR-22 on cardiac function in rats after I/R. Male Wistar rats were subjected to 30 minutes of myocardial ischemia and 24 hours of reperfusion. Cardiac function was impaired after I/R, as previously reported (Figure 1). Interestingly, overexpression of miR-22 reduced I/R induced cardiac injury and promoted cardiac function significantly which was impaired during I/R, as evidenced by a significant increase in %EF and %FS and a decrease in LVESV in the left ventricle, compared to the I/R group without pre-administration of miR-22 mimic (Figure 1A and 1B). Such cardiac-protective effect of miR-22 during I/R was further confirmed by decreased release of CK-MB, an important indicator of cardiac injury, and reduced level of LDH, a marker of cell death. Of note, an increase in the CK-MB level suggested a severe cardiac injury, and an increase in the LDH level indicated significant cell death in the heart during I/R. Further Masson’s trichrome staining of heart sections confirmed that there was significant cardiac remodeling in myocardium after I/R, while miR-22 overexpression reduced cell death and cardiac remodeling significantly (Figure 2E). Together, the study presented that overexpression of miR-22 inhibited cardiac myocyte apoptosis, reduced cardiac remodeling, and promoted cardiac function significantly during I/R.
Meanwhile, an in vitro cell model of H/R was employed as a representative process during I/R. Flow cytometry results revealed that H/R induced cardiac myocyte apoptosis significantly (Figure 3A and 3B). And the induction was suppressed significantly by overexpression of miR-22 during H/R. Of note, LDH assay confirmed the anti-apoptotic effect of miR-22 on cardiac myocytes during H/R. Interestingly, its overexpression had little effect on cardiac myocyte apoptosis under normal condition (without H/R) (Figure 3A and 3B). It may be reasoned that miR-22 overexpression inhibited detrimental changes induced by H/R.
Furthermore, it was found that eNOS activity and followed NO production was reduced during H/R (Figure 5A and 5B) or I/R (Figure 4) and such reduction was inhibited significantly by miR-22 overexpression (Figures 4 and 5). After phosphorylation at Ser1177, the activated p-eNOS catalyzes the conversion of L-arginine to L-citrulline and NO, but it also produces superoxide instead of NO under uncoupling conditions when deprived of its substrate L-arginine or its co-factors, including tetrahydrobiopterin. There is a substantial increase in ROS production during I/R [1]. Oxidative stress, overproduction of ROS, causes direct damage to cellular DNA, proteins, and lipids, which initiates a cytokine-mediated pro-inflammation. Meanwhile, over-produced ROS reacts with NO forming peroxynitrite. Peroxynitrite oxidizes eNOS cofactor tetrahydrobiopterin, resulting in eNOS uncoupling which leads to increased ROS and decreased NO production further [1]. In the study, overexpression of miR-22 was shown to upregulate NO production during I/R and H/R significantly (Figures 4A and 5A). NO scavenges ROS [25], preventing the non-specific cell damages caused by ROS. Moreover, NO inactivates caspase-3 by S-nitrosation and leads to reduced cell apoptosis [26,27]. Such decrease in caspase-3 activity was observed in rats of the I/R+miR-22 group (Figure 2B) together with an increase in eNOS activity and NO production in the serum (Figure 4A and 4B) as well as in cardiac myocytes after H/R (Figures 3D, 5A and 5B). Taken together, overexpression of miR-22 upregulated eNOS activity and the followed NO production, which inhibited cardiac myocyte apoptosis by inhibiting overproduction of ROS and inactivating caspase-3 activity during I/R or H/R. Therefore, miR-22 overexpression played an anti-apoptotic role in cardiac myocytes via upregulation of eNOS activity and NO production during I/R or H/R. In addition, previous studies showed that overexpression of miR-22 induced cardiac myocyte hypertrophy and ventricular remodeling [21,22,36]. But its anti-apoptotic effect in cardiac myocytes was not well reported, which might be due to the fact that eNOS activity and followed NO production were not changed significantly. Together, miR-22 overexpression has a favorable anti-apoptotic effect during I/R, while a long-term overexpression may cause cardiac damage.
