

Abstract
Meckel-Gruber syndrome (MKS) is a lethal autosomal recessive condition characterized by renal cysts and variably associated features, including developmental anomalies of the central nervous system (typically encephalocele), hepatic ductal dysplasia and cysts, and polydactyly. Genetic heterogeneity has been demonstrated at eleven loci, MKS1-11. Here, we present the clinical and molecular characteristics of a Chinese MKS3 family with occipital encephalocele and kidney enlargement. DNA sequencing of affected fetuses revealed a homozygous c.1645C>T substitution in exon 16 of TMEM67, leading to a p.R549C substitution in meckelin. The R549 residue is highly conserved across human, rat, mouse, zebrafish, chicken, wolf and platypus genomes. Hha I restriction analysis demonstrated that the c.1645C>T mutation was absent in 200 unrelated control chromosomes of Chinese background, supporting the hypothesis that it represents causative mutation, not rare polymorphism. Our data provide additional molecular and clinical information for establishing a better genotype-phenotype understanding of MKS.
Keywords: MKS3, TMEM67, meckelin, mutation
Introduction
Meckel-Gruber syndrome (MKS; MIM 24900) is a lethal autosomal recessive disorder characterized by various severe malformations. The minimal diagnostic criteria are cystic dysplasia of the kidneys, with fibrotic changes in the liver and occipital encephalocele or some other malformations of the central nervous system. Polydactyly is also frequently reported in MKS patients. Patients with classic MKS phenotype usually die in the perinatal period. MKS is genetically heterogenous [1,2], with eleven causative genes: MKS1 (OMIM 249000), MKS1 (OMIM 609883), 17q23; MKS2 (OMIM 603194), TMEM216 (OMIM 613277), 11q13; MKS3 (OMIM 607361), TMEM67 (OMIM 609884) 8q21.13-q22.1; MKS4 (OMIM 611134), CEP290 (OMIM 610142) 12q21.3; MKS5 (OMIM 611561), RPGRIP1L (OMIM 610937) 16q-12.2; MKS6 (OMIM 612284), CC2D2A (OMIM 612013) 4p15; and MKS7 (OMIM 267010), NPHP3 (OMIM 608002), 3q22; MKS8 (OMIM 613885), TCTN2 (OMIM 613846); MKS9 (OMIM 614209), B9D1 (OMIM 614144); MKS10 (OMIM 614175), B9D2 (OMIM 611951); and MKS11 (OMIM 615397), TMEM231 (OMIM 614949). All genes involved in MKS are associated with ciliary functions [4-7].
The TMEM67 gene encodes a component of the transmembrane protein meckelin, which has 995 amino acids and an extracellular N-terminus containing a signal peptide and a cysteine-rich domain [3]. It also has an intracellular C-terminus with a coiled-coil domain. MKS type 3 (MKS3) is characterized by occipital encephalocele and cystic dysplastic kidneys [4]. Com-pared with other MKS subtypes, MKS3 patients rarely present with polydactyly and are more likely to have milder central nervous system phenotypes with considerable variability [8-10].
To date, a total of 25 TMEM67 pathogenic mutations have been reported in TMEM67 spectrum disease (http://omim.org/entry/609884) and seven of them identified in MKS3 families. In China, patients with MKS phenotypes have been reported, but no mutation was identified. In this report, we present detailed clinical, genetic and pathological findings of four fetuses in one family who had clinical characteristics of MKS3 and exhibited pathogenic TMEM67 mutations.
Materials and methods
Ethics statement
Written informed consents were obtained from all family participants and normal controls prior to their participation in the study, and all research procedures were approved by the Research Ethics Committee of the Chinese PLA General Hospital. We also have the consent to use the tissues of the aborted normal fetus at 17 gestational week (gw) because the pregnant woman was diagnosed as primary pulmonary hypertension during her first trimester.
