

Abstract
Endothelial cells (ECs) apoptosis induced by hyperglycemia is intimately involved in the pathophysiology of diabetes and its complication. Although PGC-1α is known for its role in glucose metabolism, its role in ECs injury caused by high glucose milieu is still unclear. Therefore, this study aims to investigate whether PGC-1α participates in ECs apoptosis under high glucose condition. Human umbilical vein endothelial cells (HUVECs) were down-regulated PGC-1α expression by adenovirus-mediated PGC-1α specific siRNA (Ad-shPGC-1α) and exposed to high glucose. Cell viability, apoptosis, mitochondrial membrane permeability, apoptotic marker, reactive oxygen species (ROS), and expression of PGC-1α and VDAC isoforms were studied. Our results showed that high glucose-induced cell apoptosis was associated with an obvious decrease in PGC-1α expression. Lack of PGC-1α exacerbated high glucose-induced cell apoptosis, inner mitochondrial membrane permeabilization, mitochondrial cytochrome c release into cytoplasm and caspases activation; while further decreased cell viability and mitochondrial membrane potential. Analysis of apoptotic markers (Bcl-2, Bax), intracellular ROS and endoplasmic reticulum stress revealed that these mechanisms were not accounted for the effects of Ad-shPGC-1α on apoptosis. However, we found silencing PGC-1α further increased high glucose-induced VDAC1 expression. The pharmacological inhibition of VDAC1 with 4,4’-diisothiocyanostilbene-2,2’-disulfonic acid (DIDS) inhibited the increased apoptosis in high glucose-treated PGC-1α knockdown cells. These findings strongly suggest that PGC-1α defect is one of the major mechanisms for ECs apoptosis under high glucose condition, and provide a novel strategy to prevent endothelial dysfunction in diabetes.
Keywords: PGC-1α, high glucose, human umbilical vein endothelial cells, apoptosis, mitochondria
Introduction
Diabetes-related cardiovascular complications have been identified as the major direct cause of death in patients with diabetes mellitus, including both macrovascular and microvascular diseases [1]. In diabetes, endothelial cells (ECs) of blood vessels are characterized by the first targets [2]. ECs dysfunction is mainly caused by cell injury and apoptosis, which initially impairs endothelial integrity and blood vessel function [3]. Thus, protection against ECs injury and apoptosis may have great significance. Up until now, despite incontrovertibility of evidence implicating hyperglycemia in ECs damage, the mechanisms involved remain ambiguous. Peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1α (PGC-1α) is a key regulator in energy metabolism and mitochondrial function [4,5]. Increasing evidences suggest that PGC-1α expression is highly associated with glucose metabolism. Obvious changes in PGC-1α expression were observed in adipose tissue from insulin-resistant subjects [6], in skeletal muscle from type 2 diabetics [7], in liver of rodents in diabetic model [8]. Recent study showed that the decreased PGC-1α expression was causally related to hyperglycemia-induced vascular smooth muscle cells (VSMCs) proliferation and migration [9]. These results indicate that PGC-1α may serve as a causal factor of the pathophysiologic process in diabetes. However, the functional role of PGC-1α in endothelium remains elusive. Moreover, PGC-1α is considered to be a broad and powerful regulator of mitochondrial function and reactive oxygen species (ROS) metabolism [5,10-12]. For example, reduction in PGC-1α has been proposed to be a direct causative candidate in mitochondrial dysfunction [10]. Additionally, up-regulation of PGC-1α levels dramatically protected neural cells against oxidative stress-mediated death through induction of ROS-detoxifying enzymes [11]. Given the importance of mitochondria and ROS generation in apoptotic signaling [13,14], it is reasonable to assume that PGC-1α could exhibit a dramatic effect on ECs apoptosis induced by high glucose. Therefore, in this study, we silenced PGC-1α expression by using a recombinant adenovirus expressing PGC-1α small hairpin RNA (Ad-shPGC-1α) to determine whether PGC-1α plays a functional role in high glucose-induced apoptosis in human umbilical vein endothelial cells (HUVECs), and to further explore the precise mechanisms.
Methods
Cell culture and treatment
Human umbilical vein endothelial cells (HUVECs) were isolated and cultured as previously described [15]. The study protocol was confirmed by the declaration of Helsinki and approved by the Ethics of Committee of Shandong University. In brief, cells were harvested from umbilical cord by treatment with collagenase and cultured in M199 medium (Gibco, NY, USA) supplemented with 5.5 mmol/L glucose, 20% FBS, 2% L-glutamine, antibiotics (100 U/mL penicillin and streptomycin) and endothelial cell growth at 37°C in a 5% CO2 humidified air incubator. Cells at passages 3-8 were used. In glucose-treated experiments, HUVECs were cultured in M199 medium containing 5.5, 11, 15 or 25 mmol/L glucose, respectively. In adenovirus-infected experiments, HUVECs were processed for adenoviral infection for 24 h in prior to high glucose incubation for another 48 h.
