

Abstract
von Hippel-Lindau disease (VHLD) comprises a series of complicated clinical manifestations. We hereby described a unique case of co-existing T-cell lymphoma (TCL) and confirmed VHLD. The symptoms in this 42-year-old male included fever and pancytopenia. Overall tests and examination made an infectious process unlikely. The results of bone marrow biopsy confirmed the diagnosis. The purposes we described this case were to probe into the relationship between TCL and VHLD, which was not mentioned in previously literature. Combination of clinical, radiological, immunophenotypic, pathological, and genetic data plays an important role in improving the rate of diagnosis, particularly in the challenge for diagnosis of T cell non-Hodgkin lymphoma.
Keywords: von Hippel-Lindau disease, T-cell lymphoma, hematological neoplasm
Introduction
von Hippel-Lindau disease (VHLD) is a heritable multisystem cancer syndrome that is related to a germline mutation of the VHL tumor suppressor gene on the short arm of chromosome 3 [1]. VHLD has an incidence of 1/36,000 to 1/53,000 newborns [2]. VHL patients are predisposed to develop lesions of the central nervous system (CNS) and viscera. CNS lesions include hemangioblastomas, the most common tumor in VHL, and endolymphatic sac tumors (ELSTs). Visceral manifestations include renal carcinomas and cysts, pancreatic neuroendocrine tumors and cysts, pheochromocytomas and cystadenomas of the reproductive adnexal organs [3]. But few report covered hematological disorders, especially T-cell lymphoma.
T-cell lymphoma represents a heterogeneous group of diseases with varied clinical features, prognosis and response to treatment [4]. Their incidence seems to have increased recently, also because of an improvement in diagnostic methods. Such conditions now account for approximately 20-30% in Asia [5-7] and 5-10% in Europe and North America [8] of all lymphoid neoplasms. The low incidence of T-cell lymphoma poses real difficulties for a complete and correct assessment. The unspecified lymphomas represent a heterogeneous group, which requires additional studies to elucidate their biological and genetic bases, to separate them.
We reported an unusual case of T-cell lymphoma with von Hippel-Lindau disease. To our knowledge, it has not been reported before. It revealed the challenge and the important role of combination of clinical, radiological, immunophenotypic, pathological, and genetic technique in T-cell lymphoma diagnosis.
Case report
History and examination
A 42-year-old male South American was admitted to our ward for 6 weeks history of intermittent fever ranging from 38-39°C and pancytopenia, and 1 week history of sore throat associated with brown nasal discharge and abdominal pain. 6 days oral moxifloacin was not working. 5 weeks before admission, cell morphology of bone narrow smear showed three series decreased, mature granulocyte proliferation and increasing rate of lymphocytes.
His past medical history included post thoracic hemangioblastomas resection and von Hippel-Lindau disease for 5 years confirmed by genetic test without family history.
On our physical examination, we just found pharyngeal hyperemia but no hepatosplenomegaly or superficial lymphadenopathy.
Hematologic examination still revealed pancytopenia. Negative blood and urine cultures, negative screening tests for EBV, CMV, HIV, HCV, T spot TB test, and normal transthoracic echocardiography made an infectious process unlikely. The rheumatologic markers, such as antinuclear antibodies (ANA), anti-extractable nuclear antigen (anti-ENA) and anti-neutrophil cytoplasmic antibody (ANCA) were negative. Tumor markers showed no abnormality except for mildly increases of neuron-specific enolase (NSE) and CyFRA211, and particularly markedly ascending thymidine kinase 1 (TK1). During the admission, ultrasound found some 6×15 cm sized cervical lymph nodes which was located near bilateral vessels. An enhanced computed tomography (CT) of the abdomen showed multiple pancreatic cysts with calcification and renal cysts (Figure 1), and Chest CT revealed slight bilateral pneumonia. Nasal endoscopy, paranasal sinus CT and fundoscopy were normal.
Figure 1.
