

Abstract
3,3’-Diindolylmethane (DIM) is a natural component of cruciferous plants. Previous studies have shown that DIM has multiple physiological effects including anti-angiogenic, anti-inflammatory and anti-cancer effect. However, little is known about the role of DIM on myofibroblast differentiation and extracellular matrix (ECM) production. This study investigated the effect of DIM on myofibroblast differentiation and ECM production in neonatal rat cardiac fibroblasts induced by transforming growth factor β1 (TGF-β1). We found that DIM blunted TGF-β1 induced conversion of cardiac fibroblast into myofibroblast, and reduced the mRNA and protein expressions of α-smooth muscle actin (α-SMA). Furthermore, DIM also significantly decreased the mRNA expression of fibrosis markers (Collagen I, Collagen III, connective tissue growth factor (CTGF) in neonatal rat cardiac fibroblasts induced by TGF-β1. DIM attenuated the phosphorylation AKT and glycogen synthase kinase-3β (GSK-3β) induced by TGF-β1. Our results showed that DIM was a potential drug to attenuate myofibroblast differentiation and excessive ECM production induced by TGF-β1 through down-regulated AKT/GSK-3β signaling pathways.
Keywords: DIM, cardiac fibrosis, myofibroblast, AKT, GSK3β
Introduction
Cardiac fibrosis is characterized by accumulation of cardiac fibroblasts and excessive deposition of extracellular matrix (ECM) [1]. Pathological cardiac fibrosis can lead to abnormal myocardial stiffness, and is involved in almost all the cardiac diseases, predisposing patients to the risk of heart failure [2]. Cardiac fibroblasts are the main determinant of cardiac fibrosis [3]. Following cardiac injury, cardiac fibroblasts release cytokines, which are involved in a feed-forward loop that lead to the increased proliferation of cardiac fibroblasts, re-expression and up-regulation of the markers initially expressed in the embryonic and homeostatic cardiac fibroblasts, and eventually myofibroblast accumulation [4]. Cardiac fibroblasts activation is characterized by increased proliferation, transformation into myofibroblast, which subsequently leads to cardiac fibrosis [5,6]. The major source of pro-fibrotic cytokine TGF-β1 are cardiac fibroblasts and myofibroblasts in the heart [7], and TGF-β1, secreted by cardiac fibroblasts, can stimulate myofibroblast differentiation and excessive ECM production, which then drive cardiac fibrosis progression [8]. Recent studies pay more attention to the role of myofibroblast in cardiac fibrosis [9,10]. It is widely acknowledged that cardiac fibroblasts and myofibroblast are major targets for treating cardiac disease [3], Therefore the effective therapies and new molecular target to inhibit myofibroblast differentiation is urgent need.
DIM (3,3’-Diindolylmethane ), derived from Brassica plants, is a major metabolic product of indole-3-carbinol (I3C), which is also widely used as pharmaceutical intermediate [11]. Previous studies showed that DIM had a number of physiological effects, including anti-angiogenic [12], anti-inflammatory [13], and anti-cancer effect [14]. Moreover, recent studies have found that DIM has the effect on cardiac hypertrophy and fibrosis. Cardiac hypertrophy and fibrosis induced by pressure overload in mice was attenuated by DIM, which could contribute to the activation of 5’-adenosine monphosphate-activated protein kinase-a2 (AMPKα) and attenuate mammalian target of the rapamycin (mTOR) signaling pathways [15].
In animal experiment, previous study suggested that DIM could protect against cardiac hypertrophy and fibrosis induced by pressure overload. Moreover it was of remarkably clinical relevance that DIM could reverse pre-established cardiac dysfunction induced by chronic pressure overload. However, it was unclear that the effect of DIM on cardiac fibrosis was directly acted on cardiac fibroblasts. Therefore in this study, we established a valid model of cardiac fibroblasts proliferation and myofibroblast differentiation in neonatal rat cardiac fibroblasts induced by TGF-β1 to observe the effect of DIM on myofibroblast differentiation and ECM production, and meanwhile elucidated its molecular mechanisms.
Materials and methods
Materials
The Dulbecco’s Modified Eagle’s medium/F12 (DEME/F12) was obtained from Corning (GIBCO, C113305). The fetal bovine serum (FBS) was purchased from Hyclone (Logan, UT, USA). The cell counting kit-8 (CCK-8) was Dojindo (Kumamoto, Japan). TRIzol reagent (Invitrogen, 11667-165). The primary antibodies against total and phosphorylated AKT, GSK-3β and GAPDH were purchased from cell signaling technology (Danvers, MA, USA) and the secondary antibodies were from LI-COR Biosciences (Lincoln, NE, USA). DIM (98% purity as determined by HPLC analysis) was purchased from Shanghai Medical Technology Development Co., Ltd, Harmony.
