

Summary
Pharmaceutical inhibitors of aminoacyl-tRNA synthetases demand high species and family specificity. The antimalarial ATP-mimetic cladosporin selectively inhibits P. falciparum LysRS (PfLysRS). How the binding to a universal ATP site achieves the specificity is unknown. Here we report 3 crystal structures of cladosporin with human LysRS, PfLysRS, and a Pf-like human LysRS mutant. In all 3 structures, cladosporin occupies the class defining ATP-binding pocket, replacing the adenosine portion of ATP. Three residues holding the methyltetrahydro-pyran moiety of cladosporin are critical for cladosporin's specificity against LysRS over other class II tRNA synthetase families. The species-exclusive inhibition of PfLysRS is linked to a structural divergence beyond the active site that mounts a lysine-specific stabilizing response to binding cladosporin. These analyses reveal that inherent divergence of tRNA synthetase structural assembly may allow for highly specific inhibition even through the otherwise universal substrate binding pocket and highlight the potential for structure driven drug development.
Graphical abstract
Introduction
Aminoacyl-tRNA synthetases (aaRSs) are essential enzymes for all cellular life. They perform a central role in the translation of the genetic code by synthesizing aminoacyl-tRNAs, which are subsequently used for protein synthesis (Ibba and Soll, 2000). Because of their essential role in translation, the inhibitors of aaRSs are widely used as antimicrobial medicines or rigorously tested in clinical trials for therapeutic applications in cancer, and autoimmune diseases (Gadakh and Van Aerschot, 2012; Kim et al., 2014; Rock et al., 2007; Silvian et al., 1999; Sundrud et al., 2009; Zhou et al., 2013).
Inhibitors of aaRSs demand high species and family specificity for successful therapeutic application. For example, the natural product mupirocin inhibits bacterial isoleucyl-tRNA synthetase (IleRS) activity and is widely used as a topical treatment for skin infections (Silvian et al., 1999). Inhibiting human prolyl-tRNA synthetase (ProRS) (Keller et al., 2012), febrifugine is the active component of the Chinese herb Chang Shan (Dichroa febrifuga Lour.), which has been used for treating malaria-induced fever for about 2,000 years (Jiang et al., 2005). Halofuginone (HF), the halogenated derivative of febrifugine, has been tested in clinical trials for the treatment of cancer, fibrotic disease and recently Duchenne muscular dystrophy (Kotz, 2012; Pines et al., 2003). The boron-containing compound AN2690 binds to fungal leucyl-tRNA synthetase (LeuRS) and is licensed for the treatment of onychomycosis (Baker et al., 2006). These examples highlight the tremendous potential of aaRS inhibitors in medical applications. Understanding of the mechanism(s) for inhibition will greatly promote the design of novel therapies.
The 24 known aaRS families are divided into 2 structurally distinct classes (class I and class II), each featuring a catalytic domain with a common fold that binds ATP, amino acid, and the 3′-terminus of tRNA (Delarue and Moras, 1993; Francklyn et al., 1997; Ribas de Pouplana and Schimmel, 2001). In a common two-step reaction, the aaRS first catalyzes the activation of the amino acid by ATP, forming an aminoacyl-adenylate intermediate (aa-AMP); then the amino acid is transferred from the intermediate to either the 2′-or 3′-hydroxyl oxygen atom of the 3′-A76 tRNA nucleotide, which functions as a nucleophile in the synthesis of aminoacyl-tRNA (Ibba and Soll, 2000). A common class of aaRS inhibitors mimics aa-AMPs, with a non-hydrolyzable ester bond between amino acid and the alpha-phosphate, such as lysyl-sulfide adenylate or glutamyl-sulfide adenylate and other aminoacyl-sulfide adenylates (aa-SAs) (Belrhali et al., 1994; Crepin et al., 2006; Cvetesic et al., 2014; Fukai et al., 2000; Guo et al., 2009; Ito and Yokoyama, 2010; Iwasaki et al., 2006; Kamtekar et al., 2003; Kotik-Kogan et al., 2005; Nakanishi et al., 2005; Sakurama et al., 2009; Sankaranarayanan et al., 2000). Because these inhibitors directly occupy and block the active site for ATP and amino acid, they usually display high specificity and tight binding affinities (∼ nM) to aaRSs (Pope et al., 1998; Vondenhoff and Van Aerschot, 2011; Zhao et al., 2014). However due to the highly charged property of both amino acid and adenylate moieties, these inhibitors generally showed limited whole cell activity with poor penetration through the cell membrane, and their near identical structure to the natural aa-AMPs also causes poor selectivity against aaRSs from different species, limiting their potential application for therapy (Hurdle et al., 2005). Therefore continuous efforts have been taken to explore novel inhibitors for aaRSs with high potency and specificity.
Cladosporin (CP) is a secondary metabolite produced by various fungal genera such as Cladosporium, Eurotium, Aspergillus, and Chaetomium (Scott et al., 1971) and it exhibits potent antibiotic activity (Hoepfner et al., 2012; Kimura et al., 2012; Scott et al., 1971; Springer et al., 1981). CP shows great potential in agricultural applications for developing treatment and management of plant diseases. CP at 30 μM was shown to be able to inhibit the growth of plant pathogens Colletotrichum acutatum, Colletotrichum fragariae, Colletotrichum gloeosporioides, and Phomopsis viticola by over 80% (Wang et al., 2013). CP has even stronger growth-inhibitory activities against both blood- and liver- stage Plasmodium falciparum parasite forms with an IC50 of 40-90 nM, while having little effect on the growth of human cells (Hoepfner et al., 2012). By haploinsufficiency chemogenomic profiling in yeast, the cellular target of CP was recently identified to be cytosolic lysyl-tRNA synthetases (LysRS) (Hoepfner et al., 2012).
With a molecular weight of 292.3 Dalton and isocoumarin-containing structure that mimics the adenosine group of ATP (Figure 1A), CP possesses a high selectivity index against Plasmodium parasites compared to mammalian cells (IC50/CC50 > 111), promising great potential as a future anti-malarial (Hoepfner et al., 2012). A recent research report described a co-crystal structure of CP with P. falciparum LysRS (PfLysRS) and suggested that two residues (V328 and S344) from motif 2 determine the selectivity of CP against PfLysRS (Khan et al., 2014). However, mutagenesis of these two residues only rescued 4% of the 1000-fold specificity in yeast LysRS (Hoepfner et al., 2012). Since CP shares little in common with previously characterized aaRS inhibitors, the details of its binding mode and mechanism of selectivity, particularly to human LysRS (HsLysRS), remain unclear.
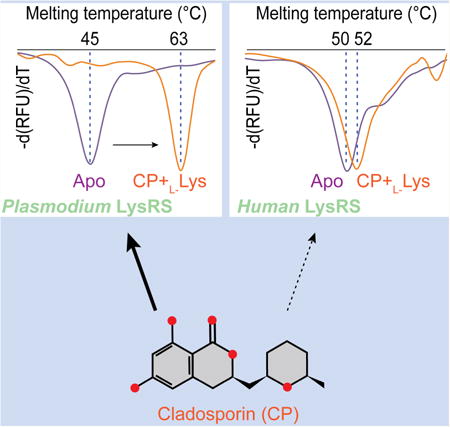
Figure 1. Specific recognition of CP by human LysRS.
A. Chemical structure of cladosporin (CP).
B. Overview of HsLysRS-CP structure determined at a resolution of 2.1 Å. A LysRS dimer is shown in cartoon and transparent surface, and CP molecues are shown in orange spheres. Anticodon binding domains (ABD) are colored cyan and black. Catalytic domains (CD) are colored green and white. The conserved motifs I, II, and III are colored yellow, blue and pink, respectively.
C. Two-dimensional presentation of CP binding in HsLysRS. CP and hydrogen-bonding residues are shown in stick representations, and other residues within 4.5Å of CP are shown in light green.