Patients
The Chinese family MKS-H01 was from Beijing, China. The proband was a 14 gw fetus (III-3) with occipital encephalocele and kidney enlargement detected by ultrasound. This was the third pregnancy of this couple. The other three pregnancies of this couple carried similar malformed fetuses. Amniotic fluid culture and chromosome analysis for the first and second fetuses were performed in another hospital and both were normal. Therapeutic termination of pregnancy was performed for the four affected fetuses after ultrasound diagnosis of MKS during the second trimester of pregnancy, followed by autopsies of the first, the third and the fourth affected fetus.
Isolation of genomic DNA and RNA
Peripheral venous blood samples (3 ml) were drawn from family participants and 200 healthy controls for genomic DNA extraction using the Genomic DNA isolation kit (Qiagen).
After abortion, fetal tissues of brain, liver and kidney from III-3 and III-4 were stored in liquid nitrogen. DNA was isolated using Gentra Puregene Mouse Tail kit (Qiagen).
The formalin-fixed, paraffin-embedded tissues of brain, liver and spleen from the III-1 fetus were applied to isolate genomic DNA using the QIAamp DNA FFPE Tissue Kit (Qiagen), following the protocol supplied by the manufacturer.
cDNA preparation
Total RNA was isolated from aborted proband’s fetal kidney using RNeasy Mini Kit (Qiagen) and cDNA was obtained by RT-PCR amplification using SuperScript™ III Reverse Transcriptase (Invitrogen) following the manufacturer’s instructions.
DNA sequencing
After gel purification, each of the amplified PCR fragments was directly sequenced in both forward and reverse directions using BigDye Terminator chemistry and ABI Prism Sequencer 3100 (Applied Biosystems).
Endonuclease digestion
In the fast assay of the c.1645C>T mutation in fetal tissues and in 200 normal controls, Hha I restriction analysis of PCR fragment covering the mutation site was applied. We designed the primers (Forward: 5’gtttttgaacaccgatgacaga 3, reverse: 5’agaaggatccagaatggtcaaa 3) to amplify a 217bp PCR product. Hha I digestion distinguishes the homozygote (217 bp) from the wildtype (130, 87 bp) and heterozygote (217, 130, 87 bp) on agarose gel electrophoresis.
Immunohistochemistry
Immunohistochemistry was conducted with the fetal tissues (liver and kidney) obtained from III-4, as well as a 17 gw fetus as a normal control. Standard procedures were used for staining tissue slides with the antibodies: primary antibodies (Rabbit polyclonal to meckelin ab76786 (Abcam) at 100 dilutions in PBS, matched to appropriate pre-immune negative control sera). The secondary antibody (general two-stage method, PV9000 Zhongshan, China) was applied at a dilution of 1:100.
Results
Clinical features
The proband fetus (III-3) was found to have occipital encephalocele and kidney enlargement by ultrasound (Figure 1) at 14 gw and was aborted one week later. The III-4 fetus was diagnosed at 13 gw by ultrasound and aborted at 14 gw. The couple had two similarly malformed fetuses (III-I, III-2) before. Four fetuses had similar clinical characteristics, including occipital encephalocele and renal cystic dysplasia, as well as some diverse phenotypes. Table 1 shows the detailed clinical features of the four affected fetuses of this Chinese MKS family. In fetus III-4, we detected parietal absence and polydactyly (Figure 1), which did not show in the first three fetuses. The absence of polydactyly is typical phenotype of MKS3.
Figure 1.