Western blot analysis
After treatment, the whole cell lysates, mitochondrial and cytosolic fractions were harvested as previously described [16]. Equal quantities of protein were separated by sodium dodecyl sulfate-polyacrylamide gels and transferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA). The blots were then probed with the following antibodies at 4°C overnight: PGC-1α, Bip/GRP78 (diluted 1:100), VDAC1, VDAC2 and VDAC3 (diluted 1:400) (Santa Cruz, CA, USA); Bcl-2, Bax, cytochrome c, COX IV, cleaved caspase-3 and cleaved caspase-9 (1:2000, Cell Signaling Technology, MA, USA); GAPDH and β-actin (diluted 1:4000, Beyotime, Jiangsu, China). After washing and incubation with the corresponding secondary antibodies (Cell Signaling Technology), membranes were visualized using enhanced chemiluminescence reagents (ECL system, Thermo Scientific, MA, USA).
Adenoviral infection of siRNA for PGC-1α
Recombinant adenovirus encoding a small hairpin shRNA of PGC-1α (Ad-shPGC-1α) was successfully constructed by Cyagen Biosciences Inc. (CA, USA). The control shRNA (Ad-Scr) containing no significant homology to any known gene was purchased from Shanghai Sunbio Bio-medical Technology (Shanghai, China) and used as a negative control. On the day before adenoviral infection, HUVECs were seeded in 6-well plates. Adenovirus were diluted in M199 medium containing 2% FBS and added to the cells for 6 h. After that, cells were transferred into complete medium and incubated for further 24 h.
Assay for cell viability
Cell viability was measured by cell counting kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s protocols. In brief, 100 μl HUVECs were seeded in 96-well plates at a density of 2×104/well. After treatment, 10 μl of CCK8 was added to each well for 2 h at 37°C. The colorimetric assay was performed with Bi o-Tek microplate reader (VT, USA) and measured at 450 nm wavelength.
Quantitative PCR (qPCR) analysis
RNA from HUVECs was isolated using RNeasy Micro Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions and was reverse-transcribed using the ReverTra ACE qPCR RT Kit (Toyobo, Osaka, Japan). PCR amplification was performed using SYBR Green PCR master mix (Invitrogen, CA, USA) with the ABI Prism 7300 Fast Real-Time PCR system (Applied Biosystem, CA, USA). Sequence-specific primers were obtained from Sangon Biothech (Shanghai, China). The primer sequences used to amplify human PGC-1α cDNA were 5’-CCTGCATGAGTGTGTGCTCT-3’ (sense) and 5’-GCACACTCGATGTCACTCCA-3’ (antisense); primers for GAPDH were 5’-GGGCACGAAGGCTCATCATT-3’ (sense) and 5’-AGAAGGCTGGGGCTCATTTG -3’ (antisense).
Apoptosis detection
HUVECs apoptosis was determined by Accuri C6 flow cytometry (BD Biosciences, CA, USA) using the FITC-Annexin V and propidium iodide (PI) double staining assay (Bender Medsystems, Vienna, Austria) according to the manufacturer’s instructions. Briefly, HUVECs were digested with trypsin and suspended in 200 μl binding buffer at density of 1×106/mL, and then added Annexin V and PI, vortexed and incubated for 20 min in dark at room temperature. Finally, binding buffer was added and cells were counted with flow cytometry.
Detection of inner mitochondrial membrane (IMM) permeabilization
IMM permeabilization was measured using calcein AM (Invitrogen) as described previously [17]. After treatment, cells were incubated with 1 μmol/L calcein AM and 1 mmol/L CoCl2 in Hanks balanced salt solution (HBSS) (135 mmol/L NaCl, 5 mmol/L KCl, 1.5 mmol/L CaCl2, 1 mmol/L MgCl2, 10 mmol/L D-glucose, 10 mmol/L HEPES, pH 7.40) for 10 min at 37°C. Cells were imaged by a laser scan confocal microscope (LSM710, ZEISS, München, Germany). The fluorescence intensity of the 96-well plates was analyzed using a microplate reader (Infinite F500, Mannedorf, Switzerland).
Detection of mitochondrial membrane potential (MMP)
Cells were staining with mitochondrial membrane potential sensitive probe, 5,5’,6,6’-Tetrachloro-1,1’,3,3’-tetraethyl-benza-midazolocarbocyanin iodide (JC-1 dye, Cayman Chemical, MI, USA) for 15 min at 37°C. Loss of MMP was observed by LSM710 laser scan confocal microscope (Zeiss). The aggregate red form indicating healthy MMP was detected at excitation/emission wavelength of 585/590 nm, while the green monomeric form indicating a loss of MMP was examined at 510/527 nm. The fluorescence intensity was calculated using ImageJ software (NIH, Maryland, USA).