D. Zoom-in view of CP localization in the conserved ATP binding site of LysRS. CP and residues within 5Å of CP are shown as sticks. The 2Fo-Fc electrodensities of CP and cobound lysine are shown as black meshes with 1.5 σ.
E. Zoom-in view of ATP localization in HsLysRS (pdb: 3BJU). ATP and residues within 5Å of ATP are shown as sticks.
(See also Figure S1)
Here we thoroughly investigated this question by determining and analyzing 3 crystal structures of CP in complex with HsLysRS, PfLysRS, and a Pf-like human LysRS mutant at resolutions of 2.1 Å, 3.4 Å and 2.9 Å, respectively. CP occupies the class defining ATP-binding pocket in the class II LysRS dimer. Three smaller residues at the bottom of the ATP binding pocket hold and interact with the methyltetrahydro-pyran group of CP, critical for its specificity against LysRS over other class II tRNA synthetase families. In contrast to the low activity against human LysRS, CP binds to both human and Plasmodium LysRS's in a nearly identical mode in the active site. The active site-mimicking Pf-like human LysRS mutant failed to replicate the significant species-specificity of CP. Since CP co-binds with lysine in all 3 of these structures (including our HsLysRS-K-CP, and Pf-like human LysRS-K-CP structures and one previous solved PfLysRS-K-CP), we determined a 4th structure of PfLysRS-CP without lysine. Analysis of this structure unveiled a lysine-dependent structural divergence of LysRS outside of the active site that may lead to a cooperative binding of CP and lysine. The hypothesis was validated by a discovery that co-binding of CP and lysine triggers a specific gain of stability for PfLysRS but not HsLysRS or the Pf-like human LysRS. Overall, this work exhibits that two features of aaRSs, the inherent divergence of tRNA synthetase structure and the cooperative binding of cognate amino acid, together could allow for highly specific inhibition through the universal pocket for ATP. This study thus provides important implications in the future drug development for tRNA synthetases.
Results
Recognition of Cladosporin by Human LysRS
CP is a composite of a (6,8)-dihydroxyl-isocoumarin group and a methyltetrahydro-pyran group, linked by a methylene at the 3′ position of the isocoumarin (Figure 1A). To determine the complex structure of HsLysRS with CP, we soaked HsLysRS crystals pre-crystallized with lysine, in a solution containing 3 mM CP and determined the structure at a resolution of 2.1 Å (Table 1). The CP molecule binds in each aminoacylation active site of the HsLysRS dimer (Figure 1B). Complete and clear densities for both CP and a co-bound lysine were observed with 100% occupancies (Figures 1C, 1D, and S1A), where CP occupies only the ATP binding site. This ternary structure of HsLysRS-K-CP (K denotes the presence of lysine) suggests that CP binds LysRS through competing the binding of ATP but not lysine.
Table 1.
Crystallographic statistics of LysRS–CP complex structures.
| HsLysRS-K-CP | Pf-like human LysRS-K-CP | PfLysRS-CP | |
|---|---|---|---|
| Data collection Space group Cell dimensions | C2221 | P1 | P21 |
| a, b, c (Å) | 96.17, 99.93, 269.78 | 51.28, 75.56, 162.99 | 70.63, 112.10, 170.80 |
| α, β, γ (°) | 90.00, 90.00, 90.00, | 90.80, 99.09, 109.83 | 90.00, 99.30, 90.00 |
| Resolution (Å) | 50.00-2.10 (2.18-2.10)* | 40.00-2.90 (3.00-2.90) | 50.00-3.40 (3.52-3.40) |
| Rsym or Rmerge (%) | 10.2 (70.9) | 8.7 (45.3) | 19.8 (53.8) |
| I/sI | 19.7 (2.7) | 9.5 (1.8) | 7.0 (2.6) |
| Completeness (%) | 99.9 (100.0) | 93.5 (90.2) | 96.5 (94.5) |
| Redundancy | 7.4 (7.6) | 2.1 (2.1) | 3.9 (3.7) |
| Refinement | |||
| Resolution (Å) | 50.00-2.10 (2.17-2.10) | 40.00-2.90 (3.00-2.90) | 50.00-3.40 (3.53-3.40) |
| No. reflections | 72043 (4465) | 44957 (2816) | 34557 (3157) |
| Rwork/Rfree (%) | 19.1/21.3 | 22.3/26.8 | 22.7/26.0 |
| No. atoms | |||
| Protein | 8288 | 15911 | 15165 |
| CP | 42 | 84 | 84 |
| L-lysine | 20 | 40 | - |
| Solvent | 700 | 9 | 7 |
| B-factors (Å2) | |||
| Protein | 31.0 | 62.6 | 59.3 |
| CP | 21.4 | 44.6 | 40.9 |
| L-lysine | 18.9 | 38.5 | - |
| Solvent | 38.1 | 28.9 | 27.8 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.006 | 0.005 | 0.006 |
| Bond angles (°) | 1.045 | 0.970 | 1.240 |
| Ramachandran plot | |||
| Most favored [%] | 98.0 | 97.0 | 96.0 |
| Additional allowed [%] | 2.0 | 3.0 | 4.0 |
Values in parentheses are for highest-resolution shell.
The previous PfLysRS-K-CP structure indicated that CP was bound in the ATP-binding site (Khan et al., 2014). Without the corresponding PfLysRS-ATP structure, however, the exact similarity between the recognitions of CP and ATP in LysRS remains undefined. Comparing this new HsLysRS-K-CP structure with our previously determined HsLysRS-K-ATP structure (Guo et al., 2008), the isocoumarin group of CP occupies a position corresponding to the adenine portion of ATP in the active site (Figures 1D, 1E, and S1B). Particularly, E325 forms a H-bond with the 6-OH group on the isocoumarin, mimicking the H-bond between E325 with the 7-OH group on the adenosine of ATP (Figures 1D and 1E). The 1-ketone group of CP forms another hydrogen bond with R553, which forms salt bridges with the γ-phosphate of ATP in the ATP-bound structure (Figures 1D and 1E). Main chain atoms of N332 form 2 additional H-bonding interactions with the 8-OH group that is not present on ATP (Figure 1D). Another shared interaction of CP with ATP is a stacking interaction with the benzene ring of F335 (Figures 1D and 1E). F335 is located on the class II characteristic Motif 2 of LysRS, together with E325 and H331, they form a conserved ATP binding loop in LysRS, and also partially conserved in the rest of other class II aaRSs (Cusack, 1997). Instead of the proposal that the entire CP mimics ATP (Hoepfner et al., 2012; Khan et al., 2014), these additional structural analysis reveal that it is the isocoumarin moiety of CP sharing key interactions of the adenosine group of ATP to bind LysRS (Figures S1C and S1D).
The rest of the CP structure interacts with LysRS in a manner distinct from ATP. A methyltetrahydro-pyran ring occupies the space for the ribose group of ATP (Figure S1C), with essentially all hydrophobic interactions, formed by surrounding residues R323, T337, E494, I495, C496, N497, G548, G550, and R553 (Figure 1D). Instead, in the ribose group of ATP, the 2′–OH group forms H-bonds with the main chain atoms of E494, I495, and G550, while the 3′-OH group forms a H-bond with the side chain of E494 (Figure 1E). The C1 and C4 atoms of the ribose ring form two other hydrophobic interactions with F335 and T337. The 3 phosphate groups of ATP are recognized by LysRS through Mg2+ coordinated interaction with the class-conserved residues E494 on motif 3 and another nearby acetic residue E487 (Figures 1E and S1B). In contrast, CP does not contain phosphate groups and thus no Mg2+ were found in the HsLysRS-K-CP electronic density (Figure S1A). Instead, E494 contributed a hydrophobic interaction with CP through the aliphatic portion (Cβ-Cδ) of its side chain. Most of the residues involved in the interactions with ATP are also found to interact with CP, with the isocoumarin group of CP replacing the adenine portion of ATP, and the methyltetrahydro-pyran group of CP replacing the ribose group of ATP (Figure 1D). Collectively, these results reveal that the inhibitory action against LysRS by CP is based on limited ATP mimicry, with partially overlapped structures and induced conformation changes of interacting residues.