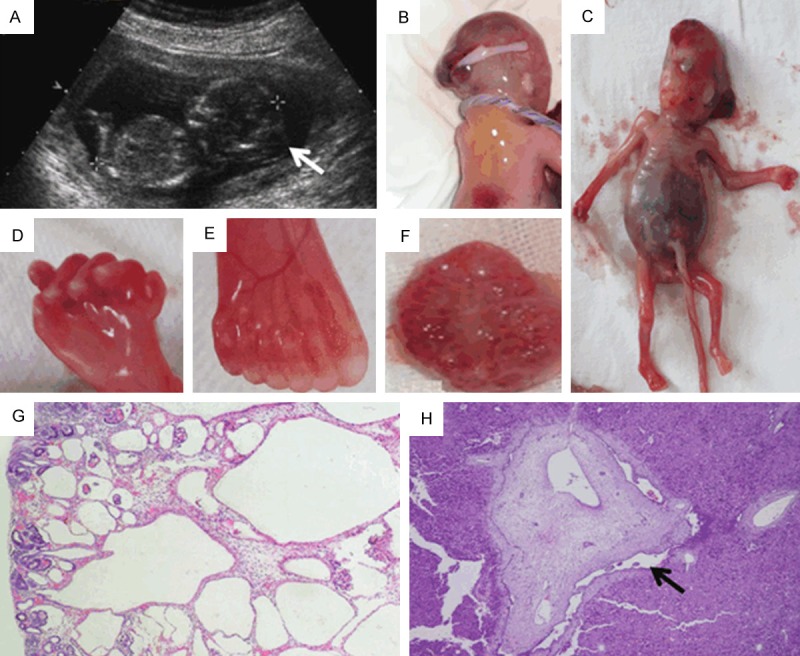
Clinical features of two affected fetuses in Chinese MKS3 family. (A) Ultrasound scans of III-3 at 14 gw. Occipital encephalocele was observed and confirmed by autopsy for III-3 (B). Parietal absence, distended abdomen due to enlarged kidney, clearly visible cysts and polydactyly of the feet were observed in III-4 fetus (C-F), which is rarely seen in MKS3 fetuses. The kidney of III-4 (F) showed diffuse multicystic renal dysplasia. Cysts are found in the deep cortex and medulla, smaller at the periphery than in the center; notice the remnant glomerulus in renal cortex (G). The liver biopsy showed hepatic ductal plate malformations. Enlarged intrahepatic bile duct is indicated by the arrow (H).
Table 1.
Clinical features observed in four MKS fetuses from the Chinese family
| Occipital encephalocele | Spina bifida | Renal cystic dysplasia | Ductal plate malformations | Polydactyly | Parietal absence | Cleft palate | Fetal hydrops | |
|---|---|---|---|---|---|---|---|---|
| III-1 | + | + | + | + | - | - | + | Unknown |
| III-2 | + | - | + | Unknown | - | - | - | Unknown |
| III-3 | + | + | + | + | - | - | - | + |
| III-4 | + | - | + | + | + | + | - | + |
+Present, -absent.
Mutation identification
Based on the clinical features of this family, we firstly chose the MKS3 candidate gene, TMEM67, for mutation screening. We performed direct sequencing of full-length cDNA extracted from fetal brain and renal tissues of proband of Chinese family MKS-H01 (III-3). A homozygous C to T transition at position 1645 in exon 16 was identified, resulting in p.R549C substitution in meckelin (Figure 2). Sequencing analysis of the formalin-fixed, paraffin-embedded tissue from III-1 and the fresh tissue from III-4 confirmed this finding (we did not have tissue from III-2). The Arg residue at 549 in meckelin is conserved across human, rat, mouse, zebrafish, chicken, wolf and platypus. Both of the parents were found to be for the carriers of this substitution. Hha I restriction analysis demonstrated that the c.1645C>T substition was absent in 200 unrelated control chromosomes with Chinese background, supporting the hypothesis that it represents a causative mutation, not a rare polymorphism.
Figure 2.