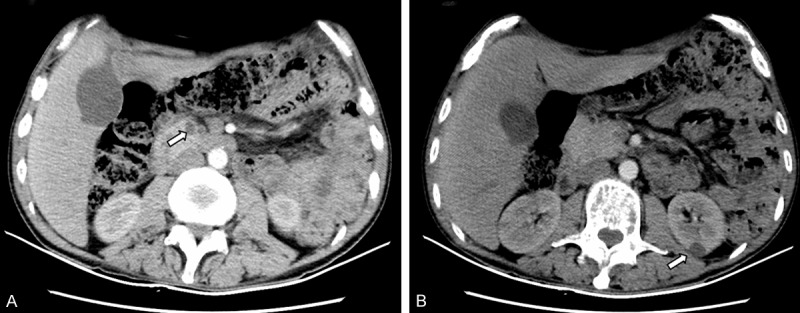
Radiologic characterization of VHLD. A. CT imaging demonstrated multiple pancreatic cysts. B. CT imaging demonstrated multiple renal cysts.
Immunophenotypic and pathological findings
After 5-day treatment of Azithromycin and Ceftazidime, pharyngalgia and abdominal pain disappeared but hyperpyrexia, leukopenia and anemia were deteriorated. Thus, we repeated bone marrow puncture. Cell morphology analysis found 2% heterocysts with myeloid and erythroid series hypoplasia. The immunophenotypic results showed high ratio of lymphocyte subsets (61.5%) in bone marrow. The following markers were positive in lymphocytes: CD38, CD2, CD3, CD5, CD7 (Table 1; Figure 2). Pathological results of bone marrow biopsy remindered a lot of abnormal hyperplastic cells with hyperchromatic nuclei and a little hemopoietic tissue and adipose tissue among bone trabeculae. Marked pancytopenia was confirmed in hemopoietic tissue. Immunohistochemical staining demonstrated proliferating T lymphocytes positive reactivity for CD5+, CD7+ and CD3+, while no B lymphocyte hyperplasia, epithelial and neuroendocrine markers expression were found (Figures 3, 4). Immunohistochemical staining confirmed T lymphocyte neoplastichyperplasia.
Table 1.
Immunophenotypic analysis by flow cytometer
| Lymphocyte subsets (61.5%) | The expression rate (%) | Monocyte subsets (19.2%) | The expression rate (%) | Granulocyte subsets (10.0%) | The expression rate (%) |
|---|---|---|---|---|---|
| CD19+ | 2.4 | CD4+ | 47.8 | CD10+ | / |
| CD20+ | 2.2 | CD15+ | 36.8 | CD15+ | / |
| CD10+ | 0 | CD11b+ | 95 | CD11b+ | / |
| CD38+ | 84 | CD14+ | 78.2 | CD16+ | / |
| CD3+ | 58.1 | CD13+ | 80.2 | CD14+ | / |
| CD3+CD4+ | 19.3 | CD33+ | 79.6 | CD33+ | / |
| CD3+CD8+ | 24.7 | HLA-DR+ | 78.8 | CD13+ | / |
| CD2+ | 91.1 | ||||
| CD5+ | 53.3 | ||||
| CD7+ | 89.9 |
Figure 2.
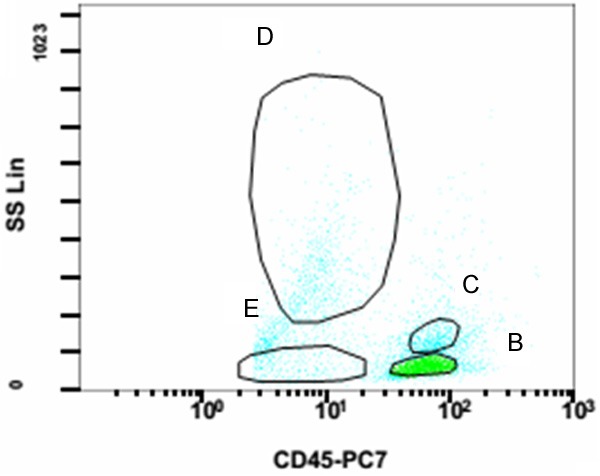
Immunophenotypic analysis by flow cytometer. CD45/SS scatter diagram showed normal CD45 subsets. B: Lymphocyte subsets (61.5%); C: Monocyte subsets (19.2%); D: Granulocyte subsets (10.0%); E: Erythrocyte subsets.
Figure 3.