Basis of the Family Specificity of Cladosporin Against LysRS
The 24 aaRS families are classified into two classes, characterized by a distinct fold of the catalytic domains (Figure S2A) (Rossmann fold for class I and 7-strand beta-sheet for class II), that each originated from a specific ATP-binding fold (Delarue and Moras, 1993; Francklyn et al., 1997; Ribas de Pouplana and Schimmel, 2001). Previous genomic screening indicated that among a collection of 5803 genes in yeast covering ∼95% of the yeast genome including all the cytoplasmic and mitochondrial aaRSs, only cytoplasmic LysRS could rescue the inhibition of CP on cells (Hoepfner et al., 2012). Since the mode of ATP binding structure is strictly conserved within each aaRS class, the fact that CP specifically binds to and inhibits LysRS rather than other class II aaRSs is astonishing, and the mechanism remains unclear.
In contrast to class I aaRSs, where ATP is held in an extended conformation by their 2 signature motifs “HIGH” and “KMSKS”, the class II aaRSs, including PfLysRS and HsLysRS, stabilize ATP in a bent form that sits between the class II characteristic motif 2 and motif 3 (Cusack, 1993; Eriani et al., 1990).
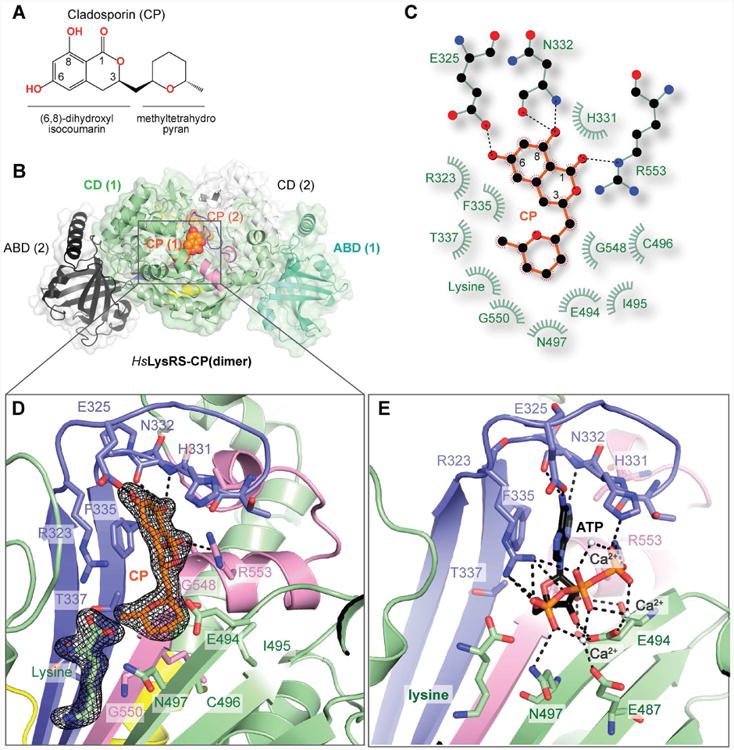
We modeled the CP molecule in 9 out of 13 class II aaRS structures, by superimposing the adenine group of ATP in aaRS structures onto the isocoumarin group of LysRS-CP structure (Figure 2 and Figure S2B). In HsLysRS enzymatic center, the ATP/adenine binding pocket residues R323, H331, F335, and R553, contribute to 58% of the total atomic interactions (111 of 191) for CP. These residues are conserved in nearly all class II aaRSs and thus may lead to a general inhibition. On the other side, T337, G548 and G550 form hydrophobic interactions with the methyltetrahydro-pyran moiety from the bottom of the ATP pocket (Figure 2A). These three residues are located 3.7 Å, 3.4 Å, and 6.0 Å away from the ribose moiety of ATP (Figure S2B). They are highly variable in other class II aaRSs (Figures 2B-2I). For example, the corresponding residues are K323, T429, and A432 in human seryl-tRNA synthetase (SerRS). They are all larger than the counterparts in LysRS, forming stereo clashes with the modeled CP (Figure 2B). Similarly, two larger-residue substitutions of the three are common in ProRS, threonyl-tRNA synthetase (ThrRS), phenylalanyl-tRNA synthetase (PheRS), glycyl-tRNA synthetase (GlyRS) and histidyl-tRNA synthetase (HisRS) (Figures 2C-2G), indicating potential steric repulsions. Asparaginyl-tRNA synthetase (AsnRS), aspartyl-tRNA synthetase (AspRS) and LysRS belong to the same sub-class (class IIb) (Ribas de Pouplana and Schimmel, 2001). Only one of the three residues is replaced in AsnRS or AspRS. In AsnRS the T337 is replaced by a H336 (Figure 2H), which clashes with the modeled CP molecule at the methyl group. While in AspRS, the T337 is replaced by a G288 (Figure 2I) resulting in a potential loss of hydrophobic interaction and binding affinity to CP. In addition, the selectivity of CP towards bacterial LysRS may also be linked to T337. In bacterial KRS, the corresponding residue to T337 is a bulkier hydrophobic methionine (Figure S2C), which makes steric hindrance with the methyl group of CP. Consistently, with the T-to-M substitution, the growth of E. coli was not affected by up to 100 μg/ml CP (Anke, 1979). In fact, these 3 residues do not always interact with ATP, nor are they conserved in their own family of aaRSs among different species (Figures S2). Therefore, the deep divergence of class II aaRSs at the ATP site allows for the strict LysRS-specific inhibition.
Figure 2. Structural basis of the selectivity of CP among class II aaRSs.
A. In human LysRS enzymatic center, the methyltetrahydro-pyran group forms hydrophobic interactions with T337, G548, and G550, which are highly variable in other class II aaRSs.
B. The corresponding residues are K323, T429, and A432 in human SerRS (wheat, pdb:4L87). The three residues are larger than those in LysRS, thus potentially form stereo clashes with CP.
C-H. Similarly, W1169 and T1276 in human ProRS (lightblue, pdb: 4HVC), Q460 and S596 in human ThrRS (paleyellow, pdb: 4P3N), Q372 and S460 in human PheRS (lightpink, pdb: 3L4G), M348 and S578 in human GlyRS (palecyan, pdb: 2ZT5), Q173 and S383 in human HisRS (lightorange, pdb: 4G84), and H336 in Brugia malayi AsnRS (bluewhite, pdb: 2XGT) prevent CP from binding by stereo repulsion.
I. The T337 residue is replaced by a G288 in human AspRS (pdb: 4J15), causing one hydrophobic interaction missing for CP binding.