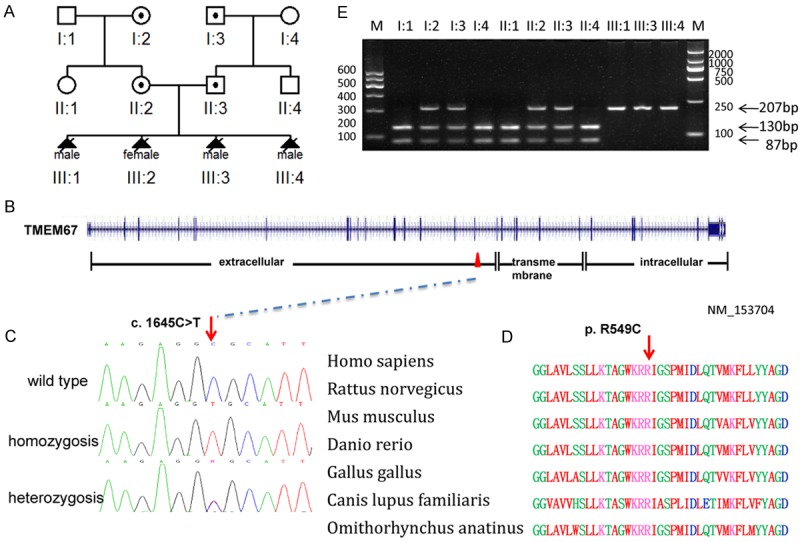
Mutation analysis of the Chinese MKS3 family. A. Pedigree of the Chinese MKS3 family. B. Genomic structure of TMEM67 (NM_153704). The 28 exons of TMEM67, encompassing 62 kb of genomic sequence, are shown as vertical bars, with their approximate sizes and positions indicated. Positions of the start codon (ATG) at nucleotide +1 and the stop codon (TAA) are indicated. Correspondence between TMEM67 exons and predicted meckelin domains is depicted at the bottom. C. Sequence chromatogram of affected individual (homozygous), carrier (heterozygous) and control (wild type). D. Conservation analysis showing that Arg549 in TMEM67 is conserved across human (Homo sapiens), rat (Rattus norvegicus), mouse (Mus musculus), zebrafish (Danio rerio), chicken (Gallus gallus), wolf (Canis lupus familiaris) and platypus, (Omithorhynchus anatinus). E. Endonuclease digestion Hha I digestion distinguish the homozygote (217 bp, III-1, III-3, III-4) from the wild-type (130, 87 bp; II-1, I-1, II-4, I-4) and heterozygote (217, 130, 87 bp; II-2, I-2, II-3, I-3) on agarose gel electrophoresis.
Immunohistochemitry of meckelin
We further visualized the localization of meckelin in paraffin-embedded fetal tissues (liver and kidney from III-4 and the normal control from a 17 gw fetus. In the normal control, moderate to high levels of meckelin were localized at the renal tubule epithelia, but not in the glomeruli. In the MKS3 fetus, intense staining was shown in the cysts epithelium of the kidney but not in the remnant glomeruli. In the liver, meckelin was expressed in the epithelial cell layer of enlarged intrahepatic bile ducts around the portal area in MKS3 fetus. In the control, the same locations were seen, but without bile ducts dilated (Figure 3).
Figure 3.
