

Abstract
Genetic mutation has served as the biomarkers for the diagnosis and treatment of glioblastoma multiforme (GBM). However, intra-tumor heterogeneity may interfere with personalized treatment strategies based on mutation analysis. This study aimed to characterize somatic mutation profiling of GBM. We collected 33 samples from 7 patients with the primary GBM associated with different Choline (Cho) to N-acetylaspartate (NAA) index (CNI) through the frameless proton magnetic resonance spectroscopy (1H-MRS) guided biopsies and investigated multiple somatic mutations profi ling using the AmpliSeq cancer hotspot panel V2. We identifi ed 53 missense or nonsense mutations in 27 genes including some novel mutations such as APC and IDH2. The mutations in EGFR, TP53, PTEN, PIK3CA genes were presented with different frequency and the majority of the mutated gene was only shared by 1-2 samples from one patient. Moreover, we found the association of CNI with histological grade, but there was no signifi cant change of CNI in the presence of TP53, EGFR and PTEN mutations. These data suggest that gene mutations constitute a heterogeneous marker for primary GBM which may be independent of intra-tumor morphological phenotypes of GBM; therefore, gene mutation markers could not be determined from a small number of needle biopsies or only confi ned to the high-grade region.
Keywords: Mutation profi ling, heterogeneity, glioblastoma multiforme, proton magnetic resonance spectroscopy, biopsy
Introduction
Glioblastoma multiforme (GBM) is one of the most aggressive primary brain malignancy and is the leading cause of death among the brain primary glioma. Despite extensive therapy developed recently, the outcome of GBM has been extremely poor with a median survival of 12-15 months [1,2]. The identification of genomic mutation has been well developed for survival prediction, disease classification and stratification of GBM [3]. However, currently the mutation analysis of tumor mainly depends on single target biopsy. Due to the existence of extensive genetic heterogeneity among GBM, intra-tumor heterogeneity may lead to sampling bias and interfere with personalized treatment strategies based on mutation analysis [4,5].
Multiple sampling has been performed to investigate the intra-tumor heterogeneity for GBM [6,7]. However, in these studies the tumor samples are obtained mainly based on the surgeon’s experience, which may not fully reflect the extent and complexity of intra-tumor heterogeneity. Although numerous genomic mutations have been identified in GBM [8], the intra-tumor somatic mutation profiling of primary GBM has been not well characterized. Stereotactic needle biopsy assisted by 1H-MRS is considered as a powerful tool for obtaining tissue samples for histopathological analysis, and multiple-voxel spectroscopy could reflect the intra-tumor heterogeneity at the level of metabolic changes [9]. In the present study, we performed multiple biopsies from patients with primary GBM guided by the neuronavigation targeting the hotspots that were showed by the proton magnetic resonance spectroscopy (1H-MRS), then the samples were checked by the AmpliSeq cancer hotspot panel V2 for identifying somatic mutations as previously reported [10,11]. We expect that the combination of stereotactic needle biopsy and molecular genetic techniques might provide novel insight into intra-tumor mutational profile and help develop a new approach to improve personalized treatment strategies for patients with primary GBM.
Materials and methods
Patients and samples
This series enrolled 7 patients who were diagnosed as primary GBM between 2010 and 2012 in Huashan Hospital. The clinical data of these patients were collected, and included 5 males and 2 females with a mean age of 45.3 years (range 16 to 64 years). None of the patients received chemotherapy or radiotherapy or had undergone surgery previously. This study was approved by the Huashan Institutional Review Board and all subjects signed informed consent.
Sample collection procedure
The procedure for sample collection has been described in detail previously (Figure 1) [12]. Briefly, each patient underwent magnetic resonance imaging (MRI) and 1H-MRS before biopsy to analyze the metabolism of the lesions and to locate appropriate target of needle biopsy. A frameless needle biopsy was performed with the assistance of real-time neuronavigation (NeuroIII-SV VISIHS Surgical System, IMRIS Inc). Three to five targets in non-eloquent regions for tumor biopsy were preoperatively determined by referring to the conventional MR images and MR spectroscopic features of the lesion, the areas with the different Choline (Cho) to N-acetylaspartate (NAA) Index (CNI) were chosen as the targets and the surgical trajectories were planned. In most situations a linear path was doted towards the tumor core region so that the biopsy needle was inserted along the same track to minimize the possible brain injury (Figure 1).