(See also Figures S2)
Structurally Indistinguishable Binding of Cladosporin to Human and P. falciparum LysRS
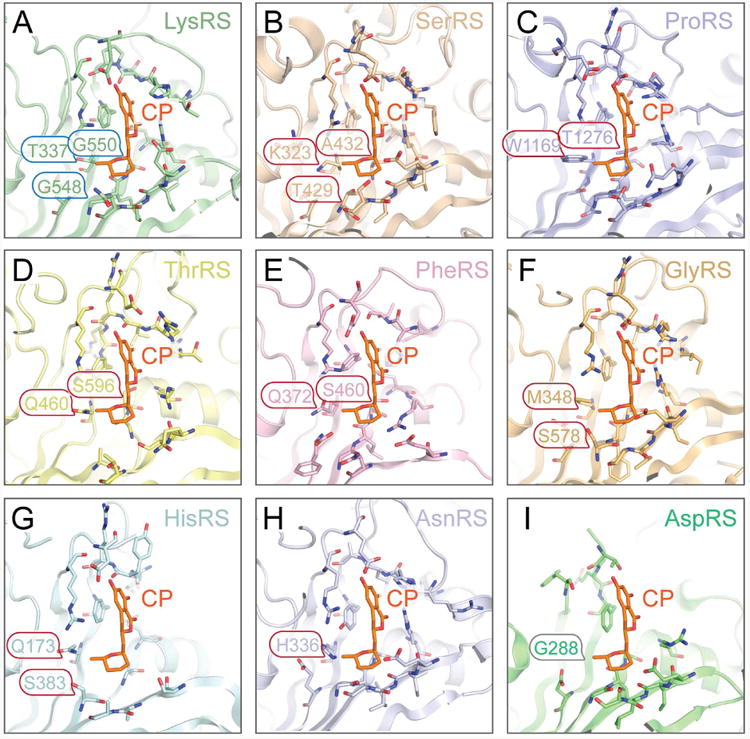
Although the above-mentioned residues differentiate LysRS from other class II aaRSs, it could not explain the strong species-specificity of CP since these 3 residues are conserved in eukaryotic LysRSs. CP inhibits PfLysRS with the strongest potency, 1000-fold higher than HsLysRS or yeast LysRS in a yeast-growth-based assay (Hoepfner et al., 2012). For 55% sequence identity between human and P. falciparum LysRS's, the structures of human and P. falciparum LysRS exhibit high similarity. An r.m.s.d. (root mean square distance) of 0.772 Å was observed for 412 superimposed Cα atoms (Figure 3A). In the active center, only 3 CP-nearby side chains are different between the two enzymes: P300, Q321, and T337 in HsLysRS (P303, Q324, and T340 in yeast LysRS) and T307, V328, and S344 in PfLysRS (Figure 3B). However, Q324V and T340S mutations of yeast LysRS partially increased the sensitivity for CP inhibition (by 40-fold, thus 4% of the total species selectivity) (Hoepfner et al., 2012). To further understand whether these amino acid substitutions are important for the high CP sensitivity in PfLysRS, we created a P. falciparum mimetic LysRS (Pf-like human LysRS) by introducing 3 mutations into human LysRS: P300T, Q321V, and T337S, so that the Pf-like human LysRS would have a PfLysRS active site. We then determined the co-crystal structure of Pf-like human LysRS-K-CP complex (Table 1).
Figure 3. Evolutionarily conserved CP-binding modes of HsLysRS and PfLysRS.
A. Superimposition of the structures of HsLysRS-K-CP (this study), PfLysRS-K-CP (PDB: 4PG3), and Pf-like human LysRS-K-CP (this study). A “K” denotes the crystal structure is obtained in the presence of lysine.
B. The only three different residues between HsLysRS and PfLysRS active sites, with their sidechains pointing the bound CP molecules are denoted and shown as sticks. The Pf-like human LysRS is made on an HsLysRS scaffold, with these three residues mutated into the PfLysRS corresponding residues.
C. The CP (black) binding pocket forming residues are shown as sticks and transparent surface. The colorings of the protein residues are same as in panel A. Numbering of residues is according to HsLysRS and the corresponding PfLysRS residues are shown in parenthesis.
D. Diagram of the atomic contacts between CP and LysRSs in the presence of lysine (K) or without the presence of lysine.
When we align the three LysRS-K-CP structures together (including our HsLysRS-K-CP, and Pf-like human LysRS-K-CP structures and one previous solved PfLysRS-K-CP), the r.m.s.d. of all the non-hydrogen atoms of the bound CPs is only 0.349 Å, 0.367 Å, and 0.364 Å, respectively (Figures 3A and 3B). The comparison failed to support that CP was better accommodated in the PfLysRS than in HsLysRS. All CP-nearby residues are in nearly identical conformation (Figure 3C). In a more quantified analysis, we calculated the atomic contacts between CP molecules and the proteins. PfLysRS-K-CP showed merely 5% more contacts than in HsLysRS for their lysine co-bound structures (Figure 3D). More surprisingly, the Pf-like human LysRS, which has an almost identical PfLysRS active site, showed even less (2%) contacts to CP than the HsLysRS (Figure 3D). Therefore, the interactive pattern observed in the Pf-specific active site could not fully explain the 1000-fold specificity of CP to PfLysRS.
Lysine Dependent Species-specific Stabilization of PfLysRS by Cladosporin
The only factor in the structures of LysRS-K-CP complexes that remained to be investigated was the co-binding of CP with lysine (K). For a rigorous comparison of the earlier PfLysRS-K-CP structure, we further solved the co-crystal structure of PfLysRS-CP complex without lysine (Table 1 and Figure S3). In the absence of lysine, the atomic contacts between CP and PfLysRS decreased about 20% relative to that in the PfLysRS-K-CP structure (Figure 3D and Figure S3B). Notably, without lysine, two regions of the PfLysRS-CP catalytic domain showed altered conformations with PfLysRS-K-CP. The region 403-457, which forms the upper side of the PfLysRS enzymatic cleft and ∼20 Å away from the bound CP and lysine, opened out by a 3-Å shift. Second, a loop next to the bound lysine (5 Å away) composed of residues 282-288 became disordered without lysine (Figure 4A). Consistent with these structures, the temperature factors (B-factors) of the PfLysRS-K-CP were 17 Å2 (∼29%) lower than the B-factors of the PfLysRS-CP, suggesting a more static structure for PfLysRS-K-CP (Figure 4B). The stabilized regions were mostly localized around the enzymatic pocket of PfLysRS, but also a general stabilizing effect (lower B-factors) throughout the entire protein. In contrast, while co-binding of lysine (K) and CP stabilized the lysine and the ATP binding residues and the mobile side of the enzymatic cleft, PfLysRS-CP showed similar B-factors as PfLysRS-apo structure thus the binding of CP alone had little effect on stabilization (Figure 4B). These lysine-induced structural changes suggest that CP preferentially binds to PfLysRS through a lysine-dependent stabilization effect.
Figure 4. Lysine-dependent specific stabilization of PfLysRS by CP.
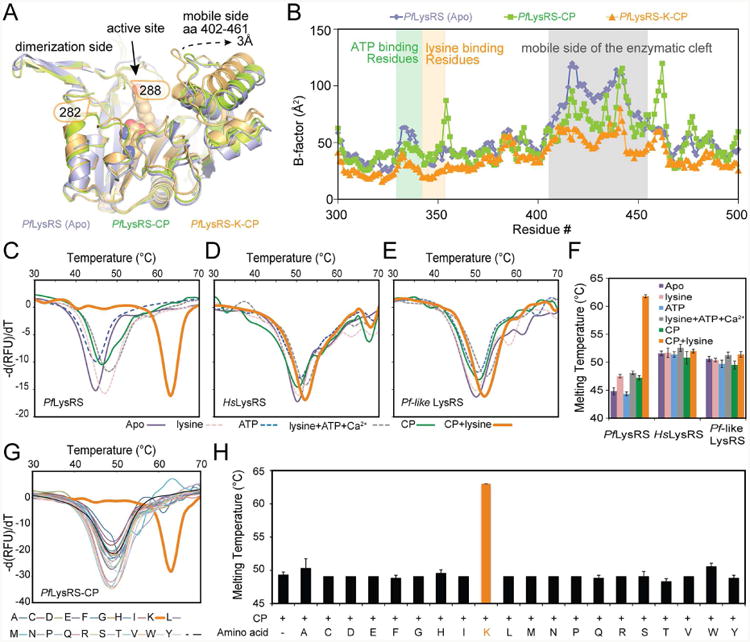
A. Superimposition of PfLysRS_Apo (light blue, pdb: 4H02), PfLysRS-CP (limon, this study), and PfLysRS-K-CP (Orange, pdb: 4PG3). Proteins are shown in cartoon, and A representative CP molecule is shown as spheres.
B. B-factors of PfLysRS_Apo (light blue, pdb: 4H02), PfLysRS-CP (limon, this study), and PfLysRS-K-CP (Orange, pdb: 4PG3) are plotted per residue.
C. Thermal melting profile of PfLysRS in the presence of different ligands.
D. Thermal melting profile of HsLysRS in the presence of different ligands.
E. Thermal melting profile of Pf-like human LysRS in the presence of different ligands. Each curve in C-E is an average of 8 measurements.