Detection of reactive oxygen species (ROS)
Intracellular ROS generation in HUVECs was visualized using 2’,7’-dichlorofluorescin diacetate (H2DCF-DA, Invitrogen) as previously described [17]. Briefly, HUVECs were seeded in 6-well plates or 96-well bottom black plates, and then applied to different treatments according to the experiment requirements. After that, H2DCF-DA (10 μmol/L) was added to each culture well and incubated for 30 min in serum-free M199 medium at 37°C. Immediately, cells were washed with HBSS and imaged with a fluorescence microscope (Olympus Corpor-ation, Tokyo, Japan) at 488 nm excitation and 525 nm emission wavelengths. The fluorescence intensity of the 96-well bottom black plates was analyzed using a microplate reader (Mannedorf).
Statistical analysis
Data analysis was performed by one-or two-way ANOVA and significant differences were determined by post hoc using the Bonferroni test. All data were given as mean ± SEM, with P < 0.05 representing to be statistically significant.
Results
PGC-1α expression in HUVECs paralleled with high glucose-decreased cell viability
Endothelial cells injury has been suggested to be a principle feature in the pathogenesis of diabetes [18]. To establish a proper experimental model of diabetes in vitro, HUVECs were treated with various concentrations of glucose, and then cell viability was examined by CCK-8 assay. Compared with control or osmotic HM (high mannitol, 25 mmol/L), the cell viability of HUVECs was significantly decreased dose-dependently. This viability inhibitory effect was more obvious at 15 mmol/L (Figure 1A). Moreover, high glucose (15 mmol/L) inhibited cell viability in a time-dependent manner (Figure 1B). Compared with control group, at 12, 24, 48, 72 and 96 h, cell viability was decreased to 92.3 ± 9.1%, 73.8 ± 8.2%, 44.7 ± 5.8%, 51.3 ± 7.5% and 47.5 ± 6.1%. The cell viability reached the minimal level at 48 h.
Figure 1.
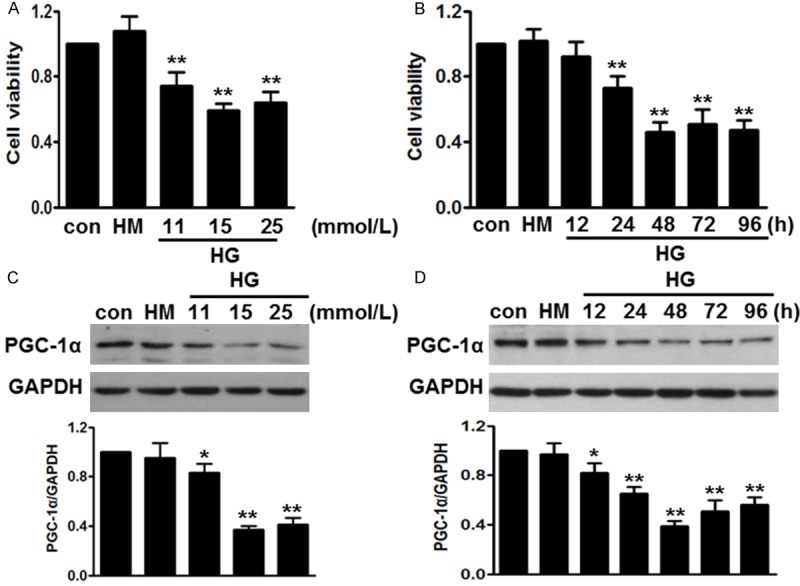
High glucose-induced cell injury was associated with decreased PGC-1α expression. A. HUVECs were incubated with different concentrations of high glucose (HG) for 48 h. Cell viability was analyzed by cell counting kit-8 (CCK-8). B. The cells were treated with high glucose (15 mmol/L) for the indicated times. CCK-8 analysis was performed. C. Western blot showed that glucose inhibited PGC-1α expression dose-dependently. D. After treatment mentioned in B, PGC-1α protein level was determined by western blot. All data were expressed as mean ± SEM. *P < 0.05, **P < 0.01 vs. control, n = 6.
Interestingly, we found PGC-1α expression paralleled with high glucose-induced cell injury. Compared to 5.5 mmol/L glucose (control group), 11 mmol/L glucose inhibited PGC-1α expression level to 74.3 ± 5.2%, and 15 mol/L glucose further inhibited it to 59.3 ± 4.7% (Figure 1C). Moreover, we also found that 15 mol/L glucose decreased PGC-1α expression in a time-dependent manner (Figure 1D). Therefore, in the following experiments, we used glucose at 15 mmol/L for 48 h.
