

Abstract
A case of epithelioid inflammatory myofibroblastic scarcoma (EIMS) developing in an 8-year-old boy who presented with a bulky intra-abdominal occupying lesion with recurrence undergoing a radical resection was reported. Histologically, the tumor cells arranged in cords, strands or sheets of round-to-epithelioid cells with a vesicular nuclear chromatin pattern, prominent nucleoli and weakly eosinophic or basophilic cytoplasm embedded in the abundant myxoid stroma with lymphocytes infiltration. They were positive for ALK, Desmin, SMA, CD30, but negative for AE1/AE3, LCA, CD2, CD3, CD5, CD7, S-100, CD34, CD31, EMA, MyoD1, and myogenin. An elevated proliferation index was demonstrated by Ki-67 comparing the first and the second lesion. Fluorescence in situ (FISH) showed the presence of chromosomal translocation involving ALK. This case show EIMS is a rare variant of inflammatory myofibroblatic tumor with aggressive biological behavior and unfavourable prognosis. To be familiar with its significant clinicopathologic characteristics could prompt us to take it into consideration when facing the relevant dieases.
Keywords: Epithelioid inflammatory myofibroblastic scarcoma, soft tissue neoplasm, ALK, FISH
Introduction
Inflammatory myofibroblastic tumors (IMT) is a mesenchymal neoplasm composed of proliferated fibroblastic or myofibroblastic cells with a promiment infiltration of chronic inflammatory cells accompanying ALK gene rearrangements. Recently, the epithelioid subset with an significantly aggressive biological behavior, proposing a nomenclature as “epithelioid inflammatory myofibroblastic scarcoma (EIMS)”, was reported by a limited literature [1]. Here, we added a case of EIMS adopting a bulky intra-abdominal occupying lesion with recurrence undergoing a radical resection to investigate the clincopathologic, immunohistochemical, and molecular features and differential diagnosis.
Clinical history
An 8-year-old boy was admitted for the right abdominal mass with recurrent hyperpyrexia as high as 39°C. A computerized tomography scan show a bulky solid lump of 156 mm in maximum diameter without definite demarcation involving partial intestinal wall and omentum majus with some enlarged lymph nodes. An eradication of the tumor and involved tissues were performed by positive resection. However, the patient experienced a dismally sinister recurrence with a returning-growing mass occupying the entire abdominal cavity and succumbed eventually, albeit a second extended resection, after 8 months.
Materials and methods
The twice removed surgical specimens were fixed in 4% formalin, embedded routinely in paraffin and stained with hematoxylin and eosin. Immuohistochemical studies were performed in selected blocks by the avidin-biotin-peroxidase complex technique using commercial antibodies in the Ventana BenchMark XT instrument (Ventana System, Tucson AZ). The antibodies included AE1/AE3, ALK, Desmin, MyoD1, myogenin, myoglobin, SMA, Caldesmon, Calponin, P63, S-100, MDM2, LCA, CD30, EMA, CD117, CD2, CD3, CD5, CD7, Ki-67 (all above from Ventana, prediluted).
Fluorescence in situ hybridization evaluation for ALK rearrangement was performed on 4 μm-thick paraffin section with LSI ALK dual color break-apart probe (Abbot Molecular, Vysis, Des Plaines, IL), based on the manufacturer’s instruction.
Results
Grossly, the first resected tumor measuring 18 cm×10 cm×9.5 cm showing a solid and gray-yellowish with myxoid appearance in cut surface (Figure 1A). An increasing mucus feeling somewhat gelatinous was appreciated in the recurrent excision (Figure 1B). Microscopically, the first and second specimens were similar, except for the latter with elevated mucus matrix. Epithelioid or round tumor cells loosely interspersed in the ample myxoid stroma as much as the extent that some areas were punctuated by the dilated irregular spaces or gland-like structure lined by neoplasm cells with scattered fall-off cells in the lumens (Figures 2, 3). And evident attenuated vascular proliferation can be seen, where there was relatively high cellularity accompanying obvious inflammation cells predominant in neutrophils (Figures 3, 4). The tumor cells were interpreted as obvious vesicular and a slightly eccentric nuclei with apparent nucleoli and eosinophilic or amphophilic cytoplasm (Figure 3). The minor spindle cell areas were hard to evaluate.
