

Abstract
Graft versus-host disease (GVHD) severely limits the application of allogeneic hematopoietic stem cell transplantation (allo-HSCT) in treating leukemia. Dendritic cells (DCs) are critical for the development. Here, we examined the effect of proteasome inhibitor Bortezomib on DCs in vitro. Primary cultured mouse DCs were treated with Bortezomib and their proliferation was observed. The expression of CD80 and CD86 and cytokine secretion of LPS-activated DCs was also quantified under Bortezomib. The ability of DCs to activate T cells was also measured by the mixed lymphocyte reaction assay. Finally the effect of Bortezomib on nuclear translocation of NF-κB was measured by EMSA. Bortezomib can inhibit the proliferation of DCs in a dose- and time-dependent manner. It also blocked the expression of co-receptors CD80 and CD86 and secretion of cytokines IL-12 and TNF-α in DCs treated with LPS. Mixed lymphocyte reaction assay suggested Bortezomib reduced the ability of DCs to activate T cells. Finally, we found Bortezomib can inhibit the nuclear translocation of NF-κB in DCs. Our findings indicated that Bortezomib blocked the functions of DCs in various aspects, and is a potential drug candidate for GVHD.
Keywords: Bortezomib, graft versus-host disease, dendritic cells, nuclear factor-κB, T cell activation
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is currently the only available curative therapy for most types of leukemia. Graft versus host disease (GVHD), an inflammatory response in which immune cells in the transplanted marrow recognize the recipient as “foreign” and mount an immune attack against it, however, severely limits allo-HSCT’s clinical applications [1]. Antigen-presenting cells (APCs) play a pivotal role in controlling immune responses by processing antigen molecules and presenting them onto the surface of other immune cells. Thus, APCs are critical for the development of GVHD and are an important target for treating various immune disorders including GVHD [1,2].
Dendritic cells (DCs), the “professional” APCs, are derived from hemopoietic bone marrow progenitor cells. These progenitor cells initially transform into immature dendritic cells, which are specialized for endocytosis and of low T-cell activation potential [3]. These cells develop into mature dendritic cells (mDCs) once they contact with antigens such as lipopolysaccharide (LPS) [4]. mDCs then migrate to the lymphoid tissue with increased APC ability and T-cell activation potential [5]. Simultaneously, cell-surface receptors that work as co-receptors in T-cell activation such as CD80 and CD86 were upregulated with greatly enhanced ability to activate T-cells [6].
Nuclear factor kappa B (NF-κB), a widely distributed transcription factor in almost all animal cells, controls the expression of a number of important genes for immunological and inflammatory responses [7]. In activated cells, the NF-κB dimers are sequestered in the cytoplasm by a family of inhibitors, called IκBs (Inhibitor of κB) [8]. Activation of NF-κB is initiated by the signal-induced degradation of IκB proteins, making the NF-κB complex enter the nucleus where it can ‘turn on’ the expression of specific genes with NF-κB-binding sites [8]. These NF-κB activated then lead to certain physiological responses including those of pathophysiological characteristics of GVHD [9]. Moreover, NF-κB pathway has also been shown to be involved in the maturation and antigen presentation of DCs [10,11].
Bortezomib (PS-341; Velcade), a selective inhibitor of the 26S proteasome, has been established as an effective drug against plasma cell myeloma [12]. Among multiple mechanisms have been reported underlying its antitumor activity, Bortezomib has been demonstrated to inhibit IκB degradation and thus subsequent activation of NF-κB [13]. In addition to antitumor effects, the inhibition of NF-κB pathway makes Bortezomib a potential drug candidate against GVHD. Evidences from animal models support the potential role of Bortezomib in treating GVHD [9,14], although the exact mechanism is still not clear. We therefore hypothesized that Bortezomib prevents GVHD by modulating DCs functions via examining the effect of Bortezomib on DCs in vitro.
Materials and methods
Animals
C57BL/6 (B6, H2b) male mice between 6 to 8 weeks old with body weights between 18 and 22 g were purchased from Slac Laboratory Animal Co. Ltd (Shanghai, China). All animals were handled in accordance with institutional and governmental directions and were approved by local authorities.