Down-regulation of PGC-1α accelerated high glucose-induced cell injury and apoptosis
GivenCo2+ that high glucose affected PGC-1α expression in HUVECs, we supposed that PGC-1α may be involved in cell injury and apoptosis. To verify this notion, HUVECs were infected with adenovirus containing PGC-1α small hairpin RNA (Ad-shPGC-1α) at multiplicity of infection (MOI) of 10, 20 and 40 for 24 h. Efficiencies of RNA interference were firstly determined. Western blot and qPCR showed that Ad-shPGC-1α at 20 MOI effectively reduced endogenous expression of PGC-1α more than 70%, respectively. Negative shRNA (Ad-Scr) had no effect on PGC-1α expression (Figure 2A, 2B). The effect of PGC-1α knockdown on high glucose-treated cell viability was tested by CCK-8. As shown in Figure 2C, Ad-Scr and Ad-shPGC-1α did not produce any significant effects on cell viability in the absence of high glucose. After treatment with 15 mmol/L glucose for 48 h, cell viability rate was significantly reduced to 48.1 ± 6.5%, compared with control group. Ad-shPGC-1α exaggerated high glucose-induced HUVECs injury, reducing the cell viability rate to 29.3 ± 5.0%. Ad-Scr did not alter cell death rate induced by high glucose. Furthermore, HUVECs apoptosis was analyzed by flow cytometry with Annexin V/PI double staining (Figure 2D). Expectedly, there was no significant difference in the apoptotic population of HUVECs between control and Ad-shPGC-1α groups. Treatment with high glucose for 48 h induced an apoptotic rate of 16.8 ± 2.0%, which could be further enhanced by the Ad-shPGC-1α (Figure 2E). These data suggest that lack of PGC-1α produces no effect on cell viability under basal conditions, but accelerates cell apoptosis induced by high glucose.
Figure 2.

Lack of PGC-1α promoted high glucose-induced cell injury and apoptosis in HUVECs. (A and B) Western blot (A) and qPCR (B) showed that infection with adenovirus containing PGC-1α small hairpin RNA (Ad-PGC-1α) at multiplicity of infection (MOI) of 10, 20 and 40 for 24 h significantly decreased PGC-1α expression, but transfection with negative shRNA (Ad-Scr) did not change endogenous PGC-1α expression. (C) HUVECs were infected with Ad-Scr or Ad-shPGC-1α in the absence or presence of high glucose. Cell viability was determined by CCK-8. (D) After treatment as described in (C), HUVECs apoptosis was determined by Annexin V/PI staining followed by flow cytometry. (E). Quantitative analysis of the percentage of apoptotic cells. *P < 0.05, **P < 0.01 vs. control, ##P < 0.01 vs. high glucose alone, n = 6.
Knockdown of PGC-1α augmented high glucose-induced alterations in mitochondrial membrane permeability
The loss of mitochondrial membrane potential (MMP) is a prominent feature during the process of apoptosis [19]. We firstly measured the inner mitochondrial membrane (IMM) permeabilization using the calcein AM staining (Figure 3A). PGC-1α deficiency had no effects on IMM permeabilization under basal conditions (data not shown). 48 h after high glucose treatment, the IMM was destroyed as it showed that calcein-dependent fluorescence was remarkably quenched by Co2+, which was further enhanced in As-shPGC-1α-infected cells (Figure 3B). Moreover, we detected the MMP by JC-1 staining (Figure 3C). After high glucose treatment, the red fluorescence of JC-1 was dramatically decreased, while the green fluorescence was increased, resulting in an attenuated ratio of red/green intensity. Knockdown of PGC-1α notably enhanced the decrease in red/green fluorescence ratio induced by high glucose, whereas Ad-Scr exhibited no effects on MMP alternations (Figure 3D).
Figure 3.
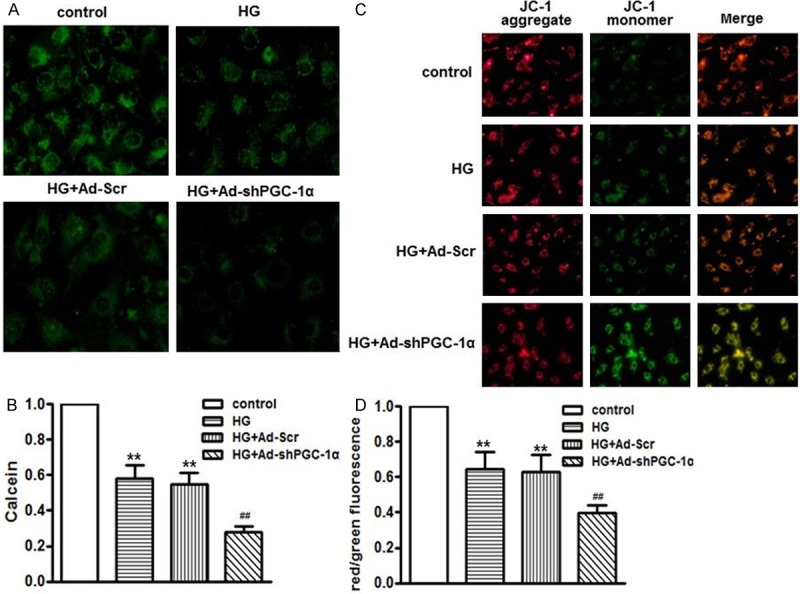
PGC-1α deficiency exacerbated high glucose-induced mitochondrial membrane dysfunction. HUVECs were infected with Ad-Scr or Ad-shp PGC-1α for 24 h, and then were cultured in high glucose medium for another 48 h. A. After treatment, cells were loaded with calcein-AM with Co2+, representative photographs of inner mitochondrial membrane permeabilization was captured by confocal microscopy. B. Quantitative measurements of calcein fluorescence intensity assessed by microplate reader. C. Loss of mitochondrial membrane potential was measured using JC-1 staining by confocal microscopy. Representative images of JC-1 derived fluorescence in HUVECs. D. Quantitative analysis of the ratio of red/green fluorescence. **P < 0.01 vs. control, ##P < 0.01 vs. high glucose alone, n = 6.
