

Abstract
Strategies that leverage bio-orthogonal interactions between small molecule ligands and genetically encoded amino acid sequences can be used to attach high-performance fluorophores to proteins in living cells. However, a major limitation of chemical protein labeling is that cells’ plasma membranes are impermeable to many useful probes and bio-labels. Here, we show that conjugation to nonaarginine, a cell penetrating peptide (CPP), enables passive cytoplasmic delivery of otherwise membrane-impermeant, small molecule protein labels. Heterodimers consisting of a luminescent Tb3+ complex, Lumi4®, linked to benzyl guanine, benzyl cytosine and trimethoprim were conjugated to the peptide CysArg9 with a reducible disulfide linker. When added to culture medium, the peptide conjugates rapidly (<30 min) enter the cytoplasm and diffuse freely throughout cells. The benzyl guanine, benzyl cytosine and trimethoprim derivatives bind selectively to fusion proteins tagged with SNAP-Tag®, CLIP-Tag® and E. coli dihydrofolate reductase (eDHFR), respectively. Furthermore, eDHFR and SNAP-Tag fusions can be labeled with Lumi4 analogs in the same cell, and this labeling can be detected using two-color, time-gated Förster Resonance Energy Transfer (FRET) microscopy. Finally, we present quantitative data showing that cytoplasmic uptake of nonaarginine-conjugated probes occurs in multiple cell types (MDCK, HeLa, NIH 3T3), most cells in a culture (>75%) are loaded with probe, and the cellular probe concentration can be controlled by varying incubation conditions. CPP-mediated delivery of Lumi4-linked protein labels will greatly increase the utility of lanthanide-based FRET microscopy. Moreover, our results strongly suggest that this approach can be adapted to deliver a wide variety of protein-targeted fluorophores or other functional probes that were previously unavailable for intracellular imaging studies.
INTRODUCTION
Various chemical biology approaches enable selective protein labeling with small molecules inside living cells.1–3 These methods entail over-expression of target proteins fused to a polypeptide that binds to an organic label. Often, the label is a heterodimer of a fluorophore or other useful functionality and a ligand moiety that interacts selectively with the polypeptide. This modular approach allows labeling with commercially available or synthetically optimized organic fluorophores that can outperform conventional fluorescent proteins in high performance microscopy applications. For example, chemical labeling systems such as SNAP-tag,4–5 HaloTag®6 and TMP-tag7–9 have been used variously for cellular single molecule,10 superresolution11–13 and fluorescence lifetime imaging.14 Ideally, exogenous small molecule probes or protein labels should rapidly enter the cytoplasm when added to culture medium, and it should be possible to control the probes’ cellular abundance by varying incubation conditions. However, many useful fluorophores will not diffuse through cell membranes, limiting their application to cell-surface studies or else requiring technically demanding cellular delivery methods like microinjection or electroporation.13, 15–16
Conjugation to cell penetrating peptides (CPPs) would seem to be a logical strategy for facilitating intracellular delivery of protein labels. CPPs are a class of short peptides (ca. 8 – 30 amino acids) that efficiently mediate entry of otherwise membrane-impermeant small and macromolecules into cells.17–19 More than 20 years of intensive study along with efforts to develop CPPs as therapeutic delivery agents have revealed that CPPs enter cells by both endocytic and non-endocytic mechanisms, and that the operative pathway depends on peptide sequence, cargo size, cell type, culture conditions and other factors.18–19 While CPP-linked macromolecules enter cells exclusively by endocytosis, there is now considerable evidence showing direct cytoplasmic entry of arginine-rich peptides conjugated to fluorophores or other relatively small cargo.19–23 Our lab recently showed that conjugation to nonaarginine or Tat-derived sequences allowed for cytoplasmic delivery of an otherwise membrane-impermeant Tb3+ complex, Lumi4-Tb.24 We further showed that CPP-coupled heterodimers of Lumi4-Tb and trimethoprim (TMP) rapidly (<30 min) entered the cytoplasm of various cell types, diffused freely throughout the cytoplasm and nucleus, and bound selectively to Escherichia coli dihydrofolate reductase (eDHFR) fusion proteins.
In this paper, we show that conjugation to nonaarginine facilitates passive, cytoplasmic delivery of Lumi4-benzyl guanine (BG-Lumi4-R9) and Lumi4-benzyl cytosine (BC-Lumi4-R9) heterodimers that bind to SNAP-Tag and CLIP-Tag fusion proteins, respectively. We also show that eDHFR and SNAP-Tag fusions can be simultaneously labeled with Lumi4 in the same cell, and this labeling can be detected using two-color, time-gated Förster Resonance Energy Transfer (FRET) microscopy. Finally, we present quantitative data showing that cytoplasmic uptake of nonaarginine-conjugated tags occurs in multiple cell types (MDCK, HeLa, NIH 3T3), most cells in a culture (>75%) are loaded with probe, and the cellular probe abundance can be controlled by varying incubation conditions. Our results suggest that CPP-mediated delivery can be extended to a wide variety of protein-targeted fluorophores or other small molecule probes that were previously unavailable for intracellular imaging studies.