F. Diagram of the melting temperatures of PfLysRS, HsLysRS, and Pf-like human LysRS in the presence of different ligands. Error bars represent standard deviations (s.d.) of 8 repeats.
G. Thermal melting profile of PfLysRS in combination of CP and different amino acids. Each curve is an average of 4 measurements.
H. Diagram of the melting temperatures of PfLysRS in combination of CP and different amino acids. Error bars represent standard deviations (s.d.) of 4 repeats.
(See also Figure S3, S4)
To further validate this hypothesis, we examined the stabilization of PfLysRS and HsLysRS in solutions by a thermal shift assay (Khan et al., 2014). CP significantly increased the melting temperature of PfLysRS by 17.1 °C only in the presence of lysine, however, we found it had little stabilization effect on HsLysRS and Pf-like human LysRS (< 2 °C) even with the presence of lysine (Figures 4C-4F). Individual or combinations of natural substrates (lysine, ATP, and metal ions) in the absence of CP only increased the melting temperature of LysRSs by less than 3 °C (Fi gures 4C-4F). Because the ligand-dependent changes in the melting temperature (ΔTm) are directly proportional to the ligand dissociation affinity (Kd) (Pantoliano et al., 2001), the Kd between CP and LysRS were thereby estimated. The presence of lysine increased the binding affinity of CP in PfLysRS by 625 fold, and that is 1375 fold tighter than in HsLysRS (Table 2), supporting the earlier biochemistry or cell-based results (Hoepfner et al., 2012). Replacing lysine with any of the other 19 non-cognate amino acids did not yield stabilization effect (Figures 4G and 4H), indicating the stabilization effect of CP to PfLysRS is also lysine specific. Finally, Pf-like human LysRS carrying a mimetic active site of PfLysRS behaved similar to HsLysRS and was not stabilized by lysine + CP (Figure 4E), indicating that the stabilization of LysRS by K-CP co-binding did not rely on the subtly different binding modes of CP in the active site. Collectively, CP stabilizes PfLysRS in a lysine-specific, Pf-specific and binding site-independent manner, where the LysRS structure scaffold beyond the active site supports the basis for this species-specific response.
Table 2.
Response of ligand /inhibitor binding on thermal shift.
| PfLysRS | HsLysRS | Pf-like human LysRS | ||||
|---|---|---|---|---|---|---|
| ΔTm (°C) | Kd (μM) | ΔTm (°C) | Kd (μM) | ΔTm (°C) | Kd (μM) | |
| Lysine | 2.7 | 34.1 | 0.1 | 84.9 | -0.2 | N.D. |
| ATP (no lysine) | -0.5 | N.D. | -0.2 | N.D. | -0.9 | N.D. |
| ATP (with lysine) | 0.6 | 2.8 | 0.9 | 2.8 | 0.9 | 2.2 |
| CP (no lysine) | 2.4 | 1.5 | -0.8 | N.D. | -1.1 | N.D. |
| CP (with lysine) | 14.4 | 0.0024 | 0.3 | 3.3 | 1.0 | 2.1 |
N.D., Not determined because of negative ΔTm.
Discussion
Specific Inhibition of tRNA Synthetases through the Universal ATP Pocket
Among the 3 substrates of aaRSs, only ATP is a universal substrate, while amino acid and tRNA are unique to each enzyme family. Because of the inherent specific recognition of amino acid by aaRSs, amino acid binding site in aaRS active center is often considered to be critical for developing aaRS specific inhibitors (Pope et al., 1998; Teng et al., 2013), with ATP site used for further binding stabilization (Keller et al., 2012; Zhou et al., 2013). The binding of natural substrate in one sub-site could also affect the binding of the inhibitor in other sub-sites. For example, the binding of HF in the proline and tRNA sub-sites in ProRS is ATP dependent (Sundrud et al., 2009; Zhou et al., 2013). Intriguingly, in a distinct manner, CP binds to LysRS solely through the conserved ATP sub-site without any occupancy of the lysine site.
The specificity of CP to LysRS but not other aaRSs in a conserved ATP binding site is surprising. The isocoumarin group of CP mimics the adenine group of ATP. The methyltetrahydopyran group of CP replaces the ribose group of ATP with pure hydrophobic interactions with LysRS instead of H-bonding interactions, thereby contributing to the specificity of CP. Three short residues from the bottom of the ATP binding pocket make CP perfectly fit in LysRS. While other class II aaRSs either have larger corresponding side chains (such as ProRS, SerRS, ThrRS, HisRS, GlyRS, AsnRS, and PheRS) or a shorter side chain (AspRS), thus cannot form stable interaction with CP. The high variability of these three residues among different aaRS families and in the same aaRS family among different species denotes their unimportance for the aminoacylation reaction. In fact, in most analyzed aaRSs, they do not directly interact with ATP or amino acid (Figure S2). Therefore, through its methyltetrahydro-pyran group, CP exploited a nonconserved patch within the overall conserved ATP binding pocket to achieve the highly specific targeting on one aaRS.
Theoretically, it is possible to modify the methyltetrahydro-pyran group to obtain specific binding to other class II aaRS other than LysRS. For example, AspRS has a G288 at the position of T337 of human LysRS. Replacement of the methyltetrahydro-pyran by an ethyltetrahydropyran would potentially clash with the LysRS T337 residue and create more interactions with AspRS, thus turning the LysRS inhibitor into an AspRS inhibitor. By uncovering the LysRS-specific recognition by CP, this study may provide a new direction to develop high affinity ATP-competitive inhibitors for other aaRSs.
Inherent Structural Divergence Endows the Specific Inhibition of PfLysRS
High species specificity is fundamental to aaRSs inhibitors for therapeutic applications. For instance, in contrast to aa-AMS, which lacks species-specificity because of a near identical mimicry of aa-AMP, mupirocin only partially mimics Ile-AMP, and has about 8000-fold selectivity for pathogenic over mammalian IleRS (Hughes and Mellows, 1980). This ultra-high specificity is mainly attributed to the pyran ring structure of mupirocin, which interacts with two bacterial/archaeal specific residues (H581 and L583) located at the C terminus of the last β-strand of the Rossmann fold and are not conserved in eukaryotic IleRSs (Nakama et al., 2001). CP inhibits PfLysRS with ∼1000 fold higher potency than HsLysRS in a yeast-growth-based assay (Hoepfner et al., 2012). The two enzymes have very minor differences within the active center as revealed by our structural studies, only involving three residues P300, Q321 and T337 (Figure 3). Further, mutagenesis of these residues from human to Plasmodium counterparts failed to increase the interactions with CP (Figure 3D). Similarly, mutagenesis of the Q324 and T340 in yeast only increased 4% sensitivity (Hoepfner et al., 2012). Interestingly, our studies demonstrate that CP together with lysine stabilized PfLysRS 7-fold stronger than CP-alone (17.1 °C increase vs. 2.4 °C), in a manner that is strictly dependent on cognate amino acid binding with no effect found for other amino acids (Figure 4). These results suggest that this high specific of inhibition to Plasmodium cells versus human cells may be facilitated by the naturally existing L-lysine in cells.