Figure 1.
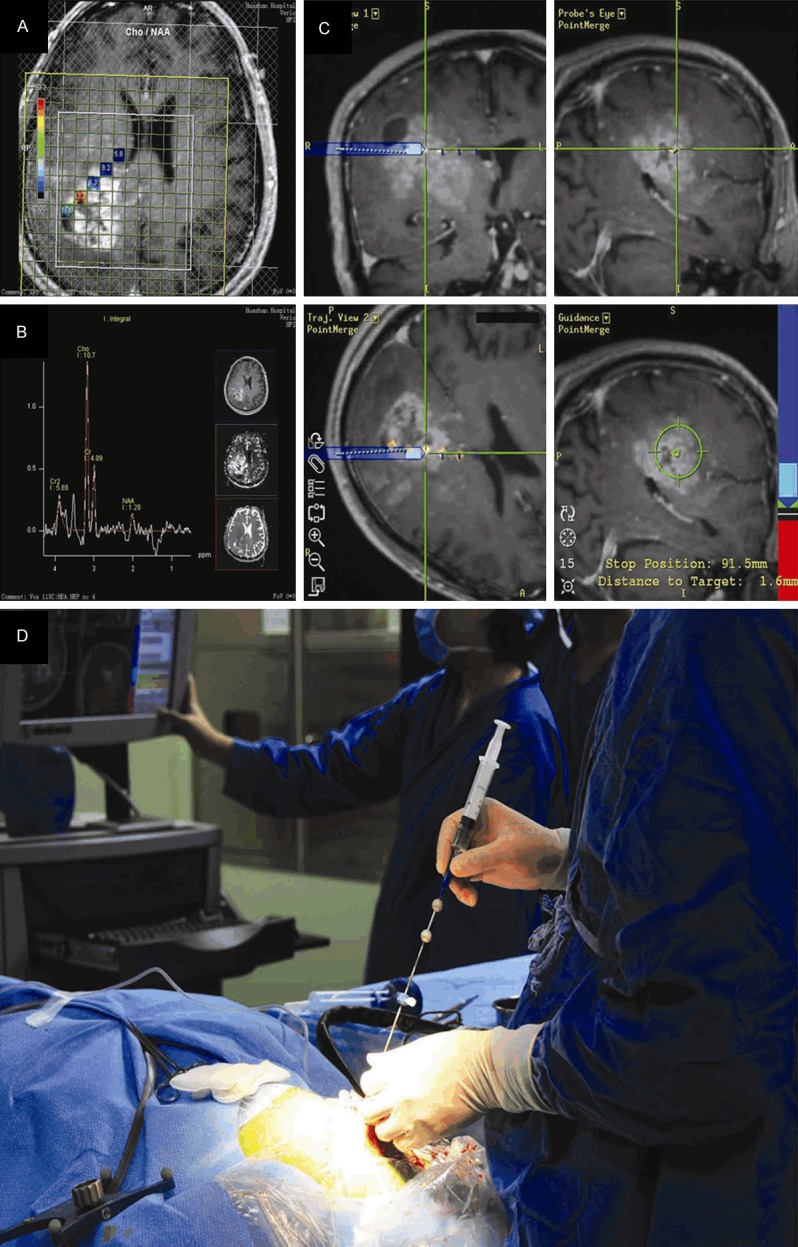
Sample collection procedure (the data of these patients was not shown in this article). (A) Contrast T1-weighted MRI superimposed with five marked targets from patient with primary GBM, a linear path was adopted towards the tumor core region (B) five targets correspond to different CNI. (C) The targets were labeled in the neuronavigation data sets, accurate sampling was guided by dynamic T1-enhanced MRI guidance. (D) Frameless and skull-screwed needle biopsy kit guarantees the accuracy of the trajectory and sampling.