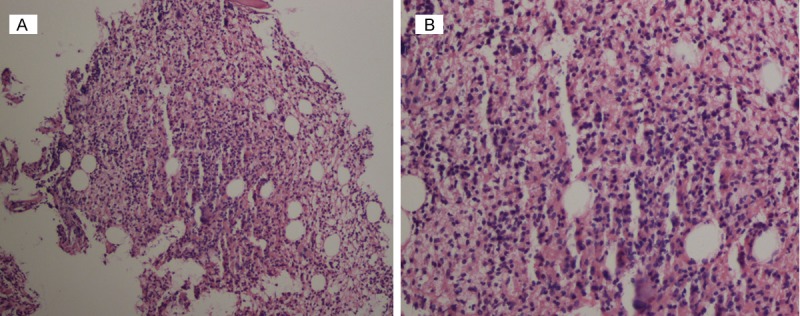
Histopathologic characterization of bone marrow biopsy. A. Histologic sections showed a little hemopoietic tissue and adipose tissue among bone trabeculae (H&E, 100×). B. A lot of abnormal hyperplastic cells with hyperchromatic nuclei appeared (H&E, 100×).
Figure 4.
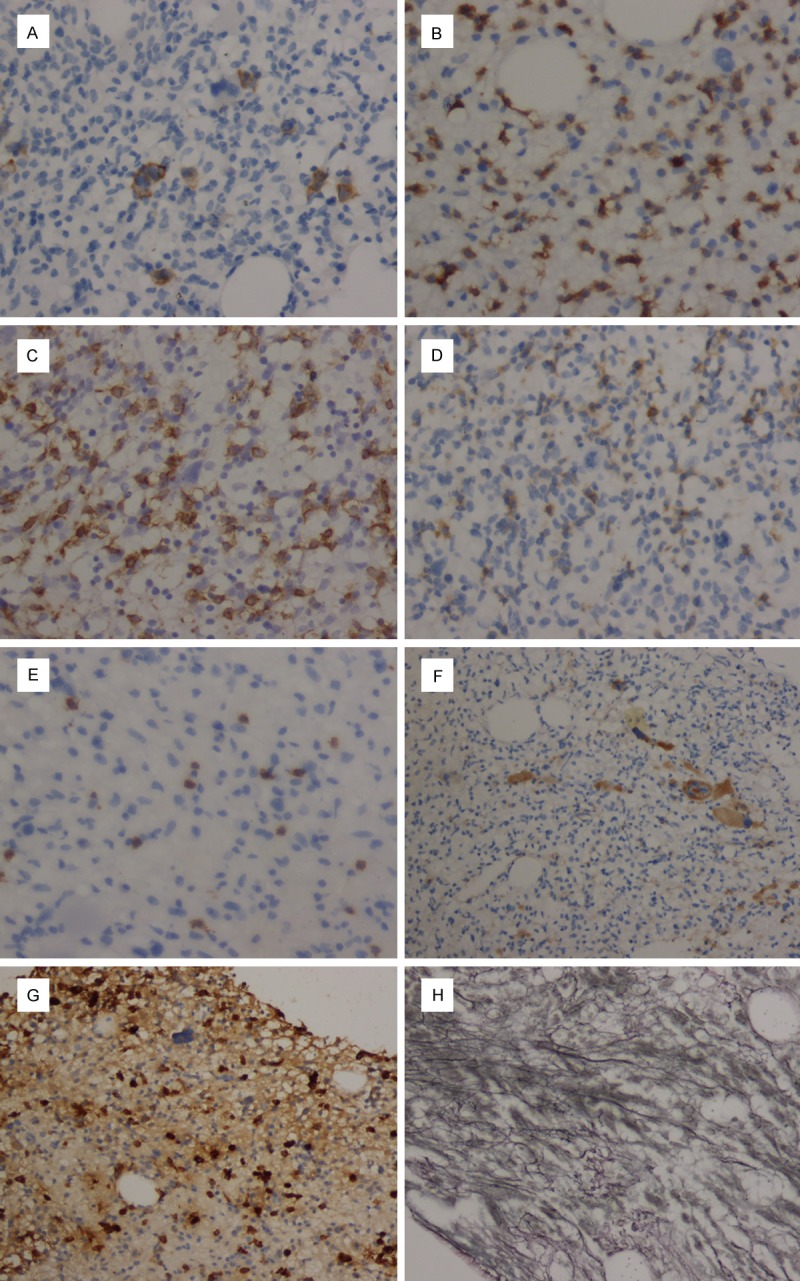
Immunohistochemical characterization of bone marrow biopsy. A. The CD30+ cells. B. The CD3+ cells. C. The CD7+ cells. D. The CD5+ cells. E. The CD20+ cells. (immunostaining, 400×). F. The CD31+ cells (megakaryocytic subsets). G. The MPO+ cells. H. The reticular fiber staining. (immunostaining, 100×).