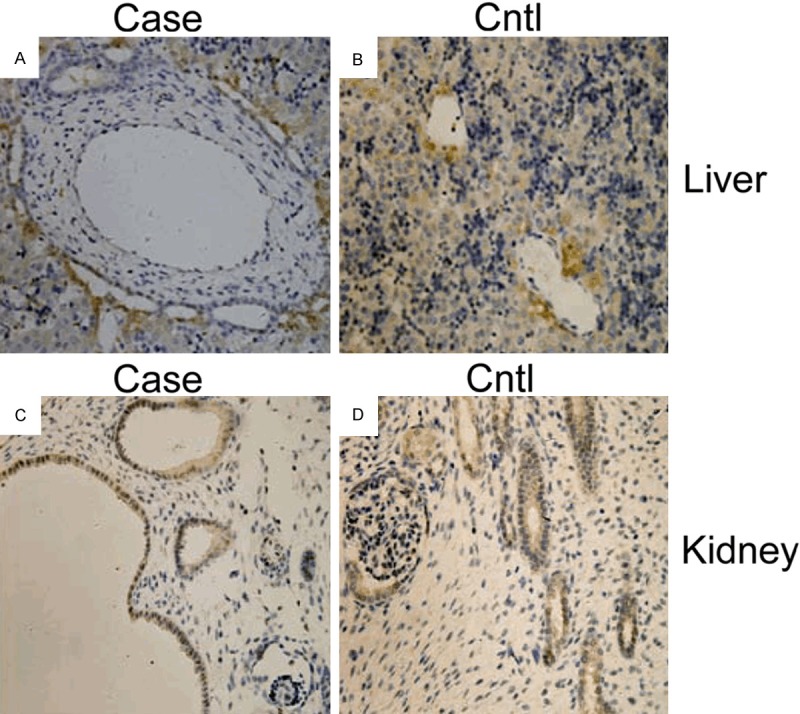
Meckelin expression detected by immunostaining. A. In the liver of the MKS3 fetus (III-4), meckelin was expressed in epithelial cells of enlarged intrahepatic bile ducts around the portal area in the MKS3 fetus. B. In the control, the same location was seen but without bile ducts dilated. Hepatocytes showed slight staining in both affected fetuses and in the control. C. In the kidney of the MKS fetus (III-4), showed intense staining was seen in cysts epithelium but not in the remnant glomerulus. D. In the control, meckelin was expressed in renal tubules but not in the glomeruli. Original magnification, ×400.
Discussion
In the present study, we report the identification of a TMEM67 mutation in a Chinese MKS3 family with four affected individuals. All affected fetuses displayed renal cystic dysplasia and occipital encephalocele. Ductal plate malformations with proliferation of bile duct were showed in the last three affected fetuses. Comparison of the clinical features of MKS3-linked cases with MKS1-linked cases suggested that polydactyly and possibly encephalocele are less common in MKS3-linked families [9,10]. The absence of polydactyly in the first three affected fetuses suggested that TMEM67 was the good candidate gene for mutation screening in this family. Sequence analysis and Hha I restriction analysis demonstrated that a homozygous c.1645C>T mutation in TMEM67 was cosegregated with the MKS phenotype in this family and the parents were heterozygous carriers. Note that the fourth affected fetus, which carried the same MEM67 mutation, had additional polydactyly which showed phenotypic heterogeneity in the Chinese MKS3 family.
TMEM67 spectrum diseases range from embryonically lethal Meckel syndrome, to less severe multisystem disorders, such as Bardet-Biedl syndrome (MIM 209900), COACH syndrome (MIM 216360), Joubert syndrome 6 (MIM 610688) and nephronophthisis 11 (MIM 613550). A systematic analysis of the phenotypic burden of TMEM67 mutations has been reported [9]. A differential distribution of mutations along the TMEM67 gene in lethal (MKS) versus non-lethal (JSRD, NPH and ARPKD-like) phenotypes is observed, particularly with regard to missense mutations. In MKS patients, most missense mutations cluster in exons 8 to 15, encoding the extracellular region of meckelin that follows the cysteine-rich domain [11]. We report a lethal missense mutation in exon 16 of the TMEM67 gene in Chinese family, which encodes the extracellular region of meckelin. This is the first report of TMEM67 mutation in Chinese population. The same substitution was reported by Katharina Hopp as a highly likely mutation [7]. It is possible that, within the range of exons 8 to 16, all encoding extracellular fragments are located just before the corresponding transmembrane fragments. Except for lethal MKS, primary cilia-related disease, including Joubert syndrome, Bardet-Biedl syndrome and COACH syndrome, were accompanied with some structural abnormality at the fetal stage, such as ‘molar tooth sign’ (MTS)-cerebellar vermis hypo-dysplasia, thickening and horizontalization of superior cerebellar peduncles and deepening of the interpeduncular fossaon MRI, Dandy-Walker malformation and polydactyly [12,13]. Because many genes are involved in these disorders, traditional methods of mutation screening are difficult to identify the causative genes. With the development of a new generation of sequencing strategies, we can test all ciliapathy-related genes simultaneously following ultrasound identification of features associated with ciliopathy by prenatal diagnosis [14].