Mechanistically, transfection of miR-22 mimic had little effect on the total protein level of eNOS, but restored the level of p-eNOS (Ser1177) which was downregulated during H/R (Figure 5B). p-eNOS is active and has the catalytic ability of NO production, while dephosphorylated eNOS is inactive. Western blot analysis demonstrated that Cav3 was involved in miR-22 induced upregulation of p-eNOS and its overexpression inhibited the upregulation of p-eNOS induced by miR-22 (Figure 5C). Further RT-PCR experiment showed that Cav3 was upregulated during H/R and it was inhibited by miR-22 at the transcript level (Figure 5D). In short, miR-22 downregulated Cav3 transcript level, promoted p-eNOS level and followed NO production during H/R. Worthy of mention, Cav3 knockout mice developed a progressive cardiomyopathy via hyperactivation of the Ras-p42/44 MAPK signaling [37]. Recent studies showed that exendin-4 and geranylgeranylacetone had a cardiac-protective effect on I/R induced injury, but the protective effect was abolished in Cav3 knockout mice [38,39]. Taken together the results in the present study, it may be reasoned that Cav3 helps maintain eNOS bioavailability and activity at a certain level which is critical for eNOS activity and NO production for normal cardiac function. Both overexpression and knockout of Cav3 have detrimental effects on cardiac function.
In summary, the present study revealed that miR-22 downregulated Cav3, leading to increased eNOS activity and NO, which inhibited cardiac myocyte apoptosis during I/R. Such anti-apoptotic effect of miR-22 is of significant importance for reducing I/R induced cardiac injury at the cellular level, given the fact that most adult cardiac myocytes are unable to divide and form new cardiac myocytes to replace those lost in I/R induced cardiac injury. Therefore, pre-administration of miR-22 mimic can prevent cardiac myocyte apoptosis, reduce cardiac remodeling, and improve cardiac function after I/R. Of clinical interest, the present study may highlight miR-22 as a potential therapeutic agent for reducing I/R induced cardiac injury.
Acknowledgements
This work was funded by Anhui Talented Doctors funding (#201302114).
Disclosure of conflict of interest
None.
References
- 1.Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat Med. 2011;17:1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frank A, Bonney M, Bonney S, Weitzel L, Koeppen M, Eckle T. Myocardial ischemia reperfusion injury: from basic science to clinical bedside. Semin Cardiothorac Vasc Anesth. 2012;16:123–132. doi: 10.1177/1089253211436350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kukreja RC, Yin C, Salloum FN. MicroRNAs: new players in cardiac injury and protection. Mol Pharmacol. 2011;80:558–564. doi: 10.1124/mol.111.073528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sluijter JP, van Mil A, van Vliet P, Metz CH, Liu J, Doevendans PA, Goumans MJ. MicroRNA-1 and -499 regulate differentiation and proliferation in human-derived cardiomyocyte progenitor cells. Arterioscler Thromb Vasc Biol. 2010;30:859–868. doi: 10.1161/ATVBAHA.109.197434. [DOI] [PubMed] [Google Scholar]
- 5.Park SY, Lee JH, Ha M, Nam JW, Kim VN. miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat Struct Mol Biol. 2009;16:23–29. doi: 10.1038/nsmb.1533. [DOI] [PubMed] [Google Scholar]
- 6.Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med. 2011;208:549–560. doi: 10.1084/jem.20101547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roy S, Khanna S, Hussain SR, Biswas S, Azad A, Rink C, Gnyawali S, Shilo S, Nuovo GJ, Sen CK. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res. 2009;82:21–29. doi: 10.1093/cvr/cvp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, Li YR, Li PF. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med. 2011;17:71–78. doi: 10.1038/nm.2282. [DOI] [PubMed] [Google Scholar]