Isolation and culture of cardiac fibroblasts
The neonatal rat cardiac fibroblasts were obtained from the Sprague-Daw rats born within three days. The neonatal rats were sacrificed because the hearts were taken. Firstly, the hearts were placed in a 100 mm dish with cold DMEM/F12, and mined to approximately 1 mm3 with scissors. The heart tissues were digested in 0.125% tryspin for 15 min at 34°C when gently shaking all the time, and the digestion was repeated five times to get single cells. All digestive fluid was collected and centrifuged at 1000 rpm for 8 minutes. The cells were resuspended and filtered through 75 µm cell strainer. Finally isolated cardiac fibroblasts were seeded on 100 mm plates for 90 min, after removing pre-seeding medium containing cardiomyocytes, the left were the cardiac fibroblasts which could be adhere to the culture plates. The cardiac fibroblasts were cultured in DMEM/F12 containing 10% FBS, penicillin (100 IU/ml) and streptomycin(100 mg/ml) stayed at 37°C in a humidified incubator (SANYO 18 M) with 5% CO2. According to experiment need, cells were seeded different densities. 96-well plates were 5×104 cells/ml, 24-well plates were 5×103 cells/ml, Cells were seeded in six-well plates and 100 mm plates at a density of 1×105/ml. All neonatal rat cardiac fibroblasts in this study were treated within three passage cultures. Before treated with TGF-β1 and DIM, Cells were cultured in 1% FBS for 12 h.
CCK-8
Cell Counting Kit-8 (CCK-8) was used to examine the cardiac fibroblast viability treated with DIM and the ability of cardiac fibroblast proliferation induced by TGF-β1. Firstly in order to test the cell toxicity of DIM, Cells (5×104 cells/ml) were seeded in 96 wells and cultured for 48 h, every concentration has 6-well microplates. The cells were cultured in 1% FBS for 12 h, and then treated with different concentrations of DIM (10 μmol/l, 20 μmol/l, 30 μmol/l and 60 μmol/l) for 48 h. Each well was added 10 μl of CCK-8 reagent for another 4 h incubation, and examined the absorbance read with a microplate reader (Bio-Tek, Synergy HT) at 450 nm (the absorption wavelength) and 630 nm (the reference wavelength). According to the same protocol, examining the ability of cardiac fibroblasts proliferation treated with TGF-β1 and (or) DIM was needed.
Immunofluorescence staining
To examine the percentage of positive cardiac fibroblasts which expressed a-SMA protein, we stained neonatal rat cardiac fibroblasts for the marker a-SMA. The cells treated as experiment design were washed three times with warm PBS, fixed with 4% paraformladehyde (Sinopharm Chemical Reagent Co., Ltd, 41533) for 15 min, permeabilized in 0.2% Triton X-100 (Amresco, 0694) in PBS, blocked with 8% goat serum, and then stained with anti-α-SMA antibody (Abcam, ab7817) at a dilution of 1:100 with 1% goat serum (GeneT-ex, GTX27481) for overnight. The cells were incubated for 1h with the secondary antibody, goat anti-Mouse lgG Alexa Fluor 488, and then mounted onto glass slides with Slow Fade Gold anti-fade reagent with DAPI. The number of positive cells was measured with a quantitative digital image analysis system (Image Pro-Plus, version 6.0).