Lack of PGC-1α exaggerated high glucose-induced cytochrome c release and caspases activation
It has been documented that MMP destabilization results in cytochrome c release and sequentially activates caspase cascades, such as caspase-3 and caspase-9 [20]. As shown in Figure 4A-D, high glucose-treated cells displayed a more than 1.5-fold increase of cytochrome c in cytoplasm compared with control group. In parallel with this, the content of cytochrome in mitochondria was significantly decreased. As expected, the increased translocation of cytochrome c was further enhanced in Ad- PGC-1α-infected cells. Next, we examined the caspases activation. Western blot showed that caspase-3 and caspase-9 were both remarkably elevated by high glucose treatment and were further enhanced by infection with Ad-shPGC-1α (Figure 4E, 4F). The cytochrome c level and caspases activation were not significantly changed by Ad-Scr.
Figure 4.
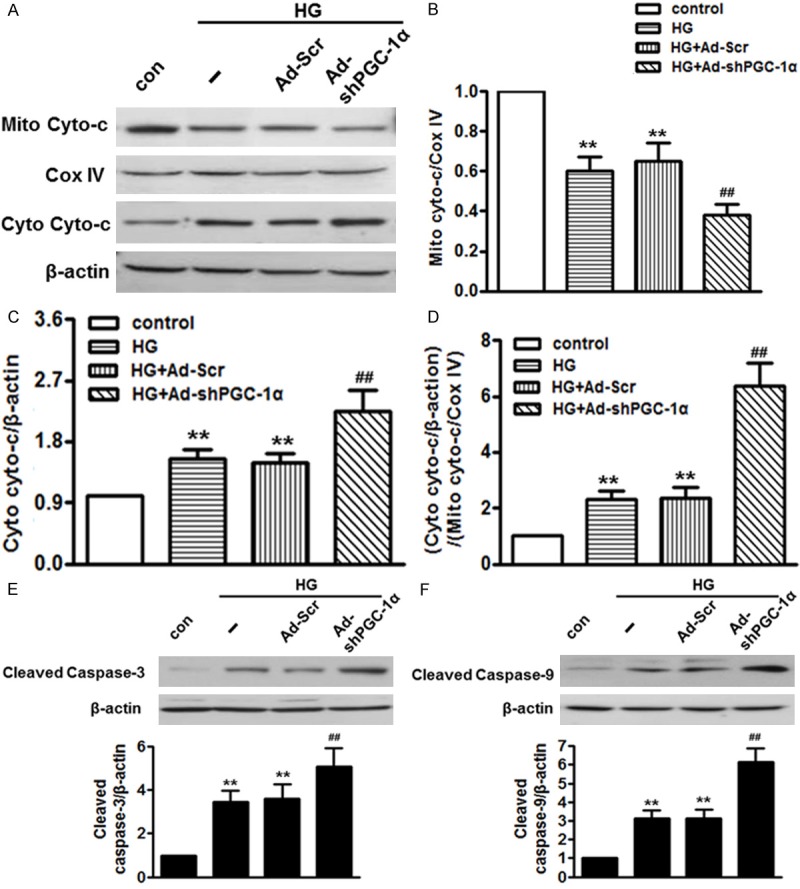
Knockdown of PGC-1α augmented high glucose-induced cytochrome c release and caspases activation. HUVECs were treated with Ad-shPGC-1α for 24 h in prior incubation with high glucose for another 48 h. (A) Western blot analysis of cytochrome c (Cyto-c) protein expression in the mitochondrial and cytosol. (B and C) Densitometric analysis of cytochrome c expression in the mitochondrial (B) and cytosol (C). (D) Densitometric analysis of cytochrome c release from mitochondria to cytoplasm. (E and F) Protein expression of caspase-3 (E) and -9 (F) were measured by western blot. **P < 0.01 vs. control, ##P < 0.01 vs. high glucose, n = 6.