The patient required to go back home for further treatment. So he was transferred to the local hospital and received chemotherapy after supportive treatment.
Discussion
VHLD is an autosomal dominant neoplasia syndrome, which was first described by the German ophthalmologist Eugene von Hippel and the Swedish pathologist Avrid Lindau at the beginning of the 20th century [3]. Its incidence is 1/36,000 to 1/53,000 of newborns [2]. VHLD is caused by a mutation in the VHL tumor suppressor gene on chromosome 3p25-26 resulting in the loss of the VHL protein (pVHL) tumor suppressor protein function [9,10]. By the age of 65 years, more than 90% of individuals with VHL will display some disease related symptoms [11]. Patients with VHL are predisposed to develop specific central nervous system (CNS) and visceral lesions [1]. Affected individuals are at risk of developing various benign and malignant tumors of the CNS, kidneys, adrenal glands, pancreas, and reproductive adnexal organs. Because of the complexities associated with management of the various types of tumors in this disease, treatment is multidisciplinary. The following three criteria or genetic testing suggest a diagnosis of VHLD: (1) one or more hemangioblastomas within the central nervous system (including retinal hemangioblastomas), typically in the cerebellum, (2) presence of visceral lesions (e.g. renal, pancreatic tumors/cysts), and (3) familial incidence (Figure 5) [12]. This patient was characterized by multiple tumors and cysts involved in spinal hemangioblastomas, pancreatic and renal cysts, as well as gene test, which was in accordance with VHL criteria.
Figure 5.
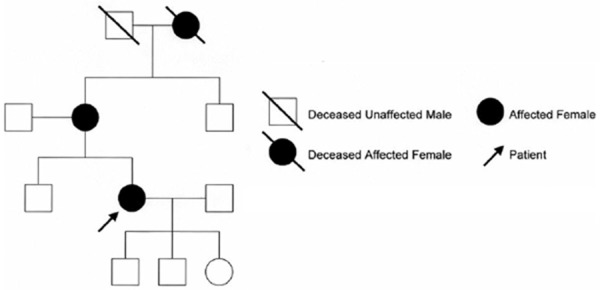
The pedigree of VHLD.
The VHL gene is widely expressed in tissues, including those not affected by VHL. Post-translation, pVHL complexes elongin B, elongin C, Rbx 1 and Cullin 2 to form an ubiquitin ligase that proteolyzes the alpha-subunit of hypoxia-inducible factor (HIF), which coordinates cellular response to hypoxia through transcriptional regulation. HIF enhances glucose uptake and increases the expression of angiogenic, growth and mitogenic factors including, vascular endothelial growth factor (VEGF), platelet derived growth factor-beta chain (PDGF-B), erythropoietin and transforming growth factor (TGF) [1,13]. With absent or abnormal pVHL function, HIF may constitutively stimulate angiogenesis and carcinogenesis by VEG, PDGF-B and TGF-α [14-16]. Besides being a potent mitogenic factor, TGF-α stimulates cellular over-expression of the epidermal growth factor receptors (EGFR, the receptors for TGF-α) creating a potential autocrine loop [15]. Dysfunction of VHL-HIF axis is the one of reasons of VHL-related carcinogenesis. But rare case reports hematological neoplasm with VHLD. We just reviewed a case report in Hodgkin’s disease [17], but no TCL. Thus, we described the special case of co-existing TCL and VHLD.