While CP and lysine can be equally accommodated in PfLysRS and human enzymes with similar amount of atomic contacts (Figure 3D), structural and sequence analysis reveals that the divergence of PfLysRS beyond the active site center may lead to the preferential inhibition of PfLysRS by CP (Figure 4). Only PfLysRS, but not human LysRS could be stabilized by K-CP for a huge (17.1 °C) increase in thermal melting test (Figure 4C). This corresponds to a Kd of ∼2.4 nM for the interaction (Table 2). Interestingly, without ligands, PfLysRS has a ∼7 °C lower melting temperature than HsLysRS (Figure 4F), suggesting PfLysRS itself may be more flexible and less stable than HsLysRS. This difference is further evidenced by the finding of destabilization in residues 518-535 of PfLysRS (Figure S4A, S4B). PfLysRS contains a specific disulfide bond (C517-C540) that encloses this region (Figure S4A, S4B) and is located 14.5 Å away from lysine and 24.3 Å away from the bound CP. This disulfide bond appears to be PfLysRS–specific and does not exist in any other solved LysRS structure so far. Particularly, the 1st cysteine (C517) is only present in Plasmodium species but substituted by a conserved leucine residue in higher eukaryotes and an arginine in lower eukaryote and bacteria (Figure S4C). In contrast, residues within the disordered region (518-535) are more conserved across different species. In fact, the 2nd cysteine (C540) is strictly conserved in eukaryotes, suggesting C540 is important for the activity of LysRS. It is plausible that the Plasmodium-specific disulfide bound may play a mechanistic role by protecting the cysteine residues from be oxidized in their highly oxidative environments such as red blood cells (Mohanty et al., 2014).
Experimental Procedures
Protein preparation
Untagged HsLysRS (70-584) was constructed in vector PET20b. The N–terminal human P38/AIMP2 sequence (1-36) was cloned into vector pET28a with a C-terminal 6×His tag. The two protein/peptide were co-expressed in BL21 (DE3) strain with 0.2 mM IPTG for 20 h at 16 °C. The cell pellet (from 4-8 l iters) was lysed in low salt buffer (150 mM NaCl, 20 mM Tris-HCl pH 8.0, and 15 mM imidazole), loaded onto a Ni-HiTrap column and washed with low salt buffer. Protein was eluted with low salt elution buffer (150 mM NaCl, 20 mM Tris-HCl pH 8.0, and 250 mM imidazole). To remove free p38, the Ni-HiTrap purified HsLysRS:p3836-His proteins were concentrated and then purified by gel filtration Superdex 200 column (GE Healthcare, 10/300 GL) with running buffer (20 mM Hepes-Na, pH 7.5, and 150 mM NaCl). The peak fractions of HsLysRS: p3836 complex were then concentrated for crystallization.
Pf-like human LysRS plasmid was made by PCR based mutagenesis from the HsLysRS plasmid, with the mutations: P300T, Q321V, and T337S. And the protein was also co-expressed and co-purified with p3836 similar as above.
PfLysRS (77-583) was constructed in vector PET20b, with a C-terminal 6×His tag. The protein was expressed in BL21 (DE3) strain with 0.2 mM IPTG for 20 h at 16 °C. The cell pellet (from 4-8 liters) was lysed in NTA-wash buffer (500 mM NaCl, 20 mM Tris-HCl pH 8.0, 15 mM imidazole), loaded onto a Ni-HiTrap column and washed with NTA-wash buffer. Protein was eluted with NTA-elution buffer (500 mM NaCl, 20 mM Tris-HCl pH 8.0, 250 mM imidazole). Then the protein was concentrated and further purified by gel filtration Superdex 200 column (GE Healthcare, 10/300 GL) with running buffer (20 mM Hepes-Na, pH 7.5, 150 mM NaCl). The peak fractions were then concentrated for crystallization.
Crystallization
All crystallizations were done by the sitting drop method. To obtain HsLysRS-K-CP complex crystals, 30 mg/ml HsLysRS:p3836 was pre-mixed with 5 mM L-lysine at 4 °C, and was crystallized by mixing 0.5 μl of protein solution with 0.5 μl of precipitant solution, containing 8% PEG20000, 0.1 M Tris pH8.0, and 0.1 M sodium chloride. After incubation at 18 °C for 3-7 days, crystals were transferred to a drop containing 8% PEG20000, 0.1 M Tris pH8.0, 0.1 M sodium chloride, and 3 mM CP. The 72-hour soaked crystals were flash-frozen in liquid nitrogen for data collection with the cryo solution containing 8% PEG20000, 0.1M Tris pH8.0, 0.1M sodium chloride, 3mM CP, and 25% glycerol.
To obtain Pf-like LysRS-K-CP complex crystals, protein (15 mg/ml) was pre-mixed with 5 mM lysine and 2 mM CP at 4 °C, and was crystallized by mixing 0.5 μl of protein solution with 0.5 μl of precipitant solution, containing 25% PEG4000, and 0.1 M MES pH6.5. After incubation at 18 °C for 3-7 days, crystals were flash-frozen in liquid nitrogen for data collection with the cryo solution containing 20% PEG4000, 0.8 M MES pH 8.0, and 20% glycerol.
To obtain PfLysRS-CP crystals, PfLysRS protein (15 mg/ml) was premixed with 2 mM CP at 4 °C, and was crystallized by mixing 0.5 μl of protein solution with 0.5 μl of precipitant solution, containing 0.2 M tri-Sodium citrate, and 20% PEG3350. After incubation at 18 °C for 3-7 days, crystals were fla sh-frozen in liquid nitrogen for data collection with the cryo solution containing 0.16 M tri-Sodium citrate, 16% PEG3350, and 20% glycerol.
Structure Determination
HsLysRS-K-CP data sets were obtained from beamline LS-CAT 21-ID-F at the Advanced Photon Source (Argonne, IL). Pf-like LysRS-K-CP and PfLysRS-CP data sets were obtained from beamline BL7-1 at the Stanford Synchrotron Radiation Lightsource (Menlo Park, CA). All data were integrated and scaled with HKL2000 (Otwinowski and Minor, 1997). The structures were determined by molecular replacement based on the human LysRS structure (PDB: 3BJU) in program Molrep (CCP4, 1994). After corrections for bulk solvent and overall B values, data were refined by iterative cycles of positional refinement and TLS refinement with PHENIX (Adams et al., 2010) and model building with COOT (Emsley et al., 2010). All current models have good geometry and no residues are in the disallowed region of the Ramachandran plot. Data collection and model statistics are given in Table 1.
Calculation of atomic contacts
The atomic contacts are calculated by CCP4 supported program CONTACT (Winn et al., 2011).
Thermal shift assay
Proteins (PfLysRS, HsLysRS, and Pf-like LysRS) were first prepared as 2× mixture, by diluting proteins at 4 μM concentration in buffer containing 20 mM Tris-HCl pH 8.0, 200 mM NaCl, 5 mM DTT, and different concentrations of ligand or inhibitors (CP: 40 μM, lysine: 1 mM, ATP: 40 μM, CaCl2: 2 mM). The 2× mixture was incubated at room temperature for 30 min, and chilled on ice for 5 min. Then the 2× Protein Thermal ShiftTM Dye (Life Technologies), which was pre-diluted in buffer containing 20 mM Tris-HCl pH 8.0, 200 mM NaCl, and 5 mM DTT, was added to the 2× mixture by 1:1 ratio on ice. After a complete mixing, the final solutions were heated from 10 °C to 90 °C at a rate of 1 °C/min and fluorescence signals were monitored by iQTM5 Real-Time PCR Detection System (Bio-Rad).
The method for Kd calculation was described earlier (Pantoliano et al., 2001). First, a nonlinear least squares fitting algorithm was used to fit the fluorescence curve using the following expression:
where y(T) is the fluorescence unfolding data. R is gas constant. And the five fitting parameters, Tm, ΔHu, ΔCpu, yf, and yu, are allowed to float according to the Levenberg-Marquardt algorithm for minimizing the sum of the squares of the residuals. Tm is the midpoint for the protein unfolding transition. ΔHu is the protein unfolding enthalpy. ΔCpu is the heat capacity change for protein unfolding. And yf and yu are the pretransitional and posttransitional fluorescence intensity levels, respectively. Then the dissociation constant at Tm (Kd™) is calculated using the following expression:
where [L]Tm is the free ligand concentration at Tm, which can be estimated by [L]Total. T0 is the midpoint for the protein unfolding transition in the absence of ligand. AHuT0 is the enthalpy of protein unfolding in the absence of ligand. ΔCpuT0 is the heat capacity change for protein unfolding in the absence of ligand. Finally the Kd at any temperature KdT (25°C /298K was used in this paper) can be estimated by the next expression:
where 62760 (J/mol) is a reasonable estimation of the ligand binding enthalpy (Pantoliano et al., 2001).