Histopathological evaluation
All biopsy specimens were immediately fixed in 10% formalin embedded in paraffin and then cut into sections for histopathological assessment. Haematoxylin and Eosin (H-E) stained sections of tissues were observed under a light microscope and classified according to the 4th edition of the WHO classification of central nervous systems tumors (2007). Immunohistochemistry was performed following routine procedures with primary antibodies for IDH1 (R132H), MIB-1, p53, Olig, GFAP, Nestin and EGFR.
DNA extraction
Tissues samples were sliced from unstrained tissue sections with an approximate size of 10 µm and deparaffinized using a series of xylene and ethanol washes. Genomic DNA was extracted from the samples using Total Nucleic Acid Isolation Kit (Ambion, Carlsbad, CA, USA) according to the manufacturer’s instruction. All DNA samples were quantified by the Qubit® 2.0 Fluorometer with the Qubit® dsDNA BR assay kit (Invitrogen, Eugene, OR, USA). After isolation, DNA samples were stored at -20°C.
AmpliSeq library preparation
Amplicon libraries were prepared using the Ion AmpliSeq Cancer Hotspot Panel V2 (Life Technologies, Carlsbad, USA) to amplify the target regions of 50 oncogenes and tumor suppressor genes from each 10 ng of genomic DNA in a single multiplex PCR reaction. Totally 207 multiplexed amplicons were treated with FuPa Reagent (Life Technologies) to partially digest the primers and phosphorylate the amplicon ends, and the products were ligated to the sequencing adapters with unique Ion Xpress Barcode (Life Technologies) according to the manufacturer’s instructions. All libraries were quantified by the Agilent 2100 Bioanalyser and Agilent High Sensitivity DNA Kit (Agilent Technologies). The final library concentrations were standardized to 100 pM in low Tris-EDTA (TE) buffer (Life Technologies).
Semiconductor sequencing
Five picomoles of each of the 33 samples were bar-coded and pooled for emulsion PCR (ePCR) on Ion Sphere Particles (ISPs) using the Ion PI Template OT2 200 Kit v3 on Ion OneTouch 2 Instrument. Following automated Ion One Touch ES enrichment of template-positive ISPs, the samples were loaded on Ion PGM318TM chip and sequenced using the Ion PGMTM sequencing 200 kit (Life Technologies). Ion Torrent reads were aligned to the human reference genome version 19 (GRCh37) using Torrent Variant Caller Plugin 4.0 (Life technologies).
Statistical analysis
SPSS 20.0 (SPSS Inc.at IBM Company, USA) was used for statistical analysis. Student’s t-test was used for comparison of means of Cho/NAA with gene mutation. When Bartlett test showed that the variance was not homogeneous, Kruskal-Wallis test was applied. In other cases, a chi-square test was applied for the comparison between grade and gene mutation profiles. P<0.05 were considered statistically significant for all tests.
Results
Patients and samples
From seven patients diagnosed as primary GBM, 33 biopsy specimens were collected for sequence analysis with the mean number of biopsy specimens per patient as 4 (range: 3 to 5). The histopathological grades for these samples were as follows: 8 samples of grade I, 9 samples of grade II, 7 samples of grade III, 9 samples of grade IV. The size of each sample was about 5 mm3.
Sequence coverage
Sequence coverage was assessed based on the number and distribution of reads across target amplicons. The distribution of reads across the 207 amplicons was consistent across all tested samples. An average of 8.9 million of the total 11 million addressable wells in the Ion 318 chip were consistently loaded with ISPs, and 8.5 million (96.5%) of these particles contained library templates. After the subtraction of multiple-templated beads and sequence reads of poor quality, an average of 8.0million reads were obtained. The individual samples averaged 762,870 mapped reads (range: 444,436 to 1,019,225). The mean read length was 154 bp (range: 111 to 187 bp), constituting an average of approximately 33 Mb of sequence per sample. After sequencing, an average depth of 3,302 reads per nucleotide position within the target region was detected. The 1×, 10×, 100× and 500× coverage were 100%, 100%, 100% and 98.13%, respectively.