RESULTS AND DISCUSSION
Synthesis of peptide conjugates
Cell-permeable analogs of Lumi4 were prepared that target three different protein tags: eDHFR, SNAP-Tag and CLIP-Tag. Lumi4 is an octadentate, macrotricyclic ligand with four, 2-hydroxyisopthalamide chelating units.25 Its Tb3+ complex exhibits highly efficient emission (Φoverall > 50%), a large extinction coefficient (εmax > 20,000 M−1cm−1 at λ = 340 nm), and long excited state lifetime (τ > 2.4 ms) in aqueous solutions. SNAP-Tag and CLIP-Tag are mutant forms of human O6-alkylguanine alkyltransferase that autocatalytically form a covalent bond with derivatives of benzyl guanine and benzyl cytosine, respectively.4–5 Both SNAP- and CLIP-targeted Lumi4 analogs were reported previously and used for time-gated FRET studies of G-protein coupled receptor oligomerization.26–27 The ~18 kDa enzyme eDHFR binds non-covalently (KD = ~1 nM) to TMP derivatives,7 and our lab previously used TMP-Lumi4 for time-gated FRET microscopy of protein-protein interactions.28 Each peptide conjugate reported here shares the same basic architecture, where a cysteine serves as a three-way bridge to couple Lumi4 to a tri- or tetra-ethyleneglycolamino derivative of one of the targeting ligands (Chart 1). The Lumi4-ligand heterodimers are in turn conjugated via a disulfide bond to CysArg9. We previously showed that the disulfide bond is reductively cleaved in the cytoplasm, freeing the heterodimer to bind to its respective target protein (Figure 1a).24 The synthesis of TMP-Lumi4-R9 was described previously,24 and the synthesis and characterization of BG-Lumi4-R9 and BC-Lumi4-R9 are provided in Supporting Information.
Chart 1. Chemical structures of the peptide conjugates used in this study.
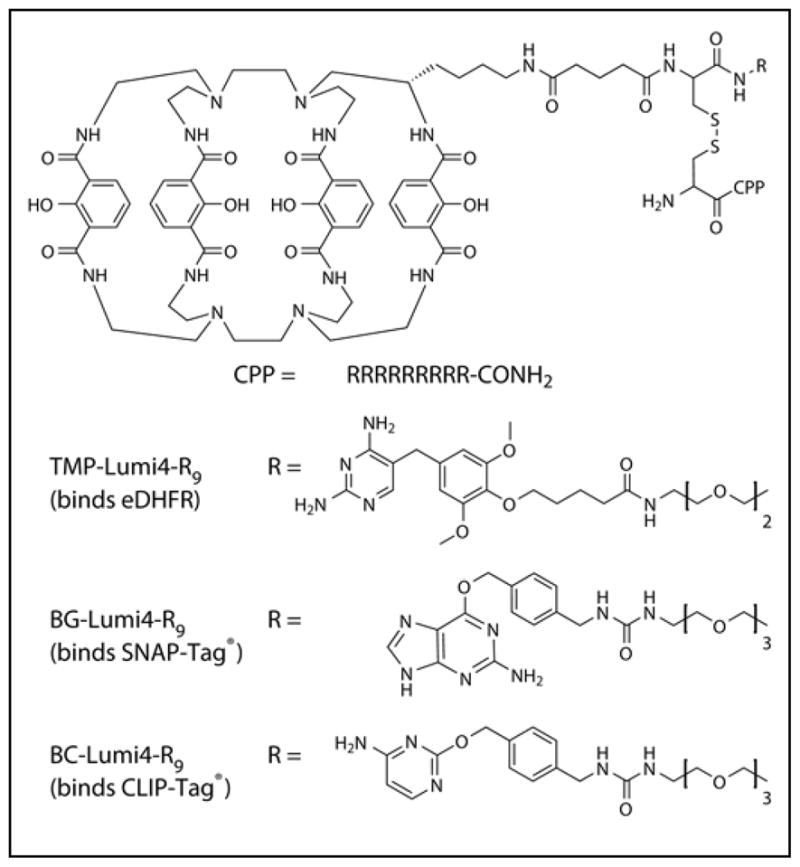
Abbreviations: CPP, cell penetrating peptide; capital letters, L-amino acids.
Figure 1. Model system for assessing CPP-mediated delivery and selective intracellular protein labeling.