Figure 1.
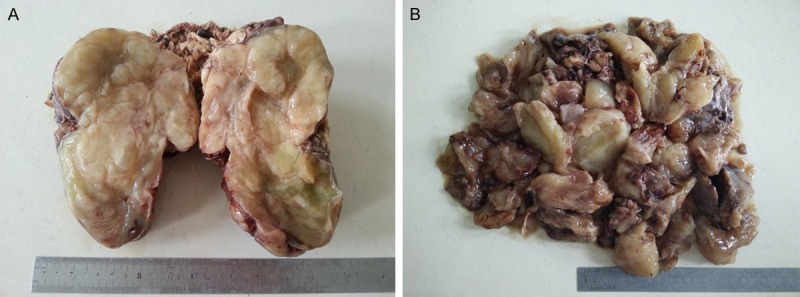
A. The cut surface of the first-resected tumor showing a solid and gray-yellowish with myxoid appearance. B. The second specimen morcellated in clinic demonstrated an increasing size and mucus feeling.
Figure 2.
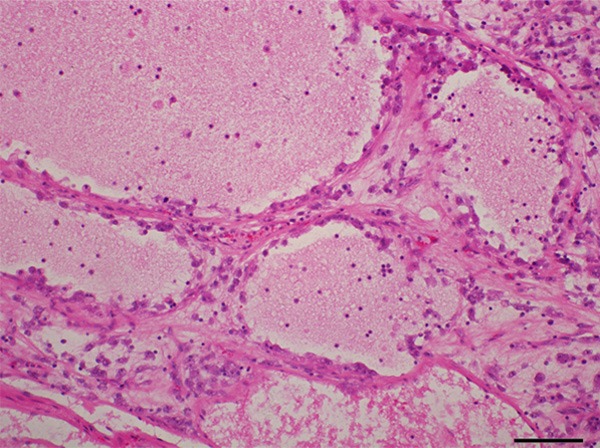
Abundant myxoid stroma contributed to evidently dilated spaces lined by tumor cells with scattered fall-off cells in the lumens.
Figure 3.
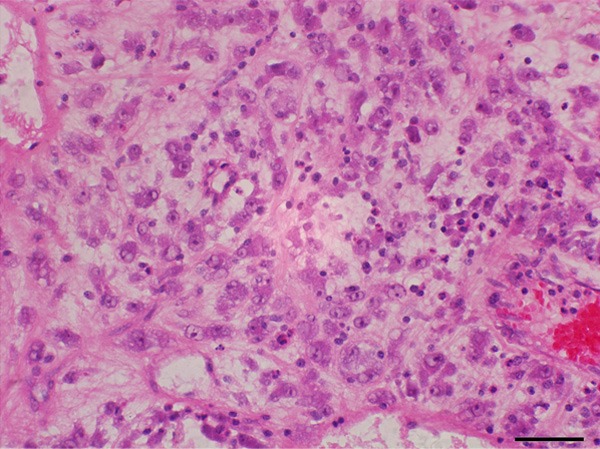
Tumor cells accentuated an epithelioid or round appearance with vesicular nuclei, apparent nucleoli, and amphophilic cytoplasm accompanying evident inflammation cells predominant in neutrophils.
Figure 4.
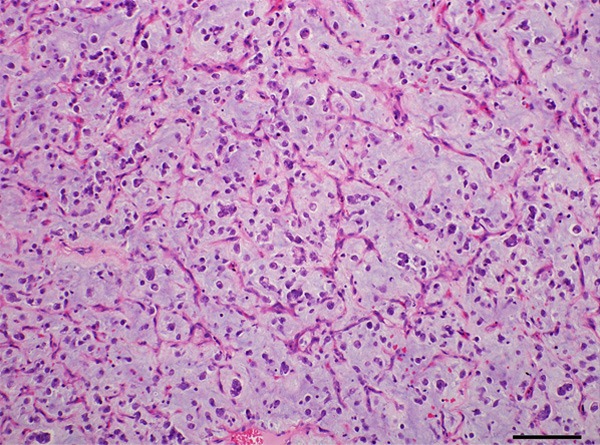
The recurrent tumor showed high density of attenuate and branching blood vessels and increasing myxoid matrix mimicking myxoid liposarcoma.