PGC-1α deficiency enhanced high glucose-induced cell apoptosis through activation of VDAC1
To investigate the mechanism by which PGC-1α regulates HUVECs apoptosis, we firstly determined the expression of anti-apoptotic protein Bcl-2 and pro-apoptotic protein Bax. Although lower Bcl-2 expression and higher Bax expression were observed following high glucose treatment, no differences were noted following infection with Ad-shPGC-1α compared to high glucose-treated cells (Figure 5A). Excessive oxygen species (ROS) and abnormal endoplasmic reticulum (ER) stress have been suggested to be major contributors for apoptosis [21,22]. As shown in Figure 5B, ROS generation was significantly increased following incubation with high glucose for 48 h. However, high glucose-induced ROS generation remained unchanged after infection with Ad-shPGC-1α (Figure 5C). Additionally, GRP78, a maker protein of endoplasmic reticulum (ER) stress, was examined by western blot. As shown in Figure 5D, lack of PGC-1α inhibited high glucose-induced GRP78 expression despite the increased apoptosis. There data indicate that ROS generation and ER stress are not responsible for the enhanced effects on high glucose-induced apoptosis in PGC-1α deficiency cells. It is well recognized that voltage-dependent anion channel (VDAC) is involved in the process of apoptosis [23]. Initially, we determined three VDAC isoforms expression levels (Figure 5E). Although VDAC2 and VDAC3 protein expression were elevated following exposure to high glucose, no significant differences were observed after knockdown of PGC-1α. Of note, merely the increased VDAC1 expression induced by high glucose were obviously enhanced following infection with Ad-shPGC-1α, indicating VDAC1 may be a key element for PGC-1α in accelerating apoptosis induced by high glucose. To verify this notion, we infected HUVECs with Ad-shPGC-1α, and then submitted the cells to high glucose medium in the presence of 4,4’-diisothiocyanostilbene-2,2’-disulfonic acid (DIDS), an inhibitor of VDAC1. Treatment with DIDS exhibited no effects on the apoptotic population of HUVECs under basal conditions, while markedly inhibited the increased apoptosis in high glucose-treated PGC-1α deficiency cells (Figure 5F, 5G).
Figure 5.

PGC-1α deficiency exaggerated ECs apoptosis through activation of VDAC1. HUVECs were pre-treated with Ad-Scr or Ad-shPGC-1α for 24 h following incubation with high glucose in the absence or presence of DIDS. A. Bcl-2 and Bax protein levels were examined by western blot. B. Representative images for cells loaded with H2DCF-DA (10 μmol/L) was captured with a fluorescence microscopy. C. Quantitative analysis of DCF fluorescence intensity assessed by microplate reader. D. Western blot showed that Ad-shPGC-1α inhibited high glucose-induced Bip/GRP78 expression. E. VDAC isoforms (VDAC1, VDAC2 and VDAC3) expression levels were determined by western blot using β-actin as an internal control. F. cell apoptosis was determined by Annexin V/PI staining. G. Quantitative analysis of the percentage of apoptotic cells. **P < 0.01 vs. control, ##P < 0.01 vs. high glucose, &&P < 0.01 vs. high glucose+Ad-shPGC-1α, n = 6.
Discussion
Dysfunction and activation of the endothelium have been implicated as critical factors in the pathogenesis of vascular disease in diabetes mellitus [18]. An understanding of the precise molecular mechanism of ECs injury and apoptosis caused by hyperglycemia is essential to clarify the contribution of ECs dysfunction in diabetic cardiovascular complication. In HUVECs, we observed that high glucose treatment decreased cell viability in a concentration and time dependent manner. Given that changes of PGC-1α expression in various tissues paralleled with the glucose level, as seen in diabetic subjects [6-8], we hypothesized that high glucose may affect PGC-1α expression in HUVECs. Here, we showed that high glucose concentration-dependently and time-dependently inhibited PGC-1α expression. Our findings were consistent with a previous study in VSMCs, which reported that the expression of PGC-1α was decreased after high glucose treatment [9]. Furthermore, reduction of PGC-1α dramatically potentiated high glucose-induced cell injury and apoptosis. These data provide the first and compelling evidence that PGC-1α plays an important role in ECs apoptosis. Apoptosis is a critical process under physiological conditions, whereas dysregulation occurs in pathological state [24]. Apoptosis can be triggered by various stimuli; however, execution occurs mainly through several pathways, including mitochondria [13], ROS generation [15], ER stress [21] and voltage-dependent anion channel (VDAC) [23]. Mitochondria have been documented to be the central executer of apoptosis through the well-known mitochondria-dependent pathway [13,25]. Mitochondrial dysfunction occurs following IMM permeabilization and loss of MMP, and consequently induces pro-apoptotic cytochrome c release into cytosol, leading to activation of caspases, such as caspase-3 