Preparation of mouse bone marrow derived DCs
Mouse DCs were prepared following Inaba et al protocol with modification [15]. Briefly, C57BL/6 mice were sacrificed by cervical dislocation. Femur and tibia were isolated aseptically and bone marrow (BM) cells were washed out using serum-free PRMI1640 medium. After removing red blood cells by the buffer (0.15 M NH4Cl, 0.01 mM KHCO3, 0.1 mM EDTANa2), BM cells were cultured in PRMI1640 medium containing 10% FBS (GIBCO, US), 100 U/ml ampicillin, 100 μg/ml Streptomycin, 2 mmol/L Glutamine and 10 ng/ml recombinant murine granulocyte-macrophage colony stimulating factor (rmGM-CSF, Clongene, China) and 1 ng/ml recombinant murine IL-4 (rmIL-4, PeproTech, US). Two days later, medium and floating cells were gently removed and the attached cells were feeded with fresh complete medium containing rmGM-CSF and rmIL-4. The medium were changed by half every second day. Suspended cells were collected at day 6 and treated with LPS or together with Bortezomib at different concentrations.
Proliferation assay
The affection of Bortezomib to the proliferation of DCs was examined with CCK-8 kit (Dojindo, Japan). When DCs were cultured at day 6, suspended cells were collected by centrifuging at 1000 g for 5 minutes. Cells were seeded in 96-well plate at a density of 5 × 105/ml and treated with serial diluted Bortezomib (in PBS, Janssen, China) for 24 hours, 48 hours and 72 hours, respectively. The proliferation of cells was evaluated by CCK-8 assay according to manufacturer’s instructions. The value of optical density (OD) was measured with a Thermomax microplate reader according to the absorbance at 450 nm.
Flow cytometry
DCs cells were seeded in 24-well plate at a density of 1 × 106/ml. Cells were treated with LPS or Bortezomib plus LPS at different concentrations. 24 hours later, cells were collected and washed twice with PBS, and then were labeled with FITC or PE conjugated antibodies against CD11C, CD80 or CD86 (BD Bioscience, US) followed by fixation and permeabilization. Labeled cells were measured on FACScan flow cytometer (Becton Dickinson, US).
Mixed lymphocyte reaction
Splenocytes were obtained from BALB/C mice and used as responder cells. G-irradiated (30 Gy cyanocobalamin 60Co) DCs cells were used as trophoblasts. Splenocytes and G-irradiated DCs were mixed at different ratios (1:1, 10:1 and 100:1) and co-cultured in 96-well plate at a density of 1 × 105 cells per well. Mixed cells were treated with 100 nM nortezomib for 5 days and cell proliferation was evaluated by CCK-8 assay described above.
Enzyme-linked immunosorbent assay (ELISA)
The supernatant of DCs treated with LPS or LPS plus Bortezomib for 24 hours were harvested. The expression level of IL-12 and TNF-α were measured using ELISA kit (Dakewe BioTech, China) according to manufacturer’s instructions.
EMSA assay for NK-κB
DNA binding of NF-κB in DCs was evaluated by EMSA. DCs treated with LPS or LPS plus Bortezomib at various concentrations for 24 hours, were harvested and their nuclear proteins were purified. The nuclear translocation of NFκB was measured by NFκB EMSA kit (Viagene Biotech, China) according to manufacturer’s instructions.
Results
Bortezomib represses the proliferation of DCs
To verify our hypothesis that Bortezomib can modulate the function of DCs, we firstly examined the proliferation of DCs treated with Bortezomib at different doses or time periods. As shown in Figure 1, the proliferation of DCs was significantly inhibited when treating with Bortezomib at 10 nM for 24 hours compared to control cells. The inhibition was significantly potentiated with higher concentrations of Bortezomib. Compared to untreated cells, less than 40% cells were detected when treated with Bortezomib at 50 nM for 24 hours. Interestingly, we found the inhibition is not linearly related to the concentrations, as cells were sensitive to Bortezomib below 20 nM. However, only little increased inhibition was observed from 20 nM to 50 nM. When cells were treated with Bortezomib at the same concentration but with different time periods, the inhibition was consistently increased when cells were treated with longer time (Figure 1). Indeed up to 80% inhibition was found when cells were treated with Bortezomib at 10 nM for 72 hours. The abovementioned dose-dependent patterns repeated when cells were treated for 48 hour and 72 hours. Together, our findings suggested that the inhibition of Bortezomib to DCs is both dose- and time-dependent.