T-cell lymphomas represent a heterogeneous group of diseases with varied clinical features, prognosis and response to treatment. There are 22 different types of T-cell and NK-cell lymphomas according to WHO Classification [18,19]. Predominantly nodal lymphoma subtypes include angioimmunoblastic T-cell lymphoma (AITL), anaplastic large-cell lymphoma (ALCL) anaplastic lymphoma kinase (ALK) -positive, anaplastic large cell lymphoma (ALCL) ALK-negative (provisional entity) and peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS). Predominantly extranodal lymphoma include extranodal NK/T-cell lymphoma nasal type, enteropathy-associated T-cell lymphoma (EATL), hepatosplenic T-cell lymphoma (HSTL), subcutaneous panniculitis-like T-cell lymphoma-alpha beta (SPTCL). Mature T-cell leukaemias include T-cell prolymphocytic leukaemia (T-PLL), T-cell large granular lymphocyte leukaemia (T-LGL), aggressive NKcell leukaemia, chronic lymphoproliferative disorder of NK-cell (provisional entity), adult T cell leukemia/lymphoma-HTLV positive (ATLL), systemic EBV positive T-cell lymphoproliferative disease of childhood, hydroa vacciniforme-like lymphoma. Cutaneous predominant subtypes include Mycosis fungoides (MF), Sezary syndrome (SS), primary cutaneous CD30-positive T-cell lymphoproliferative disorders (lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma), primary cutaneous gamma-delta T cell lymphoma, primary cutaneous CD8 positive aggressive epidermotropic cytotoxic T-cell lymphoma (provisional entity), primary cutaneous CD4 positive small/medium T-cell lymphoma (provisional entity). The diagnosis of T cell lymphomas is very laborious. Clinical, immunophenotypic, histopathological, immunohistochemical, molecular and genetic findings must be correlated as none of them is strong enough to be used alone for diagnosis. In our case, we cannot make sure the exact classification of T-cell lymphoma. But based on the specific markers for T lineage (CD2+ CD5+ CD7+ CD3+low CD4+), T lymphocyte neoplastichyperplasia was confirmed. Combined with histopathological, immunohistochemical findings in bone marrow, the specific markers for T lineage (CD5+ CD7+ CD3+) verified that the dysplastic cells were T lymphocyte.
We reported this case in order to emphasis the great real difficulties for a complete and correct assessment. The group of TCL remains a challenge for researchers. It is important to diagnose TCL by clinical, radiological, immunophenotypic, pathological, and genetic examination. It is helpful to improve the rate of diagnosis. To data no report showed TCL with VHLD. So it is the other aim of this report to study the connection of TCL and VHLD. Some previous researches show partly hematological neoplasm is related to the epigenetic changes of VHL [20,21]. Gene transfer of VHL inhibits the growth of transplanted EL-4 lymphoma cells and the HIF1 inhibitor suppresses leukaemia cell growth in association with reduced NOTCH1 expression [22,23]. But the definite mechanism is not clear now, and further research is necessary.
Conclusions
In conclusion, we reported a case of a patient with co-existing TCL and VHLD. We hope that the study would help to make correctly diagnosis for TCL in the future and contribute to a better understanding in the relation of TCL and VHLD.
Disclosure of conflict of interest
None.
References
- 1.Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH. von Hippel-Lindau disease. Lancet. 2003;361:2059–2067. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- 2.Bamps S, Calenbergh FV, Vleeschouwer SD, Loon JV, Sciot R, Legius E, Goffin J. What the neurosurgeon should know about hemangioblastoma, both sporadic and in von Hippel-Lindau disease: A literature review. Surg Neurol Int. 2013;4:145. doi: 10.4103/2152-7806.121110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butman JA, Linehan WM, Lonser RR. Neurologic manifestations of von Hippel‑Lindau disease. JAMA. 2008;300:1334–1342. doi: 10.1001/jama.300.11.1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Costello R, Sanchez C, Le Treut T, Rihet P, Imbert J, Sébahoun G. Peripheral T-cell lymphoma gene expression profiling and potential therapeutic exploitations. Br J Haematol. 2010;150:21–27. doi: 10.1111/j.1365-2141.2009.07977.x. [DOI] [PubMed] [Google Scholar]