and caspase-9 [19,20]. In the present study, PGC-1α deficiency further enhanced IMM permeabilization, decreased MMP, increased cytochrome c release into cytosol and potentiated caspases activation. The data suggest that reduction of PGC-1α enhanced mitochondrial dysfunction and thus exacerbated high glucose-induced apoptosis. It is well recognized that mitochondrial membrane permeability is controlled by the balance between anti-apoptotic protein Bcl-2 and pro-apoptotic protein Bax [22]. When this balance is disrupted, the mitochondrial membrane is destroyed, which leads to activation of downstream apoptotic signaling cascades [25,26]. Here, although some differences were observed in the levels of these two proteins following exposure to high glucose, lack of PGC-1α did not produced any effects on these alterations. ROS generation has been suggested to be a major inducer of mitochondrial dysfunction and play an important role in the regulation of ECs apoptosis [14,15]. However, the analysis of ROS generation did not support the enhanced apoptosis in PGC-1α knockdown cells as it demonstrated that high glucose-induced ROS generation was indistinguishable before and after infection with Ad-shPGC-1α. Of note, recent studies indicate that PGC-1α also plays a crucial role in the regulation of mitochondrial antioxidants and oxidative stress [27,28]. For example, PGC-1α knockout mice showed decreased antioxidants expression, increased ROS generation and enhanced susceptibility to oxidative stress-mediated cell death [11,29]. Thus, different cell type and/or different apoptotic stimuli should be noted. In addition, we also found that ER stress was not involved in the increased apoptosis in high glucose-treated PGC-1α knockdown cells because PGC-1α deficiency, even more, inhibited high glucose-induced ER stress. Therefore, our results indicate that the enhanced sensitivity to high glucose-induced ECs apoptosis in PGC-1α deficiency cells is not likely due to changes in apoptosis-associated regulatory proteins, ROS generation and ER stress. The voltage-dependent anion channel (VDAC) located in the outer mitochondrial membrane is a critical player in the regulation of many physiological and pathophysiological processes, including energy metabolism [30,31], and cell apoptosis [23,32]. Previous studies have demonstrated that induction of VDAC1 promoted cell apoptosis [33] and a plethora of apoptosis inducers was accompanied by highly increased VDAC1 expression [34]. Intriguingly, we found that PGC-1α deficiency merely augmented VDAC1 protein expression and pharmacological inhibition of VDAC1 significantly inhibited the increased apoptosis in high glucose-treated PGC-1α knockdown cells, further indicating PGC-1α protects ECs apoptosis through activation of VDAC1. In summary, our study demonstrated PGC-1α deficiency potentiated high glucose-induced VADC1 expression and activated mitochondria-dependent pathway, which resulted in the increased HUVECs apoptosis. These findings revealed that down-regulation of PGC-1α is one of the major contributors for ECs apoptosis, suggesting PGC-1α may be a rational therapeutic strategy to protect cardiovascular complications in diabetes.
Disclosure of conflict of interest
None.
References
- 1.Steiner G. Dyslipoproteinemias in diabetes. Clin Invest Med. 1995;18:282–287. [PubMed] [Google Scholar]
- 2.Bhattacharjee PS, Huq TS, Potter V, Young A, Davenport IR, Graves R, Mandal TK, Clement C, McFerrin HE, Muniruzzaman S, Ireland SK, Hill JM. High-glucose-induced endothelial cell injury is inhibited by a Peptide derived from human apolipoprotein E. PLoS One. 2012;7:e52152. doi: 10.1371/journal.pone.0052152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Triggle CR, Samuel SM, Ravishankar S, Marei I, Arunachalam G, Ding H. The endothelium: influencing vascular smooth muscle in many ways. Can J Physiol Pharmacol. 2012;90:713–738. doi: 10.1139/y2012-073. [DOI] [PubMed] [Google Scholar]
- 4.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 5.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 6.Hammarstedt A, Jansson PA, Wesslau C, Yang X, Smith U. Reduced expression of PGC-1 and insulin-signaling molecules in adipose tissue is associated with insulin resistance. Biochem Biophys Res Commun. 2003;301:578–582. doi: 10.1016/s0006-291x(03)00014-7. [DOI] [PubMed] [Google Scholar]
- 7.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 9.Zhu L, Sun G, Zhang H, Zhang Y, Chen X, Jiang X, Jiang X, Krauss S, Zhang J, Xiang Y, Zhang CY. PGC-1alpha is a key regulator of glucose-induced proliferation and migration in vascular smooth muscle cells. PLoS One. 2009;4:e4182. doi: 10.1371/journal.pone.0004182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim HK, Song IS, Lee SY, Jeong SH, Lee SR, Heo HJ, Thu VT, Kim N, Ko KS, Rhee BD, Jeong DH, Kim YN, Han J. B7-H4 downregulation induces mitochondrial dysfunction and enhances doxorubicin sensitivity via the cAMP/CREB/PGC1-alpha signaling pathway in HeLa cells. Pflugers Arch. 2014;466:2323–2338. doi: 10.1007/s00424-014-1493-3. [DOI] [PubMed] [Google Scholar]