Variants detected
A total of 815,110 nucleotides across 33 samples were sequenced. We detect 815 mutations in 40 genes with a mean of 25 variants per sample, and mutation was detected in each sample (Figure 2). Since constitutional DNA was not available to deduce germline polymorphisms, the somatic variant identification procedure was intended to minimize the false positive somatic mutation. While the coding complex mutation and frame shift-near-splice mutation were also excluded. Among the results, 53 variants were predicted to cause non-synonymous change in 27 different genes, and all patients displayed a variable number of these mutations with a mean of 2-3 variants detected per patient (Table 1). Moreover, these mutations were found across the samples with different frequency. The most frequent mutations were in EGFR (n=10), TP53 (n=7), APC (n=7), RET (n=6), PTEN (n=4), MLH1 (n=4), NOTCH (n=3) while other gene mutations were shared by total 1-2 samples. The majority of the mutated gene was only shared by 1-2 samples from one patient (Table 1).
Figure 2.
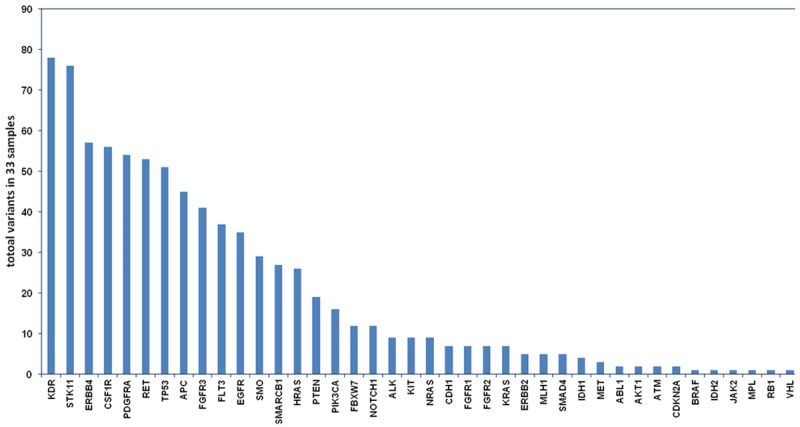
Totally 815 mutations in 40 genes were detected. Horizontal axis, mutated gene; vertical axis, total variants in 33 GBM samples.
Table 1.
Distribution of mutations across tumor samples per patient, 33 biopsy specimens were collected with 3 to 5 samples per patient (number in yellow indicate one patient

|
Among the mutated genes, 14 mutations at 6 positions were identified in EGFR gene as follows: ARG108LYS, ALA289VAL, VAL292LEU, PRO596LEU, GLY598 VAL, ALA702VAL; 9 mutations at 5 positions were identified in TP53: ARG 342 stop, GLU336LYS, ARG273CYS, ARG175HIS, PRO85LEU; 4 mutations at 2 positions were identified in PTEN: ALA126THR, ASP252ASN; 3 mutations at 3 positions were identified in PIK3CA: ARG88GLN, GLU1032 LYS, HIS 1047ARG; one mutation at one position was identified in IDH2: ARG172LYS. The status of other mutations was shown in Table 1.
Association of Cho/NAA ratio with the grade of GBM
To investigate the association of Cho/NAA ratio with histopathological grade of GBM, we measured CNI and found that it was 1.36±0.93 and 3.50±3.14, respectively, in low-grade (grade I-II) and high-grade (grade III-IV) gliomas. The samples of higher grade glioma had a significant increased CNI (P<0.05) (Figure 3A). Then we focused on the common mutations that occurred in more than 4 samples, including EGFR, TP53, and PTEN mutations. There were no significant differences in the percentage of EGFR, TP53, and PTEN mutation between the low-grade and high-grade group (P>0.05) (Table 2). We also investigated the association of CNI with gene mutations. The results showed that no significant differences in CNI were detected between samples with mutations in EGFR, TP53, PTEN and samples without these mutations (Figure 3B) (P>0.05).
Figure 3.