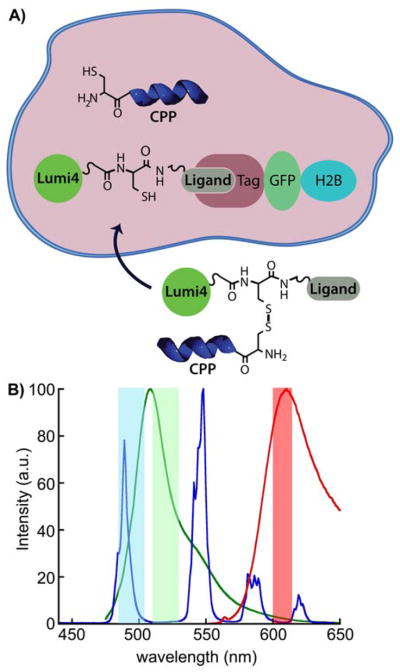
A) Following CPP-mediated delivery into the cytoplasm, disulfide reduction frees ligand-Lumi4 heterodimer to diffuse and bind to a 3-component fusion of histone 2B, fluorescent protein and tag (eDHFR, SNAP or CLIP). Selective labeling is confirmed by observation of long-lifetime, Tb3+-to-fluorescent protein emission. B) Normalized emission spectra of TMP-Lumi4(Tb3+) (blue), EGFP (green) and mCherry (red). The characteristically narrow Tb3+ emission bands enable efficient spectral separation of donor and sensitized acceptor emission signals using narrow-pass filters (colored bands).
Specific labeling of fusion proteins in live cells
Time-gated microscopic imaging of Tb3+ luminescence and Tb3+- mediated FRET was used to evaluate peptide conjugate transport into cells and subsequent intracellular labeling of target proteins. FRET is a non-radiative transfer of excitation energy from a donor fluorophore to a nearby (< 10 nm) acceptor whose absorption spectrum overlaps the donor’s emission spectrum. If the acceptor is fluorescent, excitation in the donor absorption band can be detected as sensitized acceptor emission, and intensity-based FRET measurements are widely used to analyze or image interactions between appropriately labeled biomolecules.29 However, FRET microscopy with fluorescent proteins often suffers from low signal to noise ratio because the sensitized emission signal is contaminated with donor or directly excited acceptor components. Tb3+ (or Eu3+) complexes have two key advantages when used as FRET donors in combination with conventional fluorescent acceptors. First, ms-scale Tb3+ and Tb3+-sensitized acceptor emission signals can be separated from non-specific, ns-scale fluorescent background by time-gating, where the detector is turned on some microsceconds after a brief excitation pulse. Second, multiple, narrow-line emission maxima enable detection of FRET from Tb3+ to two or more differently colored acceptors in the same sample (Figure 1b). Time-gated FRET with lanthanides has long been used for sensitive immunoassays and high-throughput screening applications, and recent years have seen substantial interest in exploiting the method’s advantages for live-cell imaging and multiplexed bio-analysis.27, 30–33
The time-gated microscope used in this study and its operation were described previously,34–35 and comprehensive details of image acquisition and analysis parameters are provided in Supporting Information. The instrument uses pulsed UV light (365 nm) to excite the Lumi4 Tb3+ complex. An intensified CCD camera (ICCD) detects long-lived signals 10 μs after the end of the excitation pulse, thereby eliminating scattering, cellular autofluorescence and directly excited acceptor fluorescence background. Plasmid DNA vectors encoding three different histone 2B (H2B) fusion proteins were prepared: 1) H2B-GFP-eDHFR; 2) H2B-GFP-SNAP; and 3) H2B-GFP-CLIP. A plasmid encoding H2B-mCherry-eDHFR was also prepared as well as MDCKII cell lines that stably express either H2B-GFP-eDHFR or H2B-mCherry-eDHFR. Visualization of Tb3+ luminescence was used to assess sub-cellular probe distribution following uptake, and detection of Tb3+- sensitized, fluorescent protein emission was used to assess specific labeling of the H2B fusion proteins (Figure 1b).
MDCKII cells that stably or transiently expressed the eDHFR, SNAP or CLIP fusion proteins were incubated for 30 min in serum-free medium containing one of the three Lumi4-peptide conjugates. After washing, Tb3+ luminescence was diffusely distributed throughout the cells, suggesting a direct transport from culture medium to cytoplasm (Figure 2a–c). Each peptide conjugate, TMP-Lumi4-R9, BG-Lumi4-R9 and BC-Lumi4-R9, specifically labeled its respective protein target as evidenced by images showing Tb3+-to-GFP sensitized emission (FRET) only in cells that expressed the H2B fusion proteins and that also showed luminescence in the Tb3+ channel (Figure 2a–c). Negligible FRET signal was observed in non-expressing cells that exhibited Tb3+ luminescence, and it was not possible to observe FRET signals when cultures of transfected cells were pre-incubated with non-luminescent conjugates that lacked Tb3+ (data not shown). Moreover, competition with excess unconjugated TMP was observed directly as a ~90% reduction of the FRET signal in TMP-Lumi4-R9-treated cells that expressed H2B-GFP-eDHFR (Supporting Figure S1). Together, these results strongly indicate that membrane-impermeant substrates of SNAP and CLIP as well as eDHFR-binding small molecules diffuse freely throughout the cytoplasm and nucleus and selectively label their targets following CPP-mediated delivery.