Quantitative real-time PCR
Total RNA was extracted from neonatal rat cardiac fibroblasts with TRIzol reagent, according to the instructions, the concentrations were detected by spectrophptpmeter and the purities were estimated using the A260/A230 and A230/A260 ratios with a Smartspec Plus Spectrophotometer (Bio-Rad). The RNA (2 μg of each sample) was reverse transcribed into cDNA using oligo (dT) primers and the Tran-scrptor First Strand cDNA Synthesis Kit (Roche, 04887352001) in 20 ul reaction volume with a 96-well thermal cycler (Applied Biosystems). We used SYBR Green PCR Master Mix (Roche, 04897030001) to quantify PCR amplifications with the Light Cycler 480 instrument (Software version 1.5, Roche) and examined the relative mRNA expression levels of Collagen I, Collagen III, CTGF and a-SMA , which were normalized to the glyceraldehydes-3-phosphate dehydrogenase (GAPDH) gene expression which was a house keeping gene. The following were PCR primers: Collagen I forward: 5’-GAGAGAGCATGACCGATGGATT-3’, and reverse: 5’-TGGACATTAGGCGCAGGAA-3’; Collagen III forward: 5’-GTGGTCCTCCAGGAGAAAATGGAAA-3’, reverse: 5’-GCACCCGCACCGCCTGGCTCAC-3’; CTGF forward: 5’-GGAAGACACATTTGGCCCTG3’, and reverse: 5’-GCAATTTTAGGCGTCCGGAT-3’; a-SMA forward: 5’-GCTATTCAGGCTGTGCTGTC-3’, and reverse: 5’-GGTAGTCGGTGAGATCTCGG-3’; GAPDH: forward: TCGGTGAGATCTCGG-3’; GAPDH: forward: 5’-GACATGCCGCCTGGAGAAAC-3’, and reverse: 5’-AGCCCAGGATGCCCTTTAGT-3’. The reaction was run at 95°C for 10min, followed by 45 cycles of 10 sec at 95°C, 10 sec at 60°C and 20 sec at 72°C.
Western blotting
The total proteins were extracted from neonatal rat cardiac fibroblasts, the protein concentrations were determined with the BCA protein assay reagent (Thermo, 23225). Proteins samples (20 μg) in a gel were separated on 10% SDS/PAGE under reducing conditions, and then transferred onto an polyvinylidene fluoride (PVDF) membrane (Millipore, Beijing, China) using a Gel Transfer Device (Invitrogen). In order to avoid the disturb of the non-specific antibody binding, PVDF membranes were blocked with 5% skim milk for 1.5 h at room temperature. After PVDF membranes were incubated with primary antibodies against phosphorylated AKT, total AKT, phosphorylated GSK3β, total GSK3β, a-SMA and GAPDH for over-night at 4°C, secondary antibodies were conjugated with primary antibodies for 1 h. The quantification of Western blotting bands was performed with the Odyssey infrared imaging system (Li-Cor Biosciences). Specific protein expression levels were normalized to the GAPDH protein level which contained total cell lysate and cytosolic proteins on the same PVDF membrane.
Statistical analysis
All quantitative data were expressed as mean ± SEM. The differences between two groups were used the Independent-Samples T test. One-way ANOVA test with a post hoc of Tukey’s test was used to compare the difference among groups. A P value of <0.05 was statistically different.
Results
The toxicity of DIM on cardiac fibroblasts
Firstly, in this study we performed different doses response of DIM in neonatal rat cardiac fibroblasts. As shown in Figure 1A, the cell toxicity of DIM was not significantly different under the concentrations of 30 μmol/l (10, 20, 30 μmol/l). Compared to control group, the cell viability in 30 umol/l group was reduced by 12.4%. The group in 60 μmol/l was reduced by 25.8%, it suggested that the concentration of 60 μmol/l on neonatal rat cardiac fibroblasts had dramatical cell toxicity. Therefore, in this study we chose the doses (10 μmol/l, 20 μmol/l and 30 μmol/l) to study changes in cardiac fibroblasts activation.
Figure 1.
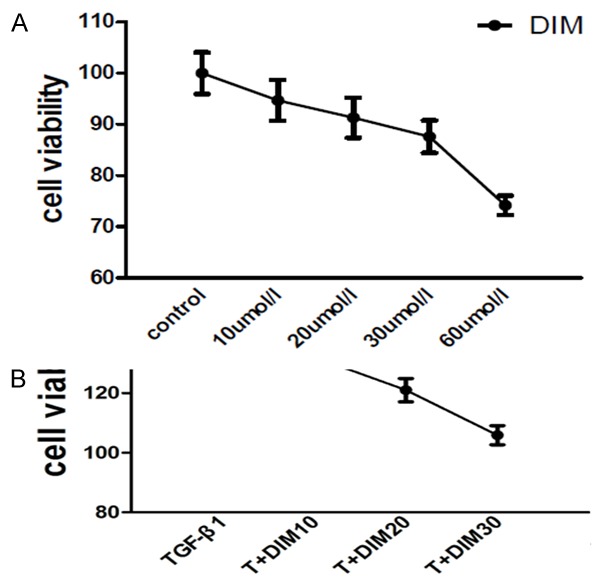
The viability of neonatal rat cardiac fibroblasts. A: The effect of DIM on the cell viability. Low dose of DIM (10, 20, 30 µmol/l) has no cell toxicity on neonatal rat cardiac fibroblasts, however, high dose of DIM (60 µmol/l) has remarkable cell toxicity. B: The proliferative response of neonatal cardiac fibroblasts induced by TGF-β1 (10 ng/ml) for 48 h have promoted, and DIM can attenuate the increased proliferative response of cardiac fibroblasts induced by TGF-β1. T is TGF-β1, OD value is examined in 450 nm (the absorption wavelength) and 630 nm (the reference wavelength), each dot represents the mean and SD.