- 10.Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, Zhang Y, Xu C, Bai Y, Wang H, Chen G, Wang Z. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486–491. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 11.Ye Y, Hu Z, Lin Y, Zhang C, Perez-Polo JR. Downregulation of microRNA-29 by antisense inhibitors and a PPAR-gamma agonist protects against myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2010;87:535–544. doi: 10.1093/cvr/cvq053. [DOI] [PubMed] [Google Scholar]
- 12.Yin C, Salloum FN, Kukreja RC. A novel role of microRNA in late preconditioning: upregulation of endothelial nitric oxide synthase and heat shock protein 70. Circ Res. 2009;104:572–575. doi: 10.1161/CIRCRESAHA.108.193250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong S, Cheng Y, Yang J, Li J, Liu X, Wang X, Wang D, Krall TJ, Delphin ES, Zhang C. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J Biol Chem. 2009;284:29514–29525. doi: 10.1074/jbc.M109.027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim HW, Haider HK, Jiang S, Ashraf M. Ischemic preconditioning augments survival of stem cells via miR-210 expression by targeting caspase-8-associated protein 2. J Biol Chem. 2009;284:33161–33168. doi: 10.1074/jbc.M109.020925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu Y, Zhang Y, Shan H, Pan Z, Li X, Li B, Xu C, Zhang B, Zhang F, Dong D, Song W, Qiao G, Yang B. MicroRNA-1 downregulation by propranolol in a rat model of myocardial infarction: a new mechanism for ischaemic cardioprotection. Cardiovasc Res. 2009;84:434–441. doi: 10.1093/cvr/cvp232. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Zhang X, Ren XP, Chen J, Liu H, Yang J, Medvedovic M, Hu Z, Fan GC. MicroRNA-494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion- induced cardiac injury. Circulation. 2010;122:1308–1318. doi: 10.1161/CIRCULATIONAHA.110.964684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn GW 2nd, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 18.Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, Willis MS, Selzman CH, Wang DZ. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest. 2009;119:2772–2786. doi: 10.1172/JCI36154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bostjancic E, Zidar N, Stajer D, Glavac D. MicroRNAs miR-1, miR-133a, miR-133b and miR-208 are dysregulated in human myocardial infarction. Cardiology. 2010;115:163–169. doi: 10.1159/000268088. [DOI] [PubMed] [Google Scholar]
- 20.Xu C, Lu Y, Pan Z, Chu W, Luo X, Lin H, Xiao J, Shan H, Wang Z, Yang B. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J Cell Sci. 2007;120:3045–3052. doi: 10.1242/jcs.010728. [DOI] [PubMed] [Google Scholar]
- 21.Huang ZP, Chen J, Seok HY, Zhang Z, Kataoka M, Hu X, Wang DZ. MicroRNA-22 regulates cardiac hypertrophy and remodeling in response to stress. Circ Res. 2013;112:1234–1243. doi: 10.1161/CIRCRESAHA.112.300682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gurha P, Abreu-Goodger C, Wang T, Ramirez MO, Drumond AL, van Dongen S, Chen Y, Bartonicek N, Enright AJ, Lee B, Kelm RJ Jr, Reddy AK, Taffet GE, Bradley A, Wehrens XH, Entman ML, Rodriguez A. Targeted deletion of microRNA-22 promotes stress-induced cardiac dilation and contractile dysfunction. Circulation. 2012;125:2751–2761. doi: 10.1161/CIRCULATIONAHA.111.044354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jovicic A, Zaldivar Jolissaint JF, Moser R, Silva Santos Mde F, Luthi-Carter R. MicroRNA-22 (miR-22) overexpression is neuroprotective via general anti-apoptotic effects and may also target specific Huntington’s disease-related mechanisms. PLoS One. 2013;8:e54222. doi: 10.1371/journal.pone.0054222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu H, Wu M, Zhao P, Huang Y, Wang W, Yin W. Neuroprotective Effects of Viral Overexpression of microRNA-22 in Rat and Cell Models of Cerebral Ischemia-Reperfusion Injury. J Cell Biochem. 2015;116:233–241. doi: 10.1002/jcb.24960. [DOI] [PubMed] [Google Scholar]