Figure 2. Conjugation to nonaarginine mediates cytoplasmic delivery of ligand-Tb3+ complex heterodimers and specific labeling of receptor fusion proteins as evidenced by time-gated imaging of Tb3+-to-GFP sensitized emission.

MDCKII cells stably expressing H2B-GFP-eDHFR or transiently expressing H2B-GFP-SNAP or H2B-GFP-CLIP were incubated with TMP-Lumi4-R9, BG-Lumi4-R9 or BC-Lumi4-R9, respectively (in DMEM w/o serum, 10 μM, 30 min), washed and imaged. Tb3+-to-GFP FRET is seen only in cells that express the target fusion protein and contain the luminescent Tb3+ complex, as indicated by arrows. A) Stable H2B-GFP-eDHFR expression; plus TMP-Lumi4-R9. B) Transient H2B-GFP-SNAP expression; plus BG-Lumi4-R9. C) Transient H2B-GFP-CLIP expression; plus BC-Lumi4-R9. Micrographs: left column, continuous wave fluorescence (λex = 480/40 nm, λem = 535/50 nm); middle column, time-gated Tb3+ luminescence (delay = 10 μs, λex = 365 nm, λem = 494/20 nm); right column, time-gated Tb3+-to-GFP FRET (delay = 10 μs, λex = 365 nm, λem = 520/20 nm). Scale bars, 10 μm. Intensity modulated displays depict the range of gray scale values in the corresponding 12-bit image.
An additional control experiment was performed to ensure that the observed FRET signals resulted from specific binding of the Lumi4 analogs to their target proteins. Non-specific FRET signals could result from bleedthrough of the Tb3+ signal into the FRET channel or because of collisions between freely diffusing Tb3+ donors and fluorescent protein acceptors. Bleedthrough into the GFP FRET channel is not significant as can be seen in Figure 2. However, substantial, diffusion-mediated energy transfer is known to occur in solution between Tb3+ donors and small molecule fluorescent acceptors even at micromolar acceptor concentrations because of the long excited state lifetimes of Tb3+ complexes.36 To determine the potential significance of collisional FRET in our experiments, we imaged MDCKII cells that stably expressed H2B-mCherry-eDHFR following loading with BG-Lumi4-R9, which does not bind to eDHFR. We then measured signal intensities in the FRET and Tb3+ channels both in the presence and absence of acceptor (Supporting Figure S2). Because Tb3+ has an emission peak centered at 620 nm that lies close to the mCherry emission filter (Figure 1b), we detected a substantial signal in the FRET channel, equal to 12.6 ± 0.1% of the measured Tb3+ signal, even in the absence of acceptor. However, the FRET signal measured when acceptor was present was only about 6% greater (13.4 ± 0.2%). Collisional FRET depends not only on acceptor concentration but also on the distance of closest approach between donor and acceptor.36 We surmise that the low levels of diffusion-mediated energy transfer seen here are due to the fact that the hydrodynamic radius of fluorescent proteins is about 2.8 nm,37 and the chromophore is located in the center of the protein.
Incubation conditions control uptake efficiency and intracellular probe abundance
We previously reported that diffuse Tb3+ luminescence may be observed when cells are incubated in growth medium containing ca. 10 μM or greater concentrations of TMP-Lumi4-R9 and other Lumi4-CPP analogs, even when incubation occurs at 4 °C.24 By contrast, when the peptide conjugate concentration in culture medium is too low, a punctate staining pattern is observed that is suggestive of endocytosis (Figure 3a). Cytoplasmic delivery is desirable as it allows for targeting the widest variety of proteins. Incubation at 37 °C in serum-free growth medium containing 10 μM of TMP-Lumi4-R9 resulted in a diffuse distribution of the probe throughout the cytoplasm and nucleus in more than 75% of cells observed. Furthermore, this level of delivery efficiency was observed in three different cell types including MDCKII epithelial cells, HeLa cells and NIH3T3 fibroblasts (Figure 3b). When the extracellular peptide concentration was lowered, the percentage of cells exhibiting diffuse luminescence decreased, and the overall abundance of probe observed in the cytoplasm decreased as well (Figure 3c). Efficient cytoplasmic delivery could be restored by co-incubating cells in medium with a low concentration (1 μM) of TMP-Lumi4-R9 and a high concentration (60 μM) of unconjugated nonaarginine.
Figure 3. Extracellular peptide concentration determines mode of uptake and intracellular abundance of cargo.