DIM attenuates TGF-β1 induced cardiac fibroblasts proliferation
Previous study reported that TGF-β1 could increase proliferation in adult cardiac fibroblasts [16]. In order to investigate the proliferative response in neonatal rat cardiac fibroblasts induced by TGF-β1, the neonatal cardiac fibroblasts were incubated with TGF-β1 for 48 h. As shown in Figure 1B, compared to control group, the proliferative rate was increased by 38.6%. DIM could blunt the increased proliferative response induced by TGF-β1. Further analysis let us identify that DIM with dose-dependence could inhibit the proliferation of neonatal cardiac fibroblasts.
DIM’s effect on myofibroblast differentiation
There was the evidence supporting a central role for TGF-β1 in cardiac fibroblast activation [3]. TGF-β1 could stimulate the transformation of cardiac fibroblasts into myofibroblast [8], and a-SMA was a typical molecular marker of myofibroblast [17]. Therefore, we stained neonatal rat cardiac fibroblasts for the marker a-SMA, and determined the percentage of cardiac fibroblasts expressing a-SMA protein. As shown in Figure 2, in control group, the percentage of cardiac fibroblasts expressing a-SMA protein was 24.05%. It suggested that the cultured neonatal rat cardiac fibroblasts in vitro had the ability of spontaneous differentiation. Compared with control group, the percentages of cardiac fibroblasts which expressed a-SMA protein were significantly increased in TGF-β1 group, while DIM could reduce percentages and the absolute cell number of cardiac fibroblasts which expressed a-SMA protein induced by TGF-β1 for 48 h. We also respectively investigated the levels of a-SMA mRNA and protein expressions with the quantitative analysis of real time-PCR and Western blotting. Compared to control group, the mRNA and protein expressions of a-SMA were promoted in the presence of TGF-β1. However, the increased levels of a-SMA mRNA and protein expressions in neonatal rat cardiac fibroblasts induced by TGF-β1 were blunted by DIM. Additionally, there was no statistical difference between DIM group and control group (Figure 2).
Figure 2.
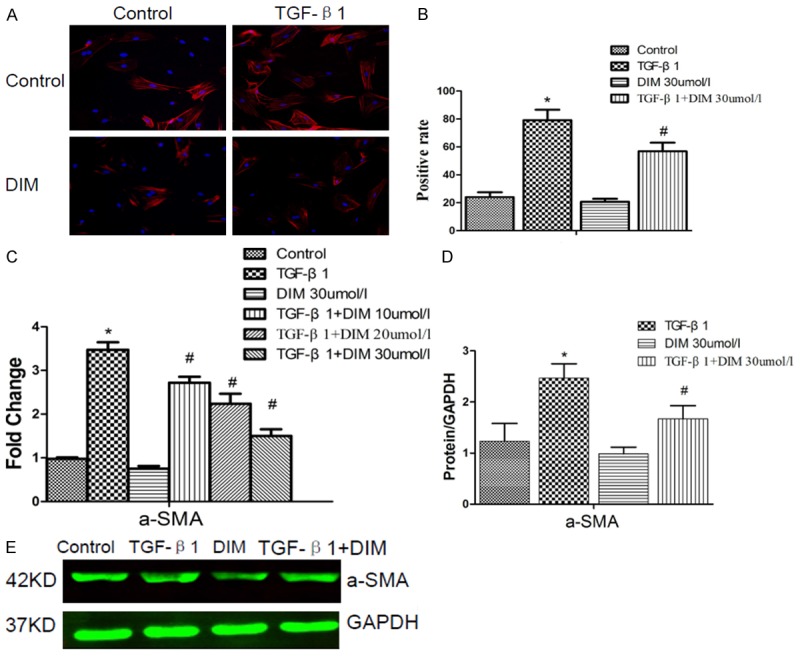
DIM attenuates cardiac myofibroblast differentiation induced by TGF-β1 for 48 h. A: DIM reduces the number of TGF-β1 induced cardiac fibroblasts which express a-SMA protein. Cardiac fibroblasts are examined with immunofluresent staining for a-SMA. Blue dot represents the nuclear of cardiac fibroblast, red represents a-SMA protein. B: Quantitative analysis of the a-SMA positive cardiac fibroblasts. C: The level of a-SMA mRNA expression is evaluated by RT-PCR. D: The image of a-SMA protein is assessed by Western blotting. E: The quantitative analysis of a-SMA protein in neonatal rat cardiac fibroblasts, the relative value is represented as normalization to GAPDH expression. The data are presented as the mean ± SD. *P < 0.05 compared to control group; #P < 0.05 compared with TGF-β1 group.