Supplementary Material
Significance.
Collectively, these studies exhibit the molecular basis for specific inhibition of a tRNA synthetase by an ATP-site competitive inhibitor, to achieve both the horizontal selectivity among all aaRS families and the longitudinal specificity across different species. CP specifically stabilized the PfLysRS in the presence of L-Lysine, and the effect is dependent on the overall structural flexibility. This study suggests that the inherent divergence of tRNA synthetase structural assembly plus the cooperative binding of amino acid may allow for highly specific inhibition of aaRS, with potential instrumental implication in drug development.
Highlights.
Cladosporin (CP) occupies the class defining ATP-binding pocket in LysRS
Three residues at the bottom of ATP pocket determine CP's family specificity
CP specifically stabilizes Plasmodium LysRS in a lysine dependent manner
Divergence beyond the active site allows for species specific AARS inhibition
Acknowledgments
We thank Robert Bacchus, Montita Sowapark, and Lindsay Placius for their assistance in plasmid construction or protein purification and Joanne Doherty for editing assistance. Use of the Advanced Photon Source is supported by the U. S. Department of Energy under Contract No. DE-AC02-06CH11357. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy under Contract No. DE-AC02-76SF00515 and by the National Institute of General Medical Sciences including P41GM103393. This work was supported by the National Institutes of Health NIEHS GM100136 and GM106134 to M.G, and National Natural Science Foundation of China No. 81071306 to H.H.
Footnotes
Author Contributions: P.F., and M.G. designed all experiments and wrote the manuscript. P.F., H.H., J.W., K.C., and X.C. performed the experiments. All authors analyzed the data and contributed to manuscript preparation.
Accession Codes: The atomic coordinates and structure factors of HsLysRS-K-CP, PfLysRS-CP, and the Pf-like human LysRS-K-CP complexes have been deposited in the Protein Data Bank (PDB) with the accession codes 4YCU, 4YCV, and 4YCW, respectively.
Competing Financial Interests: The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Pengfei Fang, Email: PFang@scripps.edu.
Min Guo, Email: GuoMin@scripps.edu.
References
- Anke H. Metabolic products of microorganisms. 184. On the mode of action of cladosporin. The Journal of antibiotics. 1979;32:952–958. doi: 10.7164/antibiotics.32.952. [DOI] [PubMed] [Google Scholar]
- Baker SJ, Zhang YK, Akama T, Lau A, Zhou H, Hernandez V, Mao W, Alley MR, Sanders V, Plattner JJ. Discovery of a new boron-containing antifungal agent, 5-fluoro-1,3-dihydro-1-hydroxy-2,1- benzoxaborole (AN2690), for the potential treatment of onychomycosis. Journal of medicinal chemistry. 2006;49:4447–4450. doi: 10.1021/jm0603724. [DOI] [PubMed] [Google Scholar]
- Belrhali H, Yaremchuk A, Tukalo M, Larsen K, Berthet-Colominas C, Leberman R, Beijer B, Sproat B, Als-Nielsen J, Grubel G, et al. Crystal structures at 2.5 angstrom resolution of seryl-tRNA synthetase complexed with two analogs of seryl adenylate. Science. 1994;263:1432–1436. doi: 10.1126/science.8128224. [DOI] [PubMed] [Google Scholar]
- Crepin T, Yaremchuk A, Tukalo M, Cusack S. Structures of two bacterial prolyl-tRNA synthetases with and without a cis-editing domain. Structure. 2006;14:1511–1525. doi: 10.1016/j.str.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Crompton PD, Moebius J, Portugal S, Waisberg M, Hart G, Garver LS, Miller LH, Barillas-Mury C, Pierce SK. Malaria immunity in man and mosquito: insights into unsolved mysteries of a deadly infectious disease. Annual review of immunology. 2014;32:157–187. doi: 10.1146/annurev-immunol-032713-120220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusack S. Sequence, structure and evolutionary relationships between class 2 aminoacyl-tRNA synthetases: an update. Biochimie. 1993;75:1077–1081. doi: 10.1016/0300-9084(93)90006-e. [DOI] [PubMed] [Google Scholar]
- Cusack S. Aminoacyl-tRNA synthetases. Current opinion in structural biology. 1997;7:881–889. doi: 10.1016/s0959-440x(97)80161-3. [DOI] [PubMed] [Google Scholar]
- Cvetesic N, Palencia A, Halasz I, Cusack S, Gruic-Sovulj I. The physiological target for LeuRS translational quality control is norvaline. The EMBO journal. 2014;33:1639–1653. doi: 10.15252/embj.201488199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delarue M, Moras D. The aminoacyl-tRNA synthetase family: modules at work. BioEssays : news and reviews in molecular, cellular and developmental biology. 1993;15:675–687. doi: 10.1002/bies.950151007. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriani G, Delarue M, Poch O, Gangloff J, Moras D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature. 1990;347:203–206. doi: 10.1038/347203a0. [DOI] [PubMed] [Google Scholar]
- Francklyn C, Musier-Forsyth K, Martinis SA. Aminoacyl-tRNA synthetases in biology and disease: new evidence for structural and functional diversity in an ancient family of enzymes. Rna. 1997;3:954–960. [PMC free article] [PubMed] [Google Scholar]
- Fukai S, Nureki O, Sekine S, Shimada A, Tao J, Vassylyev DG, Yokoyama S. Structural basis for double-sieve discrimination of L-valine from L-isoleucine and L-threonine by the complex of tRNA(Val) and valyl-tRNA synthetase. Cell. 2000;103:793–803. doi: 10.1016/s0092-8674(00)00182-3. [DOI] [PubMed] [Google Scholar]
- Gadakh B, Van Aerschot A. Aminoacyl-tRNA synthetase inhibitors as antimicrobial agents: a patent review from 2006 till present. Expert opinion on therapeutic patents. 2012;22:1453–1465. doi: 10.1517/13543776.2012.732571. [DOI] [PubMed] [Google Scholar]
- Guo M, Chong YE, Shapiro R, Beebe K, Yang XL, Schimmel P. Paradox of mistranslation of serine for alanine caused by AlaRS recognition dilemma. Nature. 2009;462:808–812. doi: 10.1038/nature08612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Ignatov M, Musier-Forsyth K, Schimmel P, Yang XL. Crystal structure of tetrameric form of human lysyl-tRNA synthetase: Implications for multisynthetase complex formation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2331–2336. doi: 10.1073/pnas.0712072105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoepfner D, McNamara CW, Lim CS, Studer C, Riedl R, Aust T, McCormack SL, Plouffe DM, Meister S, Schuierer S, et al. Selective and specific inhibition of the plasmodium falciparum lysyl-tRNA synthetase by the fungal secondary metabolite cladosporin. Cell host & microbe. 2012;11:654–663. doi: 10.1016/j.chom.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J, Mellows G. Interaction of pseudomonic acid A with Escherichia coli B isoleucyl-tRNA synthetase. The Biochemical journal. 1980;191:209–219. doi: 10.1042/bj1910209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurdle JG, O'Neill AJ, Chopra I. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrobial agents and chemotherapy. 2005;49:4821–4833. doi: 10.1128/AAC.49.12.4821-4833.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibba M, Soll D. Aminoacyl-tRNA synthesis. Annu Rev Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- Ito T, Yokoyama S. Two enzymes bound to one transfer RNA assume alternative conformations for consecutive reactions. Nature. 2010;467:612–616. doi: 10.1038/nature09411. [DOI] [PubMed] [Google Scholar]
- Iwasaki W, Sekine S, Kuroishi C, Kuramitsu S, Shirouzu M, Yokoyama S. Structural basis of the water-assisted asparagine recognition by asparaginyl-tRNA synthetase. Journal of molecular biology. 2006;360:329–342. doi: 10.1016/j.jmb.2006.04.068. [DOI] [PubMed] [Google Scholar]