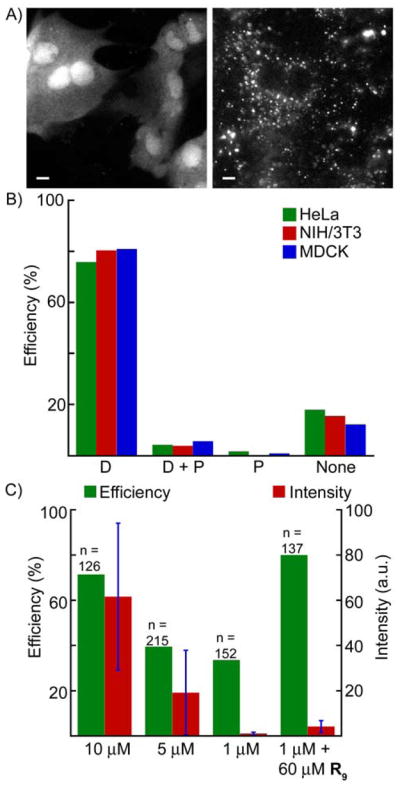
A) Representative images of diffuse (left) and punctuate (right) staining patterns. B) Various cell types were incubated in serum-free culture medium containing TMP-Lumi4-R9 (10 μM, 30 min, 37 °C), washed and imaged. Bar chart shows percentage of cells observed (n > 100 for each type) that exhibit indicated luminescence phenotypes: D, diffuse; D+P, diffuse and punctate; P, punctate; None, no signal. C) Percentage of HeLa cells (n = sample size) exhibiting diffuse luminescence and the normalized luminescence intensity (arbitrary units, see Supporting Information; error bars, sd) observed following incubation (30 min, 37 °C) in serum-free medium containing indicated concentrations of TMP-Lumi4-R9 and/or nonaarginine (R9).
Interestingly, we found that it was necessary to incubate stably transformed MDCKII cells (e.g., Figure 2a) at room temperature (~22 °C) in order to achieve consistent delivery of nonaarginine conjugates into the cytoplasm. Incubation of stably transformed MDCKII cells 37 °C resulted in punctate luminescence in nearly all cells observed. We have also observed that cytoplasmic uptake efficiency diminishes in MDCKII and other cell lines as passage number increases. We did not rigorously quantify this phenomenon, but for these experiments we only used cells that had been passaged fewer than ten times following thawing from frozen stock. Clearly, passage number, temperature and probably other culture conditions influence the mechanism of transduction, and these factors warrant future study. As a practical matter, our results show that by varying only two parameters, extracellular peptide concentration and incubation temperature, it is possible to favor direct translocation of probes into the cytoplasm and to control their ultimate cellular abundance.
The diffuse luminescence staining pattern suggests that a non-endocytic mode of entry is operative above a threshold peptide concentration, whereby nonaarginine mediates direct transduction of the probe molecules from extracellular growth medium into the cytoplasm. Such concentration-dependent, direct transduction of small molecule fluorophores conjugated to Tat or oligoarginine has been described by Brock and others.21–23 A hallmark of transduction is that entry into the cytoplasm originates from localized regions of the plasma membrane, dubbed nucleation zones by Brock.22 We observed such localized entry when we captured time-lapsed images of NIH3T3 fibroblasts mounted in medium containing TMP-Lumi4-R9 (Supporting Figure S3). Our observations and those reported by others show that conjugation to nonaarginine mediates transduction of various, membrane-impermeant small molecules, and the results also show that direct transduction may be favored over endocytosis by manipulating labeling conditions.
Multiplex protein labeling and FRET imaging
The availability of three cell-permeable Lumi4 conjugates that can target different fusion proteins makes it possible to perform multiplexed FRET imaging studies, where Lumi4-Tb is used as a FRET donor and differently colored fluorescent proteins are used as acceptors. Multi-component labeling and imaging could be used, for example, to monitor two or even three distinct protein-protein interactions in the same cell. In order to demonstrate multi-component protein labeling and imaging, we transiently transfected MDCKII cells that stably expressed H2B-mCherry-eDHFR with DNA encoding H2B-GFP-SNAP. Following transfection, the cells were incubated in growth medium containing both TMP-Lumi4-R9 and BG-Lumi4-R9 (10 μM, 30 min, RT). In cells that expressed both fusion proteins and that were loaded with the Lumi4 conjugates (as evidenced by time-gated Tb3+ emission at 494 nm), a long-lived signal was observed at both GFP and mCherry emission wavelengths (Figure 4). As noted above, Tb3+ has an emission peak centered at 620 nm that lies close to the mCherry emission channel (Figure 1b and Supporting Figure S2), and it was necessary to correct time-gated, Tb3+-to-mCherry FRET signals (at 605 nm) for bleedthrough of Tb3+ donor luminescence. Following bleedthrough correction, the Tb3+-to-mCherry FRET signal that results from specific labeling of H2B-mCherry-eDHFR with TMP-Lumi4-R9 became apparent (Figure 4).
Figure 4. Co-incubation with TMP-Lumi4-R9 and BG-Lumi4-R9 allows simultaneous labeling of H2B-mCherry-eDHFR and H2B-GFP-SNAP in the same cell and 2-color FRET detection.