Effect of DIM on pro-fibrotic gene expression
In cardiac fibroblasts, TGF-β1 directly induced ECM gene expression, and resulted in ECM deposition [18]. The increased mRNA levels of Collagen I, Collagen III and CTGF in cardiac fibroblasts were a hallmark of cardiac fibrosis, which was considered as abnormal cardiac fibroblasts proliferation and transformation into myofibroblasts. Cardiac fibroblasts were incubation with TGF-β1 for 48 h, and there were a significant increase in mRNA expression levels of Collagen I, Collagen III and CTGF (P<0.05), compared to control group (Figure 3). However, the increased mRNA expression levels of Collagen I, Collagen III and CTGF were significantly inhibited by DIM. In addition, compared with control group, the mRNA expression levels of cardiac fibroblasts treated with DIM has no significant difference.
Figure 3.
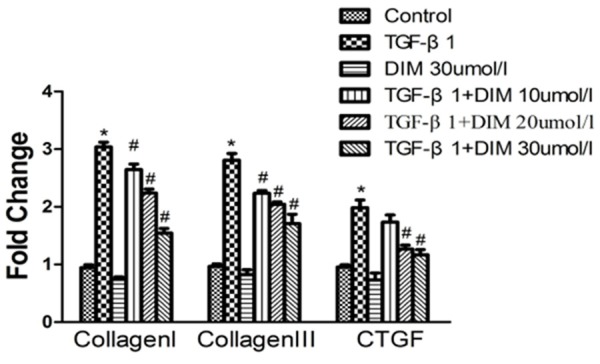
DIM blunts pro-fibrotic mRNA levels of Collagen I, Collagen III and CTGF in neoatal rat cardiac fibroblasts induced by TGF-β1. Quantitative data of CollagenI, Collagen III and CTGF in neonatal rat cardiac fibroblasts are evaluated by real time PCR. *P<0.05, compared to control group; #P<0.05, compared with TGF-β1 stimulation.
DIM’s effect on TGF-β1-induced AKT/GSK3β phosphorylation
Recent study found that GSK-3β was involved in regulating the transformation of cardiac fibroblast into myofibroblast [19], GSK-3β was the downstream target of AKT. Therefore, AKT/GSK3β signal pathways may be involved in the effect of DIM on myofibroblast differentiation induced by TGF-β1. Next, we investigated the expression levels of AKT and GSK3β. As shown in Figure 4, compared with control group, the results of western blot analysis suggested that the levels of phosphorylated AKT and GSK3β in neonatal rat cardiac fibroblasts induced by TGF-β1 were remarkably increased. While the increased proteins induced by TGF-β1 were significantly decreased by DIM. In addition, the levels of phosphorylated AKT and GSK3β between control group and DIM group was not significantly different.
Figure 4.
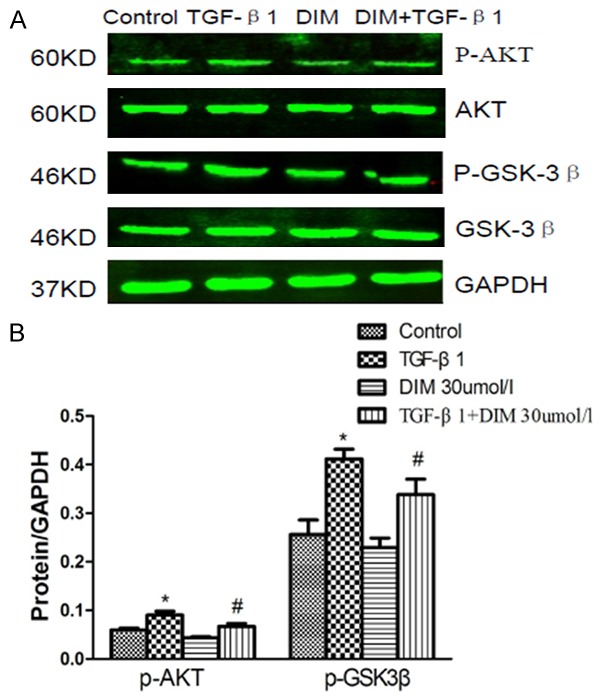
Effect of DIM on AKT/GSK3β signaling pathways. A: The images of Western blot represent the levels of phosphorylated, total AKT and GSK3β in neonatal rat cardiac fibroblasts. B: The quantitative analysis of Western blot. *P<0.05 compared to control group; #P<0.05 compared with TGF-β1 group.