The tumor cells were positive for Desmin (Figure 5), SMA, CD30 (Figure 6) and ALK (Figure 7) in nuclear membrane pattern of staining, but negative for AE1/AE2, LCA, CD2, CD3, CD5, CD7, S-100, CD34, CD31, EMA, MyoD1, Myogenin. Proliferative index Ki-67 was approximately 15%, whereas increased as high as 20% in the recurrent specimen.
Figure 5.
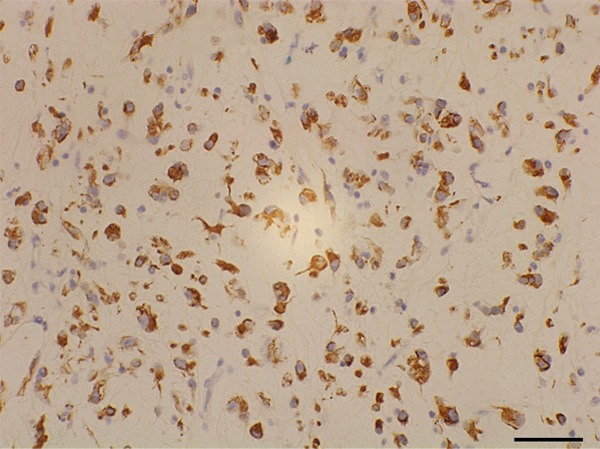
Tumor cells were positive for desmin.
Figure 6.
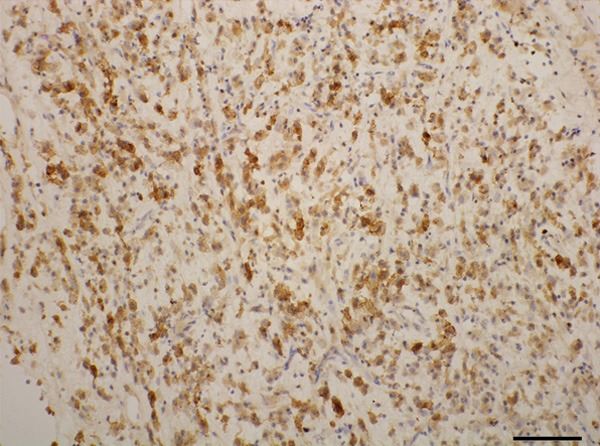
CD30 was strongly positive.
Figure 7.
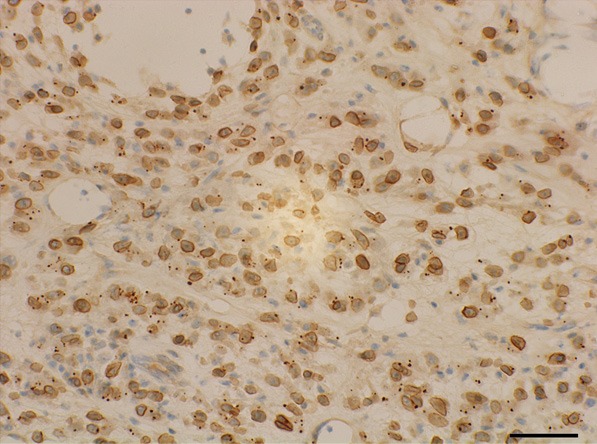
Immunohistochemistry for ALK protein distinctively exhibited nuclear membrane pattern of staining.
FISH analysis illustrated the arrangement signal in 80 out of 100 tumor nuclei, which typically demonstrated one juxtaposed green and red signal and an obvious separated, red and green signal per nucleus respectively, indicating the presence of ALK gene translocation (Figure 8).
Figure 8.