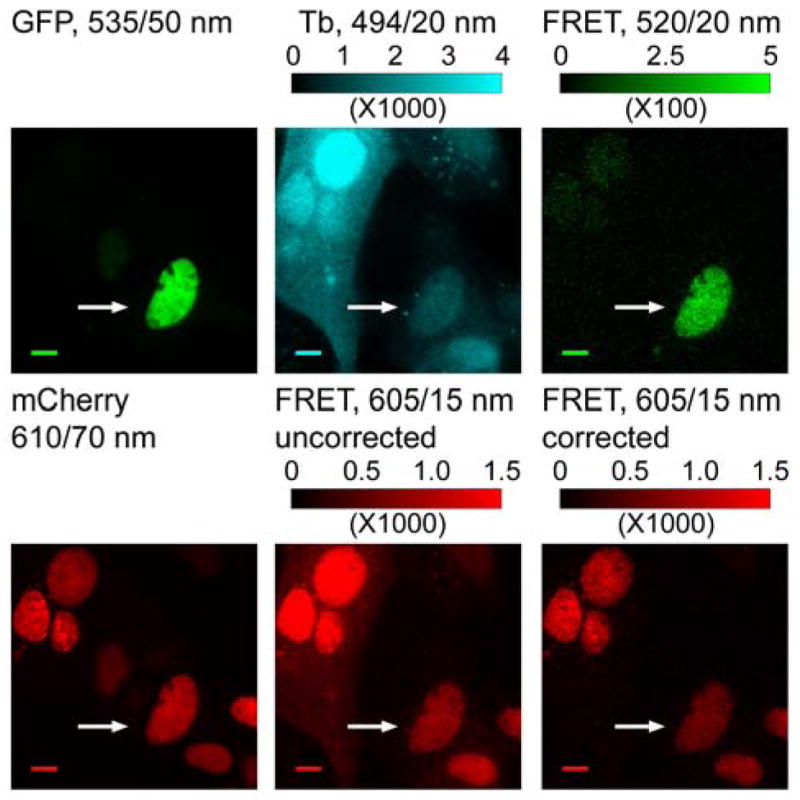
MDCKII cells stably expressing H2B-mCherry-eDHFR were transiently transfected with DNA encoding H2B-GFP-SNAP. The cells were incubated in DMEM (w/o serum, 30 min., RT) containing both TMP-Lumi4-R9 and BG-Lumi4-R9 (10 μM), washed and imaged. Tb3+-senstized emission of both GFP and mCherry can be observed in cell that expresses both fusion proteins and that is loaded with probes (arrows). Micrographs: left, continuous wave fluorescence of GFP (λex = 480/20 nm) and mCherry (λex = 535/15 nm) at the indicated emission wavelengths (center wavelength/bandwidth); middle and right, time-gated luminescence (delay = 10 μs, λex = 365 nm) at the indicated emission wavelengths; bottom middle and bottom right images are presented at identical contrast and show time-gated luminescence in the mCherry emission channel before and after bleedthrough correction, respectively. Scale bars, 10 μm. Intensity modulated displays depict range of gray scale values in corresponding 12-bit image.
CONCLUSION
Despite their varied structure and relatively large size (e.g., 1.8 kDa for TMP-Lumi4), Lumi4 derivatives can be rapidly and efficiently loaded into the cytoplasm of different cell types. The experimental simplicity, high loading efficiency (>75%) and ability to control intracellular label abundance afforded by CPP conjugation marks a substantial improvement over prior lanthanide probe delivery methods used in our laboratory. We previously reported streptolysin O-mediated membrane permeabilization and osmotic lysis of pinocytic vesicles as viable methods for delivering lanthanide labels into the cytoplasm.28 Both of those techniques required multiple experimental steps, and delivery efficiency varied widely between experiments, rarely exceeding 20%. The concentration-dependent cytoplasmic uptake described here is similar to that reported for conjugates of other fluorophores to oligoarginine or Tat.20–23 By co-incubating cells in medium containing TMP-Lumi4-R9 at low concentration (1 μM) and a large excess (60 μM) of unconjugated nonaarginine, it was possible to retain highly efficient cytoplasmic delivery. This feature would be important for single molecule studies where it would be necessary to deliver nanomolar concentrations of probes into the cytoplasm. By delivering TMP and benzylguanine derivatives into the same cell, it was possible to simultaneously label both eDHFR and SNAP-tag fusion proteins. Together, these results suggest that conjugation to arginine-rich CPPs should be a generally applicable strategy for delivering a wide variety of small molecule protein labels and sensors into cells and enhancing the utility of chemical protein labeling methods.
EXPERIMENTAL PROCEDURES
Details of peptide conjugate synthesis, plasmid DNA preparation, cell handling protocols, microscopy parameters and image analysis are available in Supporting Materials and Methods.