We also visualized the localization of meckelin in the MKS fetus and normal controls. As previously reported there was strong staining in the epithelial cell layer of the renal tubule but not in the glomerulus [6]. Our results showed intense staining of renal cysts in the MKS3 fetus. Dawe et al. reported that meckelin immunostaining was absent in the renal tissue of the MKS3 fetus who carried a homozygous c.1127A>C (p.Q376P) mutation in TMEM67 [6]. In our study, meckelin staining was present in both kidney tissues of affected fetus with homozygous c.1645C>T (p.R549C) mutation in TMEM67 and normal control.
In summary, we have identified a homozygous TMEM67 mutation in a Chinese family exhibiting clinical characteristics of MKS3. Statistically, this couple would be predicted to have a 25% chance of producing an affected embryo. However, four previous natural pregnancies of this couple all turned to be affected MKS3 fetuses detected by ultrasound, and the couple repeatedly opted to terminate the pregnancies by artificial abortions. The identification of the causative mutation of TMEM67 in this family provided a ground for PGD procedure for this family. Further efforts will be focused on developing a PGD protocol to the couples at risk of conceiving a pregnancy affected with MKS3 and other known monogenic diseases.
Acknowledgements
This work was supported by the Key Project of National Natural Science Foundation of China to Huijun Yuan (No. 81030017), National Science Fund for Distinguished Young Scholars to Huijun Yuan (No. 81125008). Science Fund from Chinese PLA General Hospital to Yanping Lu (No. 2012FC-CXYY-1001). We sincerely thank the family members for their participation and support in this study.
Disclosure of conflict of interest
None.
References
- 1.Alexiev BA, Lin X, Sun CC, Brenner DS. Meckel-Gruber syndrome: pathologic manifestations, minimal diagnostic criteria, and differential diagnosis. Arch Pathol Lab Med. 2006;130:1236–1238. doi: 10.5858/2006-130-1236-MS. [DOI] [PubMed] [Google Scholar]
- 2.Barker AR, Thomas R, Dawe HR. Meckel-Gruber syndrome and the role of primary cilia in kidney, skeleton, and central nervous system development. Organogenesis. 2014;10:96–107. doi: 10.4161/org.27375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kyttala M, Tallila J, Salonen R, Kopra O, Kohlschmidt N, Paavola-Sakki P, Peltonen L, Kestilä M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat Genet. 2006;38:155–157. doi: 10.1038/ng1714. [DOI] [PubMed] [Google Scholar]
- 4.Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S, Morgan NV, Goranson E, Gissen P, Lilliquist S, Aligianis IA, Ward CJ, Pasha S, Punyashthiti R, Malik Sharif S, Batman PA, Bennett CP, Woods CG, McKeown C, Bucourt M, Miller CA, Cox P, Algazali L, Trembath RC, Torres VE, Attie-Bitach T, Kelly DA, Maher ER, Gattone VH 2nd, Harris PC, Johnson CA. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Genet. 2006;38:191–196. doi: 10.1038/ng1713. [DOI] [PubMed] [Google Scholar]