- 5.Yoon SO, Suh C, Lee DH, Chi HS, Park CJ, Jang SS, Shin HR, Park BH, Huh J. Distribution of lymphoidneoplasms in the Republic of Korea. Analysis of 5318 cases according to the World Health Organization classification. Am J He- matol. 2010;85:760–764. doi: 10.1002/ajh.21824. [DOI] [PubMed] [Google Scholar]
- 6.Chen WL, Tsai WC, Chao TY, Sheu LF, Chou JM, Kao WY, Chen YC, Ho CL. The clinicopathological analysis of 303 cases with malignant lymphoma classified according to the World Health Organization classification system in a single institute of Taiwan. Ann Hematol. 2010;89:555–562. doi: 10.1007/s00277-009-0870-z. [DOI] [PubMed] [Google Scholar]
- 7.Ameen R, Sajnani K, Albassami A, Refaat S. Frequencies of non-Hodgkin’s lymphoma subtypes in Kuwait: comparisons between different ethnic groups. Ann Hematol. 2010;89:179–184. doi: 10.1007/s00277-009-0801-z. [DOI] [PubMed] [Google Scholar]
- 8.Abouyabis AN, Shenoy PJ, Lechowicz MJ, Flowers CR. Incidence and outcomes of the peripheral T cell-lymphoma subtypes in the United States. Leuk Lymphoma. 2008;49:2099–2107. doi: 10.1080/10428190802455867. [DOI] [PubMed] [Google Scholar]
- 9.Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 10.Wait SD, Vortmeyer AO, Lonser RR, Chang DT, Finn MA, Bhowmick DA, Pack SD, Oldfield EH, Zhuang Z. Somatic mutations in VHL germline deletion kindred correlate with mild phenotype. Ann Neurol. 2004;55:236–240. doi: 10.1002/ana.10807. [DOI] [PubMed] [Google Scholar]
- 11.Maher ER, Iselius L, Yates JR, Littler M, Benjamin C, Harris R, Sampson J, Williams A, Ferguson-Smith MA, Morton N. von Hippel-Lindau disease: a genetic study. J Med Genet. 1991;28:443–447. doi: 10.1136/jmg.28.7.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT, Ferguson-Smith MA. Clinical features and natural history of von Hippel-Lindau disease. Q J Med. 1990;77:1151–1163. doi: 10.1093/qjmed/77.2.1151. [DOI] [PubMed] [Google Scholar]
- 13.Kaelin WG Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002;2:673–682. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 14.Chan CC, Collins AB, Chew EY. Molecular pathology of eyes with von Hippel-Lindau (VHL) Disease: A review. Retina. 2007;27:1–7. doi: 10.1097/01.iae.0000244659.62202.ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niu X, Zhang T, Liao L, Zhou L, Lindner DJ, Zhou M, Rini B, Yan Q, Yang H. The von Hippel-Lindau tumor suppressor protein regulates gene expression and tumor growth through histone demethylase JARID1C. Oncogene. 2012;31:776–786. doi: 10.1038/onc.2011.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 17.D’hondt R, Thomas J, Van Oosterom AT, Dewolf-Peeters C. Hodgkin’s disease in a patient with von Hippel-Lindau disease. A case report. Acta Clin Belg. 2000;55:276–278. doi: 10.1080/17843286.2000.11754310. [DOI] [PubMed] [Google Scholar]
- 18.Sabattini E, Bacci F, Sagramoso C, Pileri SA. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008: an overview. Pathologica. 2010;102:83–87. [PubMed] [Google Scholar]
- 19.Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasm and beyond: evolving concepts and practical applications. Blood. 2011;117:5019–5032. doi: 10.1182/blood-2011-01-293050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amara K, Trimeche M, Ziadi S, Laatiri A, Hachana M, Korbi S. Prognostic significance of aberrant promoter hypermethylation of CpG islands in patients with diffuse large B-cell lymphomas. Ann Oncol. 2008;19:1774–1786. doi: 10.1093/annonc/mdn374. [DOI] [PubMed] [Google Scholar]
- 21.Hatzimichael E, Dranitsaris G, Dasoula A, Benetatos L, Stebbing J, Crook T, Bourantas KL. von Hippel-Lindau Methylation Status in Patients with Multiple Myeloma: A Potential Predictive Factor for the Development of Bone Disease. Clin Lymphoma Myeloma. 2009;9:239–242. doi: 10.3816/CLM.2009.n.047. [DOI] [PubMed] [Google Scholar]
- 22.Sun XY, Wang JL, Tang B, Liu FJ, Qiao HQ, Jiang HC. Gene transfer of von Hippel-Lindau inhibits the growth of transplanted solid tumors. Zhonghua Wei Chang Wai Ke Za Zhi. 2005;8:241–244. [PubMed] [Google Scholar]
- 23.Yonekura S, Itoh M, Okuhashi Y, Takahashi Y, Ono A, Nara N, Tohda S. Effects of the HIF1 inhibitor, echinomycin, on growth and NOTCH signalling in leukaemia cells. Anticancer Res. 2013;33:3099–3103. [PubMed] [Google Scholar]