Supplementary Material
Acknowledgments
Funding support was provided by the NIGMS (R01GM081030) and the NSF (1152688). Lumi4® is a registered trademark of Lumiphore, Inc.
Footnotes
“Supporting Information Available: synthesis and characterization of peptide conjugates, cell culture protocols, microscopy and image analysis details, supporting figures S1–S3. This material is available free of charge via the Internet at http://pubs.acs.org.”
References
- 1.Hinner MJ, Johnsson K. How to obtain labeled proteins and what to do with them. Curr Opin Biotechnol. 2010;21:766–776. doi: 10.1016/j.copbio.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Chen Z, Cornish VW, Min W. Chemical tags: inspiration for advanced imaging techniques. Curr Opin Chem Biol. 2013;17:637–643. doi: 10.1016/j.cbpa.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 3.Jung D, Min K, Jung J, Jang W, Kwon Y. Chemical biology-based approaches on fluorescent labeling of proteins in live cells. Mol Biosyst. 2013;9:862–872. doi: 10.1039/c2mb25422k. [DOI] [PubMed] [Google Scholar]
- 4.Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 5.Gautier A, Juillerat A, Heinis C, Correa IR, Jr, Kindermann M, Beaufils F, Johnsson K. An engineered protein tag for multiprotein labeling in living cells. Chem Biol. 2008;15:128–136. doi: 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 7.Miller LW, Cai Y, Sheetz MP, Cornish VW. In vivo protein labeling with trimethoprim conjugates: a flexible chemical tag. Nat Methods. 2005;2:255–257. doi: 10.1038/nmeth749. [DOI] [PubMed] [Google Scholar]
- 8.Gallagher SS, Sable JE, Sheetz MP, Cornish VW. An in vivo covalent TMP-tag based on proximity-induced reactivity. ACS Chem Biol. 2009;4:547–556. doi: 10.1021/cb900062k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Z, Jing C, Gallagher SS, Sheetz MP, Cornish VW. Second-generation covalent TMP-tag for live cell imaging. J Am Chem Soc. 2012;134:13692–13699. doi: 10.1021/ja303374p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Appelhans T, Richter CP, Wilkens V, Hess ST, Piehler J, Busch KB. Nanoscale organization of mitochondrial microcompartments revealed by combining tracking and localization microscopy. Nano Lett. 2012;12:610–616. doi: 10.1021/nl203343a. [DOI] [PubMed] [Google Scholar]
- 11.Hein B, Willig KI, Wurm CA, Westphal V, Jakobs S, Hell SW. Stimulated emission depletion nanoscopy of living cells using SNAP-tag fusion proteins. Biophys J. 2010;98:158–163. doi: 10.1016/j.bpj.2009.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wombacher R, Heidbreder M, van de Linde S, Sheetz MP, Heilemann M, Cornish VW, Sauer M. Live-cell super-resolution imaging with trimethoprim conjugates. Nat Methods. 2010;7:717–719. doi: 10.1038/nmeth.1489. [DOI] [PubMed] [Google Scholar]
- 13.Banala S, Maurel D, Manley S, Johnsson K. A caged, localizable rhodamine derivative for superresolution microscopy. ACS Chem Biol. 2012;7:289–293. doi: 10.1021/cb2002889. [DOI] [PubMed] [Google Scholar]
- 14.Gatzogiannis E, Chen Z, Wei L, Wombacher R, Kao YT, Yefremov G, Cornish VW, Min W. Mapping protein-specific micro-environments in live cells by fluorescence lifetime imaging of a hybrid genetic-chemical molecular rotor tag. Chem Commun (Camb) 2012;48:8694–8696. doi: 10.1039/c2cc33133k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maurel D, Banala S, Laroche T, Johnsson K. Photoactivatable and photoconvertible fluorescent probes for protein labeling. ACS Chem Biol. 2010;5:507–516. doi: 10.1021/cb1000229. [DOI] [PubMed] [Google Scholar]
- 16.Jones SA, Shim SH, He J, Zhuang X. Fast, three-dimensional super-resolution imaging of live cells. Nat Methods. 2011;8:499–508. doi: 10.1038/nmeth.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van den Berg A, Dowdy SF. Protein transduction domain delivery of therapeutic macromolecules. Curr Opin Biotechnol. 2011;22:888–893. doi: 10.1016/j.copbio.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Milletti F. Cell-penetrating peptides: classes, origin, and current landscape. Drug Discov Today. 2012;17:850–860. doi: 10.1016/j.drudis.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Brock R. The uptake of arginine-rich cell-penetrating peptides: putting the puzzle together. Bioconjug Chem. 2014;25:863–868. doi: 10.1021/bc500017t. [DOI] [PubMed] [Google Scholar]