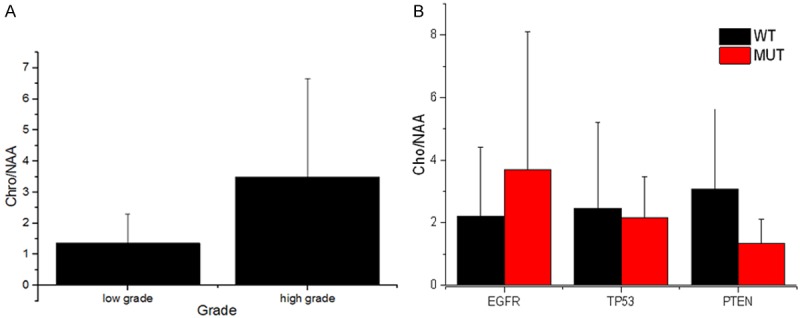
(A) Comparison of CNI between low-grade and high-grade gliomas. The samples with higher grade had a significant increased CNI (**P<0.05 vs low-grade group) (A). (B) No significant changes of CNI were observed in the presence of EGFR, TP 53 and PTEN mutations (B).
Table 2.
Association of gene mutation with samples grading
| Genetic mutation | Sample grade | P value | Total | |
|---|---|---|---|---|
|
| ||||
| Low grade (n=17) | High grade (n=16) | |||
| EGFR | ||||
| present | 8 | 5 | 0.353 | 13 (39.4%) |
| absent | 9 | 11 | 20 (60.6%) | |
| TP53 | ||||
| present | 3 | 4 | 0.606 | 7 (21.2%) |
| absent | 14 | 12 | 26 (78.8%) | |
| PTEN | ||||
| present | 2 | 2 | 0.948 | 4 (12.1%) |
| absent | 15 | 14 | 29 (87.9%) | |
There is no significantly different percentage in the EGFR, TP53, PTEN mutation between the low grade and high grade gliomas.
Discussion
It is predicted that 53 variants would cause non-synonymous change in 27 genes and induce functional consequences to contribute to the development and progression of GBM. Indeed, mutation analysis has shown that TP53, EGFR, PTEN, and PIK3CA mutations are frequently detected in primary GBM samples. Due to the Ion Ampliseqre-sequencing strategy here with sequencing depth of more than 1500-folds and multiple tumor tissues within the same tumor were obtained, rare low-frequency and potential mutations were detected in the GBM cases, in this study we found a number of novel missense mutations including APC, RET, MLH1, NOTCH1 and IDH2 etc. Among these mutations, APC gene mutations occurred in 13% of case with mutation frequency of about 14.5%. APC encodes a tumor suppressor protein that acts as an antagonist of the Wnt signaling pathway. Disease-associated APC mutations tend to be clustered in a small region designated the mutation cluster region and result in a truncated protein product [13]. Interestingly, APC mutations were not previously reported in primary GBM. Obviously, further studies are needed to explore the clinical value of these mutations in primary GBM.
In this study we demonstrated intra-tumoral homogeneity of these gene mutations, 6 variants in CSF1R, FGFR3, FLT3, PDGFRA, RET and STK11 were found across all the samples (data not shown), but all of these variants prove not to be involved in tumor initiation and progression. For the disease-associated mutations, most of them were heterogeneous and not detectable in every sequenced region from one patient. EGFR, TP53, and PTEN mutations are shared by relatively more cases. As multiple-site molecular profiling pattern could reveal evolutionary course of mutational events [4,14], we postulate that these mutations occur early in cancer development. IDH1 R132 mutations in the low-grade gliomas and secondary GBM is a defining marker and key oncogenic event for secondary GBM, but it is relatively exclusive from primary GBM and reported as only 5% in primary GBM [15]. Actually, in our cohort, 4 samples harbored validated mutation in IDH1 mutation (COSM28746, ARG132HIS), but they are coding complex mutation but not missense or stop-gained mutation. It has been reported that IDH2 gene mutation was presented in 3-5% of gliomas, while no IDH2 mutations were reported in GBM [8,15]. However, in our cohort, one sample from one patient carried mutated IDH2 (ARG172LYS). PI3KCA is involved in one of the major molecular pathways that promote the tumor genesis of high-grade glioblastoma [16]. In this study we only detected PIK3CA mutation in 2 samples, which suggests that PI3CA mutation occur lately in cancer development. Although these relatively rare mutations could not contribute to the early stages of malignant transformation, it is possible that they become common genetic mutation during treatment.