- 11.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 12.Fernandes RO, Bonetto JH, Baregzay B, de Castro AL, Puukila S, Forsyth H, Schenkel PC, Llesuy SF, Brum IS, Araujo AS, Khaper N, Bello-Klein A. Modulation of apoptosis by sulforaphane is associated with PGC-1alpha stimulation and decreased oxidative stress in cardiac myoblasts. Mol Cell Biochem. 2015;401:61–70. doi: 10.1007/s11010-014-2292-z. [DOI] [PubMed] [Google Scholar]
- 13.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 14.Chen H, Gao W, Yang Y, Guo S, Wang H, Wang W, Zhang S, Zhou Q, Xu H, Yao J, Tian Z, Li B, Cao W, Zhang Z, Tian Y. Inhibition of VDAC1 prevents Ca(2)(+)-mediated oxidative stress and apoptosis induced by 5-aminolevulinic acid mediated sonodynamic therapy in THP-1 macrophages. Apoptosis. 2014;19:1712–1726. doi: 10.1007/s10495-014-1045-5. [DOI] [PubMed] [Google Scholar]
- 15.Wang C, He Y, Yang M, Sun H, Zhang S, Wang C. Safflor yellow B suppresses angiotensin II-mediated human umbilical vein cell injury via regulation of Bcl-2/p22(phox) expression. Toxicol Appl Pharmacol. 2013;273:59–67. doi: 10.1016/j.taap.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 16.Chandra D, Liu JW, Tang DG. Early mitochondrial activation and cytochrome c up-regulation during apoptosis. J Biol Chem. 2002;277:50842–50854. doi: 10.1074/jbc.M207622200. [DOI] [PubMed] [Google Scholar]
- 17.Noubade R, Wong K, Ota N, Rutz S, Eidenschenk C, Valdez PA, Ding J, Peng I, Sebrell A, Caplazi P, DeVoss J, Soriano RH, Sai T, Lu R, Modrusan Z, Hackney J, Ouyang W. NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature. 2014;509:235–239. doi: 10.1038/nature13152. [DOI] [PubMed] [Google Scholar]
- 18.Morohoshi M, Fujisawa K, Uchimura I, Numano F. Glucose-dependent interleukin 6 and tumor necrosis factor production by human peripheral blood monocytes in vitro. Diabetes. 1996;45:954–959. doi: 10.2337/diab.45.7.954. [DOI] [PubMed] [Google Scholar]
- 19.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 20.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 21.Rasheva VI, Domingos PM. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis. 2009;14:996–1007. doi: 10.1007/s10495-009-0341-y. [DOI] [PubMed] [Google Scholar]
- 22.Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox regulation of cell survival. Antioxid Redox Signal. 2008;10:1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsujimoto Y, Shimizu S. The voltage-dependent anion channel: an essential player in apoptosis. Biochimie. 2002;84:187–193. doi: 10.1016/s0300-9084(02)01370-6. [DOI] [PubMed] [Google Scholar]
- 24.Dam AD, Mitchell AS, Quadrilatero J. Induction of mitochondrial biogenesis protects against caspase-dependent and caspase-independent apoptosis in L6 myoblasts. Biochim Biophys Acta. 2013;1833:3426–3435. doi: 10.1016/j.bbamcr.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 25.Mignotte B, Vayssiere JL. Mitochondria and apoptosis. Eur J Biochem. 1998;252:1–15. doi: 10.1046/j.1432-1327.1998.2520001.x. [DOI] [PubMed] [Google Scholar]
- 26.Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12:913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 27.Lu Z, Xu X, Hu X, Fassett J, Zhu G, Tao Y, Li J, Huang Y, Zhang P, Zhao B, Chen Y. PGC-1 alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid Redox Signal. 2010;13:1011–1022. doi: 10.1089/ars.2009.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang C, Li Ji L. Role of PGC-1alpha signaling in skeletal muscle health and disease. Ann N Y Acad Sci. 2012;1271:110–117. doi: 10.1111/j.1749-6632.2012.06738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci U S A. 2009;106:20405–20410. doi: 10.1073/pnas.0911570106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Shoshan-Barmatz V, Israelson A, Brdiczka D, Sheu SS. The voltage-dependent anion channel (VDAC): function in intracellular signalling, cell life and cell death. Curr Pharm Des. 2006;12:2249–2270. doi: 10.2174/138161206777585111. [DOI] [PubMed] [Google Scholar]
- 31.Saks V, Guzun R, Timohhina N, Tepp K, Varikmaa M, Monge C, Beraud N, Kaambre T, Kuznetsov A, Kadaja L, Eimre M, Seppet E. Structure-function relationships in feedback regulation of energy fluxes in vivo in health and disease: mitochondrial interactosome. Biochim Biophys Acta. 2010;1797:678–697. doi: 10.1016/j.bbabio.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 32.Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med. 2010;31:227–285. doi: 10.1016/j.mam.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 33.Keinan N, Tyomkin D, Shoshan-Barmatz V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol Cell Biol. 2010;30:5698–5709. doi: 10.1128/MCB.00165-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keinan N, Pahima H, Ben-Hail D, Shoshan-Barmatz V. The role of calcium in VDAC1 oligomerization and mitochondria-mediated apoptosis. Biochim Biophys Acta. 2013;1833:1745–1754. doi: 10.1016/j.bbamcr.2013.03.017. [DOI] [PubMed] [Google Scholar]