- 25.Wink DA, Hanbauer I, Krishna MC, DeGraff W, Gamson J, Mitchell JB. Nitric oxide protects against cellular damage and cytotoxicity from reactive oxygen species. Proc Natl Acad Sci U S A. 1993;90:9813–9817. doi: 10.1073/pnas.90.21.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Billiar TR, Talanian RV, Kim YM. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem Biophys Res Commun. 1997;240:419–424. doi: 10.1006/bbrc.1997.7672. [DOI] [PubMed] [Google Scholar]
- 28.Sbaa E, Frerart F, Feron O. The double regulation of endothelial nitric oxide synthase by caveolae and caveolin: a paradox solved through the study of angiogenesis. Trends Cardiovasc Med. 2005;15:157–162. doi: 10.1016/j.tcm.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 29.Mineo C, Shaul PW. Regulation of eNOS in caveolae. Adv Exp Med Biol. 2012;729:51–62. doi: 10.1007/978-1-4614-1222-9_4. [DOI] [PubMed] [Google Scholar]
- 30.Roth DM, Patel HH. Role of caveolae in cardiac protection. Pediatr Cardiol. 2011;32:329–333. doi: 10.1007/s00246-010-9881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA, Michel T. Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J Biol Chem. 1996;271:22810–22814. doi: 10.1074/jbc.271.37.22810. [DOI] [PubMed] [Google Scholar]
- 32.Garcia-Cardena G, Oh P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc Natl Acad Sci U S A. 1996;93:6448–6453. doi: 10.1073/pnas.93.13.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiong J, Du Q, Liang Z. Tumor-suppressive microRNA-22 inhibits the transcription of E-box-containing c-Myc target genes by silencing c-Myc binding protein. Oncogene. 2010;29:4980–4988. doi: 10.1038/onc.2010.241. [DOI] [PubMed] [Google Scholar]
- 34.Xu D, Takeshita F, Hino Y, Fukunaga S, Kudo Y, Tamaki A, Matsunaga J, Takahashi RU, Takata T, Shimamoto A, Ochiya T, Tahara H. miR-22 represses cancer progression by inducing cellular senescence. J Cell Biol. 2011;193:409–424. doi: 10.1083/jcb.201010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu XD, Song XW, Li Q, Wang GK, Jing Q, Qin YW. Attenuation of microRNA-22 derepressed PTEN to effectively protect rat cardiomyocytes from hypertrophy. J Cell Physiol. 2012;227:1391–1398. doi: 10.1002/jcp.22852. [DOI] [PubMed] [Google Scholar]
- 36.Gurha P, Wang T, Larimore AH, Sassi Y, Abreu-Goodger C, Ramirez MO, Reddy AK, Engelhardt S, Taffet GE, Wehrens XH, Entman ML, Rodriguez A. microRNA-22 promotes heart failure through coordinate suppression of PPAR/ERR-nuclear hormone receptor transcription. PLoS One. 2013;8:e75882. doi: 10.1371/journal.pone.0075882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woodman SE, Park DS, Cohen AW, Cheung MW, Chandra M, Shirani J, Tang B, Jelicks LA, Kitsis RN, Christ GJ, Factor SM, Tanowitz HB, Lisanti MP. Caveolin-3 knock-out mice develop a progressive cardiomyopathy and show hyperactivation of the p42/44 MAPK cascade. J Biol Chem. 2002;277:38988–38997. doi: 10.1074/jbc.M205511200. [DOI] [PubMed] [Google Scholar]
- 38.Tsutsumi YM, Tsutsumi R, Horikawa YT, Sakai Y, Hamaguchi E, Ishikawa Y, Yokoyama U, Kasai A, Kambe N, Tanaka K. Geranylgeranylacetone protects the heart via caveolae and caveolin-3. Life Sci. 2014;101:43–48. doi: 10.1016/j.lfs.2014.02.019. [DOI] [PubMed] [Google Scholar]
- 39.Tsutsumi YM, Tsutsumi R, Hamaguchi E, Sakai Y, Kasai A, Ishikawa Y, Yokoyama U, Tanaka K. Exendin-4 ameliorates cardiac ischemia/reperfusion injury via caveolae and caveolins-3. Cardiovasc Diabetol. 2014;13:132. doi: 10.1186/s12933-014-0132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]