Clinically, some GBM such as diffusive brain stem gliomas, functional area gliomas are frequently non-resectable which necessitate stereotactic biopsies evaluation of the molecular marker and for histopathological analysis. Currently, collecting GBM samples from different sites of the tumor mostly depends on the surgeon’s experience. In addition, the obtained samples may not fully reflect the extent and complexity of intra-tumor heterogeneity and lead to sampling bias. In order to obtain a representative set of samples, we performed multiple sampling through the frameless 1H-MRS guided biopsies. Cho and NAA are usually reliably quantified in 1H-MRS evaluation and considered as two of the most important indexes for glioma examination. CNI has been suggested to be helpful to differentiate low-grade and high-grade astrocytomas [17,18]. In the present study, we found that there was a favorable correlation of CNI with histopathological grades. These data suggest that MRS is a helpful method to target brain biopsy for histopathological analysis.
We further explored the relationship between the intra-tumor gene mutation and metabolic profiling detected by MRS. It is known that genomic mutation may alter the metabolism [19,20] and oncogenes have been involved in the regulation of different metabolic pathways [21]. However, in this study we did not find significant change of CNI in the presence of EGFR, TP53 and PTEN mutation. This could be partly explained by that not all gene mutations lead to cellular metabolic changes or changes enough to be detected by MRS. On the other hand, because CNI has been correlated to the histopathological diagnosis [9,17], it might suggest the intra-tumor genetic mutation are distributed independently of morphological phenotype. Actually, there were no significant differences in the mutation rates in EGFR, TP53 and PTEN genes between low-grade and high-grade groups. Some prognostic indicators for GBM such as MGMT promoter hypermethylation has been reported to likely occur in the tumor region with contrast enhancement in MRI [22]. However, in the present study, we advocated that multiple sampling for mutation analysis should include the low-grade region or without contrast enhancement in the tumor. It was noteworthy that two hotspot mutations in TP53 (Arg273CYS) and EGFR (ALA289VAL) were detected in almost of all samples from one patient. Nevertheless, whether these mutations represent a homogenous marker for GBM needs further evaluation because of our small-sized patients.
Because our study only included 7 primary GBM patients, it remains to be determined how representative our results of mutation analysis will be in a larger sample. Furthermore, more mutation need to be further investigated because the panel used in the present study is not specific for GBM. Despite these limitations, our study demonstrated that intra-tumor heterogeneity exists within the individual primary GBM samples at the level of genomic mutation. Our data suggest that gene mutation markers could not be determined from small-sized needle biopsies and they may be independent of intra-tumor morphological phenotypes of GBM. Detection of rare mutations would be also possible when multiple cancer tissues within the same tumor was sequenced at a greater sequencing depth. The combination of multiple target needle biopsy and molecular genetic techniques might provide novel insight into intra-tumor mutational profiling and help develop new approach to improve personalized treatment strategies for primary GBM.
Acknowledgements
This work was supported by National Clinical Key Subject of China (to Liangfu Zhou), the Project for Science and Technology Commission of Shanghai Municipality Grant, 13JC1408000 (to Liangfu Zhou); General Financial Grant from the China Postdoctoral Science Foundation, 2014M550217 (to Chao Tang).
Disclosure of conflict of interest
None.