- 5.Dawe HR, Smith UM, Cullinane AR, Gerrelli D, Cox P, Badano JL, Blair-Reid S, Sriram N, Katsanis N, Attie-Bitach T, Afford SC, Copp AJ, Kelly DA, Gull K, Johnson CA. The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet. 2007;16:173–186. doi: 10.1093/hmg/ddl459. [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, García-Verdugo JM, Katsanis N, Hildebrandt F, Reiter JF. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43:776–784. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hopp K, Heyer CM, Hommerding CJ, Henke SA, Sundsbak JL, Patel S, Patel P, Consugar MB, Czarnecki PG, Gliem TJ, Torres VE, Rossetti S, Harris PC. B9D1 is revealed as a novel Meckel syndrome (MKS) gene by targeted exon-enriched next-generation sequencing and deletion analysis. Hum Mol Genet. 2011;20:2524–34. doi: 10.1093/hmg/ddr151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S, Ozilou C, Faivre L, Laurent N, Foliguet B, Munnich A, Lyonnet S, Salomon R, Encha-Razavi F, Gubler MC, Boddaert N, de Lonlay P, Johnson CA, Vekemans M, Antignac C, Attie-Bitach T. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet. 2007;80:186–194. doi: 10.1086/510499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Consugar MB, Kubly VJ, Lager DJ, Hommerding CJ, Wong WC, Bakker E, Gattone VH 2nd, Torres VE, Breuning MH, Harris PC. Molecular diagnostics of Meckel-Gruber syndrome highlights phenotypic differences between MKS1 and MKS3. Hum Genet. 2007;121:591–599. doi: 10.1007/s00439-007-0341-3. [DOI] [PubMed] [Google Scholar]
- 10.Khaddour R, Smith U, Baala L, Martinovic J, Clavering D, Shaffiq R, Ozilou C, Cullinane A, Kyttälä M, Shalev S, Audollent S, d’Humières C, Kadhom N, Esculpavit C, Viot G, Boone C, Oien C, Encha-Razavi F, Batman PA, Bennett CP, Woods CG, Roume J, Lyonnet S, Génin E, Le Merrer M, Munnich A, Gubler MC, Cox P, Macdonald F, Vekemans M, Johnson CA, Attié-Bitach T SOFFOET (Société Française de Foetopathologie) Spectrum of MKS1 and MKS3 mutations in Meckel syndrome: a genotype-phenotype correlation. Hum Mutat. 2007;28:523–524. doi: 10.1002/humu.9489. [DOI] [PubMed] [Google Scholar]
- 11.Iannicelli M, Brancati F, Mougou-Zerelli S, Mazzotta A, Thomas S, Elkhartoufi N, Travaglini L, Gomes C, Ardissino GL, Bertini E, Boltshauser E, Castorina P, D’Arrigo S, Fischetto R, Leroy B, Loget P, Bonnière M, Starck L, Tantau J, Gentilin B, Majore S, Swistun D, Flori E, Lalatta F, Pantaleoni C, Penzien J, Grammatico P International JSRD Study Group. Dallapiccola B, Gleeson JG, Attie-Bitach T, Valente EM. Novel TMEM67 mutations and genotype-phenotype correlates in meckelin-related ciliopathies. Hum Mutat. 2010;31:E1319–E1331. doi: 10.1002/humu.21239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, Hove MT, Fennell EB, Booth-Jones M, Ringdahl DM, Yachnis AT, Creel G, Frerking B. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997;12:423–430. doi: 10.1177/088307389701200703. [DOI] [PubMed] [Google Scholar]
- 13.Saleem SN, Zaki MS, Soliman NA, Momtaz M. Prenatal magnetic resonance imaging diagnosis of molar tooth sign at 17 to 18 weeks of gestation in two fetuses at risk for Joubert syndrome and related cerebellar disorders. Neuropediatrics. 2011;42:35–38. doi: 10.1055/s-0031-1275739. [DOI] [PubMed] [Google Scholar]
- 14.Otto EA, Ramaswami G, Janssen S, Chaki M, Allen SJ, Zhou W, Airik R, Hurd TW, Ghosh AK, Wolf MT, Hoppe B, Neuhaus TJ, Bockenhauer D, Milford DV, Soliman NA, Antignac C, Saunier S, Johnson CA, Hildebrandt F GPN Study Group. Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J Med Genet. 2011;48:105–116. doi: 10.1136/jmg.2010.082552. [DOI] [PMC free article] [PubMed] [Google Scholar]