- 20.Ziegler A, Nervi P, Durrenberger M, Seelig J. The cationic cell-penetrating peptide CPP(TAT) derived from the HIV-1 protein TAT is rapidly transported into living fibroblasts: optical, biophysical, and metabolic evidence. Biochemistry. 2005;44:138–148. doi: 10.1021/bi0491604. [DOI] [PubMed] [Google Scholar]
- 21.Tunnemann G, Martin RM, Haupt S, Patsch C, Edenhofer F, Cardoso MC. Cargo-dependent mode of uptake and bioavailability of TAT-containing proteins and peptides in living cells. FASEB J. 2006;20:1775–1784. doi: 10.1096/fj.05-5523com. [DOI] [PubMed] [Google Scholar]
- 22.Duchardt F, Fotin-Mleczek M, Schwarz H, Fischer R, Brock R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic. 2007;8:848–866. doi: 10.1111/j.1600-0854.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- 23.Kosuge M, Takeuchi T, Nakase I, Jones AT, Futaki S. Cellular internalization and distribution of arginine-rich peptides as a function of extracellular peptide concentration, serum, and plasma membrane associated proteoglycans. Bioconjug Chem. 2008;19:656–664. doi: 10.1021/bc700289w. [DOI] [PubMed] [Google Scholar]
- 24.Mohandessi S, Rajendran M, Magda D, Miller LW. Cell-penetrating peptides as delivery vehicles for a protein-targeted terbium complex. Chem -- Eur J. 2012;18:10825–10829. doi: 10.1002/chem.201201805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu J, Corneillie TM, Moore EG, Law GL, Butlin NG, Raymond KN. Octadentate Cages of Tb(III) 2-Hydroxyisophthalamides: A New Standard for Luminescent Lanthanide Labels. J Am Chem Soc. 2011;133:19900–19910. doi: 10.1021/ja2079898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Comps-Agrar L, Kniazeff J, Brock C, Trinquet E, Pin JP. Stability of GABAB receptor oligomers revealed by dual TR-FRET and drug-induced cell surface targeting. FASEB J. 2012;26:3430–3439. doi: 10.1096/fj.12-203646. [DOI] [PubMed] [Google Scholar]
- 27.Zwier JM, Bazin H, Lamarque L, Mathis G. Luminescent lanthanide cryptates: from the bench to the bedside. Inorg Chem. 2014;53:1854–1866. doi: 10.1021/ic402234k. [DOI] [PubMed] [Google Scholar]
- 28.Rajapakse HE, Gahlaut N, Mohandessi S, Yu D, Turner JR, Miller LW. Time-resolved luminescence resonance energy transfer imaging of protein-protein interactions in living cells. Proc Natl Acad Sci U S A. 2010;107:13582–13587. doi: 10.1073/pnas.1002025107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Medintz IL, Hildebrandt N. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2013. p. 816. [Google Scholar]
- 30.Butler SJ, Lamarque L, Pal R, Parker D. EuroTracker dyes: highly emissive europium complexes as alternative organelle stains for live cell imaging. Chemical Science. 2014;5:1750–1756. [Google Scholar]
- 31.Delbianco M, Sadovnikova V, Bourrier E, Mathis G, Lamarque L, Zwier JM, Parker D. Bright, highly water-soluble triazacyclononane europium complexes to detect ligand binding with time-resolved FRET microscopy. Angew Chem Int Ed Engl. 2014;53:10718–10722. doi: 10.1002/anie.201406632. [DOI] [PubMed] [Google Scholar]
- 32.Geissler D, Linden S, Liermann K, Wegner KD, Charbonniere LJ, Hildebrandt N. Lanthanides and quantum dots as Forster resonance energy transfer agents for diagnostics and cellular imaging. Inorg Chem. 2014;53:1824–1838. doi: 10.1021/ic4017883. [DOI] [PubMed] [Google Scholar]
- 33.Hildebrandt N, Wegner KD, Algar WR. Luminescent terbium complexes: Superior Forster resonance energy transfer donors for flexible and sensitive multiplexed biosensing. Coordination Chemistry Reviews. 2014;273:125–138. [Google Scholar]
- 34.Gahlaut N, Miller LW. Time-resolved microscopy for imaging lanthanide luminescence in living cells. Cytometry A. 2010;77:1113–1125. doi: 10.1002/cyto.a.20964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin D, Lu Y, Leif RC, Yang S, Rajendran M, Miller LW. How to build a time-gated luminescence microscope. Curr Protoc Cytom. 2014;67(Unit 2):22. doi: 10.1002/0471142956.cy0222s67. [DOI] [PubMed] [Google Scholar]
- 36.Thomas DD, Carlsen WF, Stryer L. Fluorescence energy transfer in the rapid-diffusion limit. Proc Natl Acad Sci U S A. 1978;75:5746–5750. doi: 10.1073/pnas.75.12.5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liarzi O, Epel BL. Development of a quantitative tool for measuring changes in the coefficient of conductivity of plasmodesmata induced by developmental, biotic, and abiotic signals. Protoplasma. 2005;225:67–76. doi: 10.1007/s00709-004-0079-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.