References
- 1.Clarke J, Butowski N, Chang S. Recent advances in therapy for glioblastoma. Arch Neurol. 2010;67:279–83. doi: 10.1001/archneurol.2010.5. [DOI] [PubMed] [Google Scholar]
- 2.Bai RY, Staedtke V, Riggins GJ. Molecular targeting of glioblastoma: Drug discovery and therapies. Trends Mol Med. 2011;17:301–12. doi: 10.1016/j.molmed.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN. Integrated genomic analysis identifi es clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonavia R, Inda MM, Cavenee WK, Furnari FB. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res. 2011;71:4055–60. doi: 10.1158/0008-5472.CAN-11-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavare S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A. 2013;110:4009–14. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grasbon-Frodl EM, Kreth FW, Ruiter M, Schnell O, Bise K, Felsberg J, Reifenberger G, Tonn JC, Kretzschmar HA. Intratumoral homogeneity of MGMT promoter hypermethylation as demonstrated in serial stereotactic specimens from anaplastic astrocytomas and glioblastomas. Int J Cancer. 2007;121:2458–64. doi: 10.1002/ijc.23020. [DOI] [PubMed] [Google Scholar]
- 8.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bulik M, Jancalek R, Vanicek J, Skoch A, Mechl M. Potential of MR spectroscopy for assessment of glioma grading. Clin Neurol Neurosurg. 2013;115:146–53. doi: 10.1016/j.clineuro.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 10.Liu X, Mody K, de Abreu FB, Pipas JM, Peterson JD, Gallagher TL, Suriawinata AA, Ripple GH, Hourdequin KC, Smith KD, Barth RJ, Colacchio TA, Tsapakos MJ, Zaki BI, Gardner TB, Gordon SR, Amos CI, Wells WA, Tsongalis GJ. Molecular profi ling of appendiceal epithelial tumors using massively parallel sequencing to identify somatic mutations. Clin Chem. 2014;60:1004–11. doi: 10.1373/clinchem.2014.225565. [DOI] [PubMed] [Google Scholar]
- 11.Tabone T, Abuhusain HJ, Nowak AK, Erber WN, McDonald KL. Multigene profi ling to identify alternative treatment options for glioblastoma: a pilot study. J Clin Pathol. 2014;67:550–5. doi: 10.1136/jclinpath-2014-202173. [DOI] [PubMed] [Google Scholar]
- 12.Guo J, Yao C, Chen H, Zhuang D, Tang W, Ren G, Wang Y, Wu J, Huang F, Zhou L. The relationship between Cho/NAA and glioma metabolism: implementation for margin delineation of cerebral gliomas. Acta Neurochir (Wien) 2012;154:1361–70. doi: 10.1007/s00701-012-1418-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boman BM, Fields JZ. An APC: WNT counter-current-like mechanism regulates cell division along the human colonic crypt axis: a mechanism that explains how apc mutations induce proliferative abnormalities that drive colon cancer development. Front Oncol. 2013;3:244. doi: 10.3389/fonc.2013.00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet. 2012;13:795–806. doi: 10.1038/nrg3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–73. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weber GL, Parat MO, Binder ZA, Gallia GL, Riggins GJ. Abrogation of PIK3CA or PIK3R1 reduces proliferation, migration, and invasion in glioblastoma multiforme cells. Oncotarget. 2011;2:833–49. doi: 10.18632/oncotarget.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Porto L, Kieslich M, Franz K, Lehrnbecher T, Zanella F, Pilatus U, Hattingen E. MR spectroscopy differentiation between high and low grade astrocytomas: a comparison between paediatric and adult tumours. Eur J Paediatr Neurol. 2011;15:214–21. doi: 10.1016/j.ejpn.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 18.Magalhaes A, Godfrey W, Shen Y, Hu J, Smith W. Proton magnetic resonance spectroscopy of brain tumors correlated with pathology. Acad Radiol. 2005;12:51–7. doi: 10.1016/j.acra.2004.10.057. [DOI] [PubMed] [Google Scholar]
- 19.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander HM, Su SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vander HM, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weise LM, Harter PN, Eibach S, Braczynski AK, Dunst M, Rieger J, Bahr O, Hattingen E, Steinbach JP, Plate KH, Seifert V, Mittelbronn M. Confounding factors in diagnostics of MGMT promoter methylation status in glioblastomas in stereotactic biopsies. Stereotact Funct Neurosurg. 2014;92:129–39. doi: 10.1159/000360582. [DOI] [PubMed] [Google Scholar]