Figure 1.
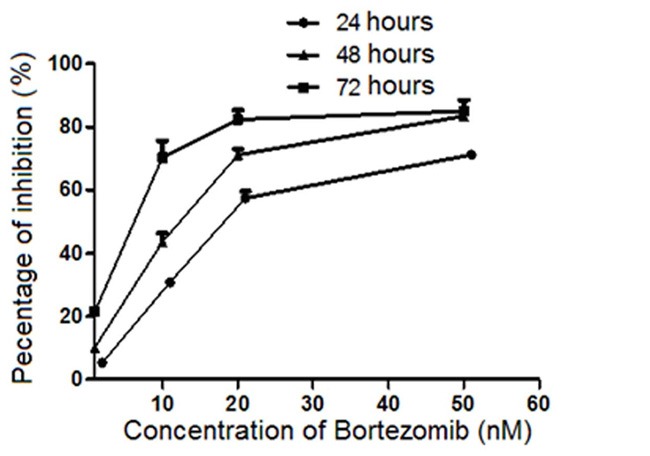
Proliferation inhibition analysis of DCs. was treated with Bortezomib at different concentrations and time. The proliferation inhibition of DCs was measured with a Thermomax microplate reader at 450 nm absorbance.
Bortezomib inhibits the expression of cell surface co-receptors in DCs
Upregulated expression of cell-surface receptors is one of hallmarks of mature DCs and is critical for its ability to activate T-cells during immune response [Carreno BM and Collins M. 2002]. To evaluate the effect of Bortezomib on the expression of co-receptors on DCs’ cell surface, flow cytometry was used to detect the expression of CD80 and CD86 on DCs treated with LPS or LPS plus Bortezomib at different doses for 24 hours. As shown in Figure 2, expressions of co-receptors CD80 and CD86 were strongly increased in DCs treated with LPS only compared to untreated cells, suggesting that DCs isolated from mouse bone marrow can be efficiently transformed into mature DCs by LPS in vitro. When DCs were treated with LPS plus Bortezomib, expressions of CD80 and CD86 were dose-dependently decreased. Comparing to cells treated with only LPS, less than half of CD80 and CD86 were detected when DCs were co-treated with LPS and Bortezomib at 100 nmol/L, suggesting that Bortezomib can strongly inhibit the upregulation of co-receptors CD80 and CD86 in LPS-activated DCs.
Figure 2.
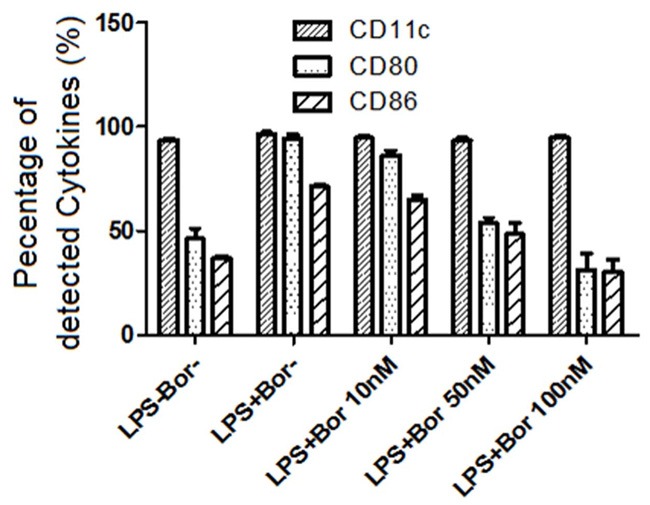
Bortezomib inhibit the upregulation of CD80 and CD86 in LPS-activated DCs. CD11c was used as a control cytokine. The expression of CD11C, CD80 and CD86 were measured by ELISA.