Discussion
In this study, the data demonstrated that DIM could attenuate the cardiac fibroblast proliferation and myofibroblast differentiation induced by TGF-β1. Moreover, DIM could reduce excessive ECM production. We also found that the effect of DIM on myofibroblast differentiation and excessive ECM production was involved in AKT/GSK-3β signaling pathways.
Recent studies had reported that TGF-β1 could promote myofibroblast differentiation, and myofibroblast phenotype was characterized by expression of aSMA protein [20]. The level of a-SMA protein induced by TGF-β was increased [21]. However, DIM could reduce the increased positive percentage of cardiac fibroblasts induced by TGF-β1, which expressed a-SMA protein. Furthermore, DIM also could decrease the increased protein and mRNA expressions of a-SMA induced by TGF-β1. Collectedly, the results implied that DIM could attenuate myofibroblast differentiation.
Profibrotic cytokines TGF-β1 was involved in cell proliferation, differentiation and ECM production. The synthesis of collagen was increas-ed in cardiac fibroblasts induced by TGF-β1, and ultimately leaded to deposition of ECM [22,23]. Collagen I, Collagen III and CTGF, mainly produced by fibroblasts, were the important ECM proteins, and abnormal synthesis of these proteins indicated cardiac fibroblast activation [24]. Our data showed that the increased pro-fibrotic mRNA levels of Collagen I, Collagen III and CTGF in neonatal rat cardiac fibroblasts induced by TGF-β1 were reduced by DIM. Taken together, the data in our study showed that DIM could block the excessive deposition of extracellular matrix, it was consistent with the animal experiment [15].
AKT/GSK-3β signaling pathways was associated with cardiac hypertrophy [25], epithelial to mesenchymal transition [26,27], and cell cycle [28]. GSK-3β, the downstream target of AKT, had proved that the roles of GSK-3β in cardiac myocyte [29]. Recent the study found that GSK-3β was involved in myofibroblast differentiation has [30]. Therefore, AKT/GSK-3β signaling pathways may be involved in regulating the effect of DIM on myofibroblast differentiation, and eventually improved cardiac fibrosis. In this study, the phosphorylation of AKT and of GSK-3β was increased in neonatal rat cardiac fibroblasts induced by TGF-β1, and DIM could obviously decrease the expression levels of phosphorylation AKT and GSK-3β. The data indicated that the role of DIM on myofibroblast differentiation and ECM production was mediated by AKT/GSK-3β signaling pathways, and eventually achieved anti-fibrotic effect.
In conclusion, our studies showed that in-creased activation of neonatal rat cardiac fibroblasts induced by TGF-β1 were attenuated by DIM, AKT/GSK-3β signaling pathways was involved in the effect of DIM on myofibroblast differentiation and excessive ECM production, strongly indicating the potential role of DIM on cardiac fibrosis.
Acknowledgements
The project was supported by grants from National Natural Science Foundation of China (No. 81300104), Specialized Research Fund for the Doctoral Program of Higher Education of China (No. 20130141120042), and Natural Science Foundation of Hubei Province, China (No. 2013CFB303). We also sincerely thank Cardiovascualar Research Institute of Wuhan University, and Central Laboratory of Renmin Hospital of Wuhan University for providing the experimental conditions.
Disclosure of conflict of interest
None.