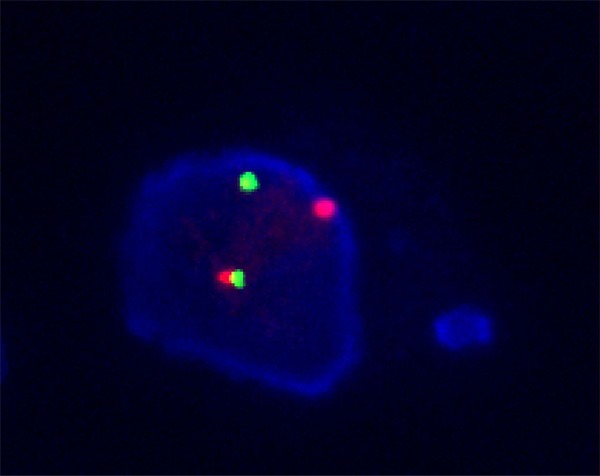
An image of ALK rearrangement. The tumor cell displayed 1 intact fused signal and a separated green and red signal indicating the translocation of one copy of the ALK per nucleus.
Discussion
First described by Marino-Enriquez [1], an extremely rare IMT with a prominent epithelioid or round cell morphology is different from the conventional IMT owing to their distinct clinicopathologic and cytogenetic features and therefore was proposed a brand new designation as “epithelioid inflammatory myofibroblastic sarcoma (EIMS)”, which has not yet been included in 2013 WHO classification of soft tissue tumors.
Clinically, the tumor occurs mainly in children and adolescents, although the overall range in age very wide (from 7 months to 63 years, with median age of 39 years) [1]. Compared to extensive anatomic distribution and female predominance of the conventional IMT, EIMS has a male predilection and almost always involves in abdominal cavity, except for in pleural cavity by two cases [2,3]. A bulky lesion is usually represented depending on its unique anatomic location with or without visceral involvement. And even more important, as an intermediate neoplasm, IMT has a tendency to local recurrence, but rarely undergoes malignant transformation [4]. However, the biological behavior of EIMS seems to be overtly aggressive, with rapid local recurrence, lymph node or remote metastases, particularly involving in liver according to the limited literature [1,5,6]. Accordingly, the patient of our case also suffered from a sinister duration and a surprisingly quick recurrence, even if a complete resection in clinic, and died eventually. Grossly, tumors usually abut on bowel tissues, and the cut surface tends to be solid and, regardless of microscopy, relative demarcation, with a fleshy appearance accompanying some mucoid areas. Histologically, EIMS exhibits a frankly epithelioid or round tumor cells embedded in abundant myxoid matrix haphazardly and loosely separating from the conventional IMT, albeit minor areas composed of spindle cells. The neoplastic cells bear vesicular nuclei, distinct nucleoli and amphophilic cytoplasm admixed with a prominent lymphocytes infiltration predominant in neutrophils aggregation [3,6]. Immunochemically, the vast majority of tumor cells show a consistent immunophnotype, including ALK, CD30, Desmin, with some cells labeled by SMA, but absence of AE1/AE3, EMA, S-100, caldesmon, myogenin [1]. Typically, immunostain of ALK mainly located in nuclear membrane, occasionally imparting a cytoplasmic pattern with perinuclear accentuation. Although a collection of ALK fusion partner genes were subsequently appreciated, including TMP-3/4, CLTC, ATIC, CARS, RANBP2, LMNA and PRKAR1A [7], EIMS is characteristic of a constant gene fusion involving in exon18 of RANPB2 at 2q13 and exon 20 of ALK at 2p23, frequently contributing to unfavorable prognosis [3]. Two literature reveal that administration of crizotinib, an anaplastic kinase inhibitor, resulted in tumor shrinkage, however, in one of which patients quickly relapsed and resisted to targeted therapeutic drugs [6,8]. Similarly, the tumor of our case displayed a full lethal features with prominently aggressive behavior. Several entities with epithelioid cells or myxoid stroma sheared a constellation of morphologic characteristics should be taken into consideration in the differential diagnosis such as myxofibrosarcoma, myoxid/round cell liposarcoma, rabdomysarcoma, high-grade epithelioid leiomyosarcoma and melanoma. EIMS features child or adolescence predominance, overwhelmingly abdominal cavity involved in clinical representation, epithelioid cells bathed in the abundant myxoid stroma with significant inflammatory cells predominant in distinct neutrophils aggregation and positive for ALK, CD30, Desmin, SMA, but negative for S-100, Caldesmon, myogenin and HMB45 in immunohistochimistry without arrangement involving DDIT3 gene could easily tell them apart [1]. In addition, the most challenges encountered when differentiating anaplastic large cell lymphoma (ALCL) from EMIS. However, the former rarely expresses ALK protein in nuclear membrane or perinuclear accentuation staining pattern, and is also deficient of expression of Desmin and SMA [3]. A distinctive rearrangement showing ALK-RANBP2 fusion which rarely appreciated in ALCL can be diagnostically helpful.