Bortezomib modulates the secretion of IL-12 and TNF-α
Stimulating DCs in vivo with microbial extracts causes the dendritic cells to rapidly begin producing and secreting cytokines such as IL-12 and TNF-α [16]. IL-12 is a signal that helps to transform naive CD4 T cells towards Th1 phenotype, which leads to priming and activation of the immune system for attacking antigens which the dendritic cell presents on its surface. To evaluate the effect of Bortezomib on the secretion of cytokines, we used ELISA to detect IL-12 and TNF-α in the supernatant of cultured DCs that were treated with LPS or LPS plus Bortezomib. As shown in Figure 3, low level of IL-12 and TNF-α were detected in untreated DCs. When cells were treated with LPS, expressions of both IL-12 and TNF-α were strongly increased up to 10 folds, suggesting that DCs can be efficiently stimulated to secrete cytokines by LPS in vitro. When DCs were treated with both LPS and Bortezomib, IL-12 and TNF-α showed dose-dependent decrease expressions. There was no significant change of detected cytokine levels when cells were treated with Bortezomib at 10 nM. Strongly decreased IL-12 and TNF-α, however, were found when the concentration of Bortezomib increased to 50 nM. The expression level of IL-12 and TNF-α were even lower than untreated cells at 100 nM Bortezomib at, suggesting that Bortezomib inhibits the secretion of IL-12 and TNF-α in LPS-activated DCs.
Figure 3.
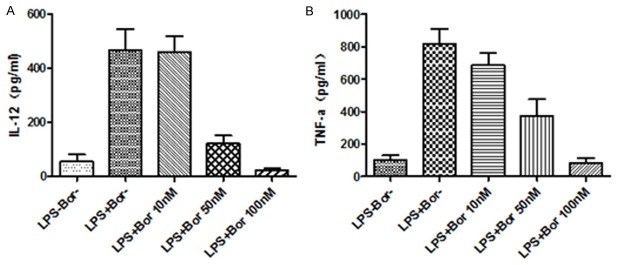
Bortezomib inhibits the secretion of IL-12 and TNF-α in LPS-activated DCs. The expression level of IL-12 (A) and TNF-α (B) were examined by ELISA as described in Methods.
Bortezomib repressed the ability of DCs to induce T-cell proliferation
The most typical feature of DCs is their ability to induce T cell proliferation. When T cells were co-cultured with DCs at different ratios, T cells showed ratio-dependent growth as more DCs induced rapid T cells growth (Figure 4). When the co-cultured cells were treated with Bortezomib, T cells showed much lower proliferation than control cells (P<0.001), especially at 10:1 ratio. Our data demonstrated that treatment with Bortezomib reduced the capacity of DCs to induce the proliferation of T cells.
Figure 4.
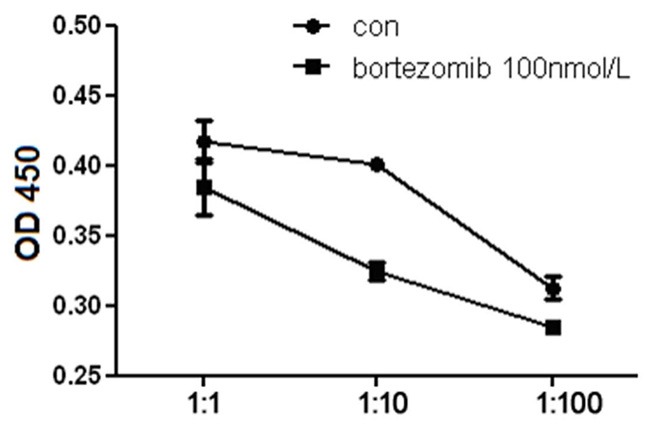
Proliferation analysis of T cells co-cultured with DCs. T cells were co-cultured with DCs with or without Bortezomib at 100 nmol/L. 5 days later, the proliferation of T cells were measured with a Thermomax microplate reader at 450 nm absorbance.
Bortezomib blocked the nuclear translocation of NF-κB in DCs
Since NF-κB targets can lead to physiological responses of GVHD [9], we asked if Bortezomib can block the activity of NF-κB in DCs. NF-κB complex in cytoplasm will enter into the nucleus when it is activated. Compared to control cells treated with only LPS, DCs treated with both LPS and Bortezomib had much lower NF-κB levels in the nuclear fraction (Figure 5). Only trace amounts of NF-κB were detected when Bortezomib was increased to 100 nM, indicating that Bortezomib blocks the nuclear translocation of NF-κB from cytoplasm, thus inhibiting the activation of NF-κB in DCs.