References
- 1.Krenning G, Zeisberg EM, Kalluri R. The Origin of Fibroblasts and Mechanism of Cardiac Fibrosis. J Cell Physiol. 2010;225:631–7. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li J, Ichikawa T, Villacorta L, Janicki JS, Brower GL, Yamamoto M, Cui T. Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. Arterioscler Thromb Vasc Biol. 2009;29:1843–50. doi: 10.1161/ATVBAHA.109.189480. [DOI] [PubMed] [Google Scholar]
- 3.Lajiness JD, Conway SJ. Origin, development, and differentiation of cardiac Fibroblasts. J Mol Cell Cardiol. 2014;70:2–8. doi: 10.1016/j.yjmcc.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bigalke B, Schwimmbeck PL, Haas CS, Lindemann S. Effect of interleukin-15 on the course of myocarditis in Coxsackievirus B3-infected BALB/c mice. Can J Cardiol. 2009;25:e248–54. doi: 10.1016/s0828-282x(09)70511-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Driesen RB, Nagaraju CK, Abi-Char J, Coenen T, Lijnen PJ, Fagard RH, Sipido KR, Petrov VV. Reversible and irreversible differentiation of cardiac fibroblasts. Cardiovasc Res. 2014;101:411–22. doi: 10.1093/cvr/cvt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olson ER, Naugle JE, Zhang X, Bomser JA, Meszaros JG. Inhibition of cardiac fibroblast proliferation and myofibroblast differentiation by resveratrol. Am J Physiol Heart Circ Physiol. 2005;288:H1131–38. doi: 10.1152/ajpheart.00763.2004. [DOI] [PubMed] [Google Scholar]
- 7.Leask A. TGF beta, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res. 2007;74:207–12. doi: 10.1016/j.cardiores.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 8.Li P, Wang D, Lucas J, Oparil S, Xing D, Cao X, Novak L, Renfrow MB, Chen YF. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ Res. 2008;102:185–92. doi: 10.1161/CIRCRESAHA.107.157677. [DOI] [PubMed] [Google Scholar]
- 9.Santiago JJ, Dangerfield AL, Rattan SG, Bathe KL, Cunnington RH, Raizman JE, Bedosky KM, Freed D, Kardami E, Dixon IM. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev Dyn. 2010;239:1573–84. doi: 10.1002/dvdy.22280. [DOI] [PubMed] [Google Scholar]
- 10.Petrov VV, van Pelt JF, Vermeesch JR, Van Duppen VJ, Vekemans K, Fagard RH, Lijnen PJ. TGF-beta1-induced cardiac myofibroblasts are non proliferating functional cells carrying DNA damages. Exp Cell Res. 2008;314:1480–94. doi: 10.1016/j.yexcr.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 11.Cho HJ, Seon MR, Lee YM, Kim J, Kim JK, Kim SG, Park JH. 3,3’-Diindolylmethane suppresses the inflammatory response to lipopolysaccharide in murine macrophages. J Nutr. 2008;138:17–23. doi: 10.1093/jn/138.1.17. [DOI] [PubMed] [Google Scholar]
- 12.Kunimasa K, Kobayashi T, Kaji K, Ohta T. Anti-angiogenic effects of indole-3-carbinol and 3,3’-diindolylmethane are associated with their differential regulation of ERK1/2 and Akt in tube-forming HUVEC. J Nutr. 2010;140:1–6. doi: 10.3945/jn.109.112359. [DOI] [PubMed] [Google Scholar]
- 13.Cho HJ, Seon MR, Lee YM, Kim J, Kim JK, Kim SG, Park JH. 3,3’-diindolylmethane suppresses the inflammatory response to lipopolsacchride in murine macrophages. J Nutr. 2008;138:17–23. doi: 10.1093/jn/138.1.17. [DOI] [PubMed] [Google Scholar]
- 14.Chen D, Banerjee S, Cui QC, Kong D, Sarkar FH, Dou QP. Activation of AMP-activated protein kinase by 3,3’-Diindolylmethane (DIM) is associated with human prostatecancer cell death in vitro and in vivo. PLoS One. 2012;7:e47186. doi: 10.1371/journal.pone.0047186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zong J, Deng W, Zhou H, Bian ZY, Dai J, Yuan Y, Zhang JY, Zhang R, Zhang Y, Wu QQ, Guo HP, Li HL, Tang QZ. 3,3’-Diindolylmethane protects against cardiac Hypertrophy via 5’-adenosine monophosphate-activated protein kinase-α2. PLoS One. 2013;8:e53427. doi: 10.1371/journal.pone.0053427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brønnum H, Eskildsen T, Andersen DC, Schneider M, Sheikh SP. IL-1β suppresses TGF-β-mediated myofibroblast differentiation in cardiac fibroblasts. Growth Factors. 2013;31:81–9. doi: 10.3109/08977194.2013.787994. [DOI] [PubMed] [Google Scholar]