In conclusion, EIMS is a distinctive rare tumor with significant clinicopathologic and cytogenetic features and aggressive biological behavior. To familiar with this entity could contribute to avoid misdiagnosis when we face the analogous diseases.
Acknowledgements
This work was financially supported by the first affiliated hospital of Zhengzhou University.
Disclosure of conflict of interest
None.
References
- 1.Marino-Enriquez A, Wang WL, Roy A, Lopez-Terrada D, Lazar AJ, Fletcher CD, Coffin CM, Hornick JL. Epithelioid inflammatory myofibroblastic sarcoma: an aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol. 2011;35:135–144. doi: 10.1097/PAS.0b013e318200cfd5. [DOI] [PubMed] [Google Scholar]
- 2.Kozu Y, Isaka M, Ohde Y, Takeuchi K, Nakajima T. Epithelioid inflammatory myofibroblastic sarcoma arising in the pleural cavity. Gen Thorac Cardiovasc Surg. 2014;62:191–194. doi: 10.1007/s11748-013-0204-x. [DOI] [PubMed] [Google Scholar]
- 3.Li J, Yin WH, Takeuchi K, Guan H, Huang YH, Chan JK. Inflammatory myofibroblastic tumor with RANBP2 and ALK gene rearrangement: a report of two cases and literature review. Diagn Pathol. 2013;8:147. doi: 10.1186/1746-1596-8-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gupta RK, Samalavicius NE, Sapkota S, Sah PL, Kafle SU. Colonic inflammatory myofibroblastic tumours: an institutional review. Colorectal Dis. 2013;15:e239–243. doi: 10.1111/codi.12149. [DOI] [PubMed] [Google Scholar]
- 5.Fujiya M, Kohgo Y. ALK inhibition for the treatment of refractory epithelioid inflammatory myofibroblastic sarcoma. Intern Med. 2014;53:2177–2178. doi: 10.2169/internalmedicine.53.3038. [DOI] [PubMed] [Google Scholar]
- 6.Kimbara S, Takeda K, Fukushima H, Inoue T, Okada H, Shibata Y, Katsushima U, Tsuya A, Tokunaga S, Daga H, Okuno T. A case report of epithelioid inflammatory myofibroblastic sarcoma with RANBP2-ALK fusion gene treated with the ALK inhibitor, crizotinib. Jpn J Clin Oncol. 2014;44:868–871. doi: 10.1093/jjco/hyu069. [DOI] [PubMed] [Google Scholar]
- 7.Lovly CM, Gupta A, Lipson D, Otto G, Brennan T, Chung CT, Borinstein SC, Ross JS, Stephens PJ, Miller VA, Coffin CM. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014;4:889–895. doi: 10.1158/2159-8290.CD-14-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butrynski JE, D’Adamo DR, Hornick JL, Dal Cin P, Antonescu CR, Jhanwar SC, Ladanyi M, Capelletti M, Rodig SJ, Ramaiya N, Kwak EL, Clark JW, Wilner KD, Christensen JG, Janne PA, Maki RG, Demetri GD, Shapiro GI. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363:1727–1733. doi: 10.1056/NEJMoa1007056. [DOI] [PMC free article] [PubMed] [Google Scholar]