- Jiang S, Zeng Q, Gettayacamin M, Tungtaeng A, Wannaying S, Lim A, Hansukjariya P, Okunji CO, Zhu S, Fang D. Antimalarial activities and therapeutic properties of febrifugine analogs. Antimicrobial agents and chemotherapy. 2005;49:1169–1176. doi: 10.1128/AAC.49.3.1169-1176.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jortzik E, Becker K. Thioredoxin and glutathione systems in Plasmodium falciparum. International journal of medical microbiology : IJMM. 2012;302:187–194. doi: 10.1016/j.ijmm.2012.07.007. [DOI] [PubMed] [Google Scholar]
- Kamtekar S, Kennedy WD, Wang J, Stathopoulos C, Soll D, Steitz TA. The structural basis of cysteine aminoacylation of tRNAPro by prolyl-tRNA synthetases. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:1673–1678. doi: 10.1073/pnas.0437911100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, Kim YJ, Lee HK, Cortese JF, Wirth DF, et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nature chemical biology. 2012;8:311–317. doi: 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Sharma A, Belrhali H, Yogavel M, Sharma A. Structural basis of malaria parasite lysyl-tRNA synthetase inhibition by cladosporin. Journal of structural and functional genomics. 2014;15:63–71. doi: 10.1007/s10969-014-9182-1. [DOI] [PubMed] [Google Scholar]
- Kim DG, Lee JY, Kwon NH, Fang P, Zhang Q, Wang J, Young NL, Guo M, Cho HY, Mushtaq AU, et al. Chemical inhibition of prometastatic lysyl-tRNA synthetase-laminin receptor interaction. Nature chemical biology. 2014;10:29–34. doi: 10.1038/nchembio.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Shimomura N, Tanigawa F, Fujioka S, Shimada A. Plant growth activities of aspyran, asperentin, and its analogues produced by the fungus Aspergillus sp. Zeitschrift fur Naturforschung C, Journal of biosciences. 2012;67:587–593. doi: 10.1515/znc-2012-11-1209. [DOI] [PubMed] [Google Scholar]
- Kotik-Kogan O, Moor N, Tworowski D, Safro M. Structural basis for discrimination of L-phenylalanine from L-tyrosine by phenylalanyl-tRNA synthetase. Structure. 2005;13:1799–1807. doi: 10.1016/j.str.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Kotz J. Halofuginone target ID. SciBX. 2012;5 [Google Scholar]
- Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002;415:673–679. doi: 10.1038/415673a. [DOI] [PubMed] [Google Scholar]
- Mohanty JG, Nagababu E, Rifkind JM. Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Frontiers in physiology. 2014;5:84. doi: 10.3389/fphys.2014.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S. Redox and antioxidant systems of the malaria parasite Plasmodium falciparum. Molecular microbiology. 2004;53:1291–1305. doi: 10.1111/j.1365-2958.2004.04257.x. [DOI] [PubMed] [Google Scholar]
- Nakama T, Nureki O, Yokoyama S. Structural basis for the recognition of isoleucyl-adenylate and an antibiotic, mupirocin, by isoleucyl-tRNA synthetase. The Journal of biological chemistry. 2001;276:47387–47393. doi: 10.1074/jbc.M109089200. [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Ogiso Y, Nakama T, Fukai S, Nureki O. Structural basis for anticodon recognition by methionyl-tRNA synthetase. Nature structural & molecular biology. 2005;12:931–932. doi: 10.1038/nsmb988. [DOI] [PubMed] [Google Scholar]
- Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. Journal of biomolecular screening. 2001;6:429–440. doi: 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- Pines M, Snyder D, Yarkoni S, Nagler A. Halofuginone to treat fibrosis in chronic graft-versus-host disease and scleroderma. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2003;9:417–425. doi: 10.1016/s1083-8791(03)00151-4. [DOI] [PubMed] [Google Scholar]
- Pope AJ, Lapointe J, Mensah L, Benson N, Brown MJ, Moore KJ. Characterization of isoleucyl-tRNA synthetase from Staphylococcus aureus. I: Kinetic mechanism of the substrate activation reaction studied by transient and steady-state techniques. The Journal of biological chemistry. 1998;273:31680–31690. doi: 10.1074/jbc.273.48.31680. [DOI] [PubMed] [Google Scholar]
- Ribas de Pouplana L, Schimmel P. Two classes of tRNA synthetases suggested by sterically compatible dockings on tRNA acceptor stem. Cell. 2001;104:191–193. doi: 10.1016/s0092-8674(01)00204-5. [DOI] [PubMed] [Google Scholar]
- Rock FL, Mao W, Yaremchuk A, Tukalo M, Crepin T, Zhou H, Zhang YK, Hernandez V, Akama T, Baker SJ, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759–1761. doi: 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- Sakurama H, Takita T, Mikami B, Itoh T, Yasukawa K, Inouye K. Two crystal structures of lysyl-tRNA synthetase from Bacillus stearothermophilus in complex with lysyladenylate-like compounds: insights into the irreversible formation of the enzyme-bound adenylate of L-lysine hydroxamate. Journal of biochemistry. 2009;145:555–563. doi: 10.1093/jb/mvp014. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan R, Dock-Bregeon AC, Rees B, Bovee M, Caillet J, Romby P, Francklyn CS, Moras D. Zinc ion mediated amino acid discrimination by threonyl-tRNA synthetase. Nat Struct Biol. 2000;7:461–465. doi: 10.1038/75856. [DOI] [PubMed] [Google Scholar]
- Scott PM, Van Walbeek W, MacLean WM. Cladosporin, a new antifungal metabolite from Cladosporium cladosporioides. The Journal of antibiotics. 1971;24:747–755. doi: 10.7164/antibiotics.24.747. [DOI] [PubMed] [Google Scholar]
- Silvian LF, Wang J, Steitz TA. Insights into editing from an ile-tRNA synthetase structure with tRNAile and mupirocin. Science. 1999;285:1074–1077. [PubMed] [Google Scholar]
- Springer JP, Cutler HG, Crumley FG, Cox RH, Davis EE, Thean JE. Plant-Growth Regulatory Effects and Stereochemistry of Cladosporin. J Agr Food Chem. 1981;29:853–855. [Google Scholar]
- Sundrud MS, Koralov SB, Feuerer M, Calado DP, Kozhaya AE, Rhule-Smith A, Lefebvre RE, Unutmaz D, Mazitschek R, Waldner H, et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science. 2009;324:1334–1338. doi: 10.1126/science.1172638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng M, Hilgers MT, Cunningham ML, Borchardt A, Locke JB, Abraham S, Haley G, Kwan BP, Hall C, Hough GW, et al. Identification of bacteria-selective threonyl-tRNA synthetase substrate inhibitors by structure-based design. Journal of medicinal chemistry. 2013;56:1748–1760. doi: 10.1021/jm301756m. [DOI] [PubMed] [Google Scholar]
- Vondenhoff GH, Van Aerschot A. Aminoacyl-tRNA synthetase inhibitors as potential antibiotics. European journal of medicinal chemistry. 2011;46:5227–5236. doi: 10.1016/j.ejmech.2011.08.049. [DOI] [PubMed] [Google Scholar]
- Wang X, Radwan MM, Tarawneh AH, Gao J, Wedge DE, Rosa LH, Cutler HG, Cutler SJ. Antifungal activity against plant pathogens of metabolites from the endophytic fungus Cladosporium cladosporioides. J Agric Food Chem. 2013;61:4551–4555. doi: 10.1021/jf400212y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta crystallographica Section D, Biological crystallography. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Meng Q, Bai L, Zhou H. In silico discovery of aminoacyl-tRNA synthetase inhibitors. International journal of molecular sciences. 2014;15:1358–1373. doi: 10.3390/ijms15011358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Sun L, Yang XL, Schimmel P. ATP-directed capture of bioactive herbal-based medicine on human tRNA synthetase. Nature. 2013;494:121–124. doi: 10.1038/nature11774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.