- 17.Leask A. Targeting the TGF, endothelin-1 and CCN2 axis to combat fibrosis in scleroderma. Cell Signal. 2008;20:1409–14. doi: 10.1016/j.cellsig.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 18.Leask A. Potential therapeutic targets for cardiac Fibrosis: TGF², angiotensin endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675–1680. doi: 10.1161/CIRCRESAHA.110.217737. [DOI] [PubMed] [Google Scholar]
- 19.Lal H, Ahmad F, Zhou J, Yu JE, Vagnozzi RJ, Guo Y, Yu D, Tsai EJ, Woodgett J, Gao E, Force T. Cardiac fibroblast glycogen synthase kinase-3β regulates ventricular remodeling and dysfunction in ischemicheart. Circulation. 2014;130:419–30. doi: 10.1161/CIRCULATIONAHA.113.008364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rohr S. Myofibroblasts in diseased hearts: new players in cardiac arrhythmias? Heart Rhythm. 2009;6:848–56. doi: 10.1016/j.hrthm.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 21.Santiago JJ, Dangerfield AL, Rattan SG, Bathe KL, Cunnington RH, Raizman JE, Bedosky KM, Freed DH, Kardami E, Dixon IM. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: Expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev Dyn. 2010;239:1573–84. doi: 10.1002/dvdy.22280. [DOI] [PubMed] [Google Scholar]
- 22.Eghbali M, Tomek R, Woods C, Bhambi B. Cardiac fibroblasts are predisposed to convert into myocyte phenotype: specific effect of transforming growth factor. Proc Natl Acad Sci U S A. 1991;88:795–799. doi: 10.1073/pnas.88.3.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leask A. TGF-β, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res. 2007;74:207–12. doi: 10.1016/j.cardiores.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 24.Snider P, Standley KN, Wang J, Azhar M, Doetschman T, Conway SJ. Origin of cardiac fibroblasts and the role of periostin. Circ Res. 2009;105:934–47. doi: 10.1161/CIRCRESAHA.109.201400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Y, Wu D, Zhang X, Jiang M, Hu C, Lin J, Tang J, Wu L. Cardiac-specific Traf2 overexpression enhances cardiac hypertrophy through AKT/GSK3β signaling. Gene. 2014;536:225–31. doi: 10.1016/j.gene.2013.12.052. [DOI] [PubMed] [Google Scholar]
- 26.Lin CH, Wang WC, Kao SH. Der p 2 promotes motility of airway epithelial cell attributing to AKT/GSK3β-associated epithelial-to-mesenchymal transition. Mol Cell Biochem. 2014;395:135–43. doi: 10.1007/s11010-014-2119-y. [DOI] [PubMed] [Google Scholar]
- 27.Liu CW, Li CH, Peng YJ, Cheng YW, Chen HW, Liao PL, Kang JJ, Yeng MH. Snail regulates Nanog status during the epithelial-mesenchymal transition via the Smad1/Akt/GSK3β signaling pathway in non-small-cell lung cancer. Oncotarget. 2014;5:3880–94. doi: 10.18632/oncotarget.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park JH, Shin YJ, Riew TR, Lee MY. The indolinone MAZ51 induces cell rounding and G2/M cell cycle arrest in glioma cells without the inhibition of VEGFR-3 phosphorylation: involvement of the RhoA and Akt/GSK3β signaling pathways. PLoS One. 2014;9:e109055. doi: 10.1371/journal.pone.0109055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woulfe KC, Gao E, Lal H, Harris D, Fan Q, Vagnozzi R, DeCaul M, Shang X, Patel S, Woodgett JR, Force T, Zhou J. Glycogen synthase kinase-3beta regulates post-myocardial infarction remodeling and stress-induced cardiomyocyte proliferation in vivo. Circ Res. 2010;106:1635–45. doi: 10.1161/CIRCRESAHA.109.211482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuda T, Zhai P, Maejima Y, Hong C, Gao S, Tian B, Goto K, Takagi H, Tamamori-Adachi M, Kitajima S, Sadoshima J. Distinct roles of gsk-3alpha and gsk-3beta phosphorylationin the heart under pressure overload. Proc Natl Acad Sci U S A. 2008;105:20900–5. doi: 10.1073/pnas.0808315106. [DOI] [PMC free article] [PubMed] [Google Scholar]