Figure 5.
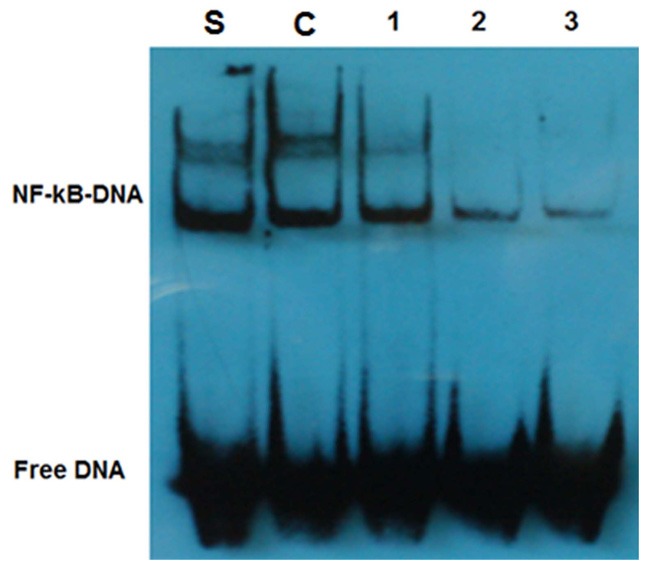
Examination of the nuclear translocation of NF-κB by EMSA. S. standard sample; C. control DCs, 1, 2 and 3, DCs treated with Bortezomib at 10 nmol/L, 50 nmol/L and 100 nmol/L, respectively.
Discussion
In the present in vitro study, we examined the effect of proteasome inhibitor Bortezomib on bone marrow derived DCs in cell proliferation, expression of co-receptors, cytokine secretions, and activation of T cells, in addition to the nuclear translocation of NF-κB. Data about plasma levels of Bortezomib in patients with advanced solid tumors have shown peak concentrations ranging between 10 and 100 nM [17], our data demonstrated that Bortezomib at 20 nM can strongly inhibit the proliferation of DCs while 100 nM Bortezomib can strongly block the expression of CD80 and CD86 in DCs treated with LPS, and Bortezomib at 50 nM can significantly block the secretion of IL-12 and TNF-α in LPS-activated DCs. Bortezomib also reduced the ability of DCs in activating T cells. All these findings indicated that Bortezomib has the potency to inhibit phenotypic maturation of imDCs, providing a basis for the immunosuppressive effects. Finally, we found that Bortezomib blocked the nuclear translocation of NF-κB in DCs, providing a possible mechanism for the functional inhibition of DCs by Bortezomib.
Bortezomib has been shown to inhibit NF-κB activity in multiple myeloma cells by blocking IκB degradation [18]. Additionally, Yoshimura et al. study found that NF-κB was an effective target for blocking DC antigen presentation and inhibiting T-cell-dependent immune responses [11], suggesting that NF-κB pathway may be the mechanism of Bortezomib-induced inhibition of DCs. This mode was supported by our findings that Bortezomib reduced the ability of DCs in activating T cells and blocked the nuclear translocation of NF-κB in DCs. Because active protein synthesis was found to represent an upstream prerequisite for Bortezomib-induced DC apoptosis , it could be speculated that observed effects in DCs with Bortezomib may be due to active NF-κB-dependent protein synthesis in imDCs upon maturation stimuli.
In summary, our data provide new insights into the functions of Bortezomib on DCs in vitro. It is worth to further examine the modulation of DCs in animal model and finally to provide a potential drug candidate for GVHD in the future.
Disclosure of conflict of interest
None.
References
- 1.Ferrara JL, Reddy P. Pathophysiology of graft-versus-host disease. Semin Hematol. 2006;43:P3–10. doi: 10.1053/j.seminhematol.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Duffner U, Lu B, Hildebrandt GC, Teshima T, Williams DL, Reddy P, Ordemann R, Clouthier SG, Lowler K, Liu C, Gerard C, Cooke KR, Ferrara JL. Role of CXCR3-induced donor T-cell migration in acute GVHD. Exp Hematol. 2003;31:897–902. doi: 10.1016/s0301-472x(03)00198-x. [DOI] [PubMed] [Google Scholar]
- 3.Sanchez-Sanchez N, Riol-Blanco L, de la Rosa G, Puig-Kröger A, García-Bordas J, Martín D, Longo N, Cuadrado A, Cabañas C, Corbí AL, Sánchez-Mateos P, Rodríguez-Fernández JL. Chemokine receptor CCR7 induces intracellular signaling that inhibits apoptosis of mature dendritic cells. Blood. 2004;104:619–25. doi: 10.1182/blood-2003-11-3943. [DOI] [PubMed] [Google Scholar]
- 4.Shakhar G, Lindquist RL, Skokos D, Dudziak D, Huang JH, Nussenzweig MC, Dustin ML. Stable T cell-dendritic cell interactions precede the development of both tolerance and immunity in vivo. Nat Immunol. 2005;6:707–14. doi: 10.1038/ni1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banchereau J, RM Steinman. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 6.Carreno BM, Collins M. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu Rev Immunol. 2002;20:29–53. doi: 10.1146/annurev.immunol.20.091101.091806. [DOI] [PubMed] [Google Scholar]
- 7.Kaisho T, Tanaka T. Turning NF-kappaB and IRFs on and off in DC. Trends Immunol. 2008;29:329–36. doi: 10.1016/j.it.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 8.Shih VF, Tsui R, Caldwell A, Hoffmann A. A single NFkappaB system for both canonical and non-canonical signaling. Cell Res. 2011;21:86–102. doi: 10.1038/cr.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vodanovic-Jankovic S, Hari P, Jacobs P, Komorowski R, Drobyski WR. NF-kappaB as a target for the prevention of graft-versus-host disease: comparative efficacy of bortezomib and PS-1145. Blood. 2006;107:827–34. doi: 10.1182/blood-2005-05-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ouaaz F, Arron J, Zheng Y, Choi Y, Beg AA. Dendritic cell development and survival require distinct NF-kappaB subunits. Immunity. 2002;16:257–70. doi: 10.1016/s1074-7613(02)00272-8. [DOI] [PubMed] [Google Scholar]
- 11.Yoshimura S, Bondeson J, Brennan FM, Foxwell BM, Feldmann M. Role of NFkappaB in antigen presentation and development of regulatory T cells elucidated by treatment of dendritic cells with the proteasome inhibitor PSI. Eur J Immunol. 2001;31:1883–93. doi: 10.1002/1521-4141(200106)31:6<1883::aid-immu1883>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 12.Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. Proteasome inhibition as a novel therapeutic target in human cancer. J. Clin. Oncol. 2005;23:630–9. doi: 10.1200/JCO.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 13.Sunwoo JB, Chen Z, Dong G, Yeh N, Crowl Bancroft C, Sausville E, Adams J, Elliott P, Van Waes C. Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin Cancer Res. 2001;7:1419–28. [PubMed] [Google Scholar]
- 14.Sun K, Welniak LA, Panoskaltsis-Mortari A, O’Shaughnessy MJ, Liu H, Barao I, Riordan W, Sitcheran R, Wysocki C, Serody JS, Blazar BR, Sayers TJ, Murphy WJ. Inhibition of acute graft-versus-host disease with retention of graft-versus-tumor effects by the proteasome inhibitor bortezomib. Proc Natl Acad Sci U S A. 2004;101:8120–5. doi: 10.1073/pnas.0401563101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reis e Sousa C, Hieny S, Scharton-Kersten T, Jankovic D, Charest H, Germain RN, Sher A. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J Exp Med. 1997;186:1819–29. doi: 10.1084/jem.186.11.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papandreou CN, Daliani DD, Nix D, Yang H, Madden T, Wang X, Pien CS, Millikan RE, Tu SM, Pagliaro L, Kim J, Adams J, Elliott P, Esseltine D, Petrusich A, Dieringer P, Perez C, Logothetis CJ. Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J. Clin. Oncol. 2004;22:2108–21. doi: 10.1200/JCO.2004.02.106. [DOI] [PubMed] [Google Scholar]
- 18.Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A, Palombella V, Adams J, Anderson KC. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277:16639–47. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]