

Summary
The unexpected diversity of the human microbiome and metabolome far exceeds the complexity of the human genome. Although we now understand microbial taxonomic and genetic repertoires in some populations, we are just beginning to assemble the computational and experimental tools to understand the metabolome in comparable detail. However, even with the limited current state of knowledge, individual connections between microbes and metabolites, between microbes and immune function, and between metabolites and immune function are being established. Here we provide our perspective on these connections. We also outline a systematic research program that could turn these individual links into a broader network that allows us to understand how these components interact. This program will allow us to exploit connections among the microbiome, metabolome, and host immune system to maintain health, and perhaps help us understand how to reverse the processes that lead to a wide range of immune and other diseases.
Introduction
The human microbiota (the collection of microbes that inhabits our bodies) and human microbiome (the collection of DNA from microbes) are remarkably, and unexpectedly, diverse. Although the Human Microbiome Project (HMP) (Group et al., 2009; Turnbaugh et al., 2007) was predicated on the assumption that there would be a large core of microbial lineages that we all share, sprinkled with a diversity of “peripheral” lineages that make each of us unique (Turnbaugh et al., 2007), this hypothesis was not validated by empirical evidence following completion of the HMP. Indeed, the first deep sequencing of multiple fecal samples from each of three individuals revealed that the differences between individuals is large. The difference between individuals is substantially greater than the difference within an individual at different sampling sites along the distal large intestine (Eckburg et al., 2005). This pattern of diversity has subsequently been reinforced in different body habitats (Costello et al., 2009; Findley et al., 2013; Grice et al., 2009; Human Microbiome Project, 2012), and with larger subject populations, especially in the gut (Qin et al., 2010; Turnbaugh et al., 2009; Yatsunenko et al., 2012). Even within healthy Western adults, studies routinely show that different people can be >90% different in terms of the populations of microbes in their gut: in other words, a microbial cell chosen from person A and from person B will be different at the species level more than 90% of the time. Additionally, the dynamic range of the most common microbes is hugely variable. Within the microbes identified by the European MetaHIT Project as “core”, meaning that they were found in at least 90% of the healthy European cohort studied, the dynamic range was several orders of magnitude different for each species-- in other words, any microbial species found with at least 10% abundance in one person was at least as rare as one cell in 1000 in another person in the cohort (Qin et al., 2010). We are at the leading edge of understanding the implications of this tremendous diversity at the metabolic level and of the interplay between the gut microbiota, metabolic function, and the immune system. To address the molecular connections between gut bacteria and immune function, a collection of synergistic technologies now exists that will allow rapid progress in untangling these complex interactions in ways that may be harnessed to promote health.
Grappling with a diverse microbiota
Although the microbiota is incredibly diverse, and much of this diversity still consists of uncharacterized species and genes, defining this diversity for some populations appears within reach based on the multitudes of microbiome profiling projects to date. Recent large-scale studies, such as the HMP and MetaHIT efforts, are beginning to saturate the gene catalog for healthy Western cohorts (Human Microbiome Project, 2012; Qin et al., 2010). This diversity is usually assessed by a technique called rarefaction: as additional subjects are examined, a curve is plotted with the number of subjects on the x-axis, the number of unique taxa or genes on the y-axis. As discovery of this microbial “parts list” becomes more complete, additional people and subsets of populations are required in order to find a new unique part, so the curve levels off, until at last it reaches an asymptote when all the parts have been discovered. Similar saturation of rarefaction curves is now being observed for several clinical conditions in which the microbiome is involved, such as obesity (Le Chatelier et al., 2013) and diabetes (Qin et al., 2012). This finding has several important implications: first, that the depth of microbial diversity is addressable using currently available technologies; second, that a reference database could be constructed, allowing interpretations of new sequences based on matching findings to what is known already rather than by computationally-driven de novo assembly and annotation procedures; and third, that markers can be discovered within this known universe and then prioritized for further laboratory characterization and experimental work, including studies in animal models such as those using gnotobiotic mice. However, a cautionary note appears needed: studies of children and of non-Western individuals reveal completely different configurations of the microbiome and the microbiota (Yatsunenko et al., 2012). Consequently, substantial additional work will be required to extend these techniques to cover the diverse complexity found in humanity, a notion that can be extended to various conditions in relevant animal models.
Another key assumption made previously that was not validated by subsequent studies is that diversity in the microbiota would correlate directly to diversity in the microbiome in terms of overall metabolic functions. This property, that different assemblages of species converge on very similar functional profiles, was first observed in the human gut (Turnbaugh et al., 2009), then more recently extended to other human body sites (Human Microbiome Project, 2012). Conceptually, this makes sense in retrospect and by analogy to larger-scale ecosystems: two grasslands might look relatively similar to each other, especially compared to two forests, even in situations where essentially none of the species in either grassland is shared (Hamady and Knight, 2009). Although particular species and functions seem to be highly individualized (Fierer et al., 2010; Schloissnig et al., 2013) and even stable over time (Caporaso et al., 2011; Faith et al., 2013), and with associations between genetic lineages of microbes and functional capacities so highly conserved that functional profiles can be predicted accurately from species distributions (Langille et al., 2013), the overall functional profiles appear remarkably static in healthy subjects overall. Although a few studies have correlated the microbiota to the microbiome (Muegge et al., 2011) and to the metabolome (Ridaura et al., 2013; Smith et al., 2013a), the relationships among the various ‘omics’ levels and to the host immune system remain largely unexplored.
Metabolite diversity may exceed even microbial diversity
In contrast to the microbiota and the microbiome, which we are now starting to saturate for some populations, the metabolome is extremely diverse and remains largely undescribed and undefined. For example, tissue culture cells grown in pure culture remain poorly characterized in terms of their metabolic profiles, underscoring the dearth of knowledge about more complex biological systems. The human genome contains approximately 20,000 protein-coding genes, but our metabolomic profile is much more complex, numbering at least 500,000 and with individual classes of compounds such as lipids and oligosaccharides potentially harboring very high levels of diversity simply on combinatorial grounds (Quehenberger et al., 2010; Shevchenko and Simons, 2010). For example, for the molecular family of 6 common triacylglycerides that use at least 20 different fatty acids provides the combinatorial capacity to make more than 40,000 different lipids (and in reality there are many more fatty acids, leading to further combinatorial explosion). This calculation does not take in account the many modifications that fatty acids undergo, and the many modifications triacylglycerides undergo themselves. This is just one description of one subfamily of one molecular family. Furthermore, there are many specialized metabolites including secondary metabolites, natural products, quorum sensors and small molecule virulence factors that nearly all microbes produce. Some organisms have the metabolic capacity to make as many as 50 of these. These molecules are optimized to regulate the surrounding environment and to control and alter biology at the host-microbe interface, by driving microbial community composition and host immune regulation (Figure 1). In turn, these molecules drive complex physiological feedback loops (Nicholson et al., 2005). These microbial molecules have been exploited in the clinic as cholesterol-lowering drugs, immune regulators, or antibiotics. Based on the genome sequences now available, there are strong indications that thousands more of these microbial effector molecules remain to be discovered in the human microbiota. Finally, given that the diversity of diet-derived cells and microbial cells present in the gut dwarf human cell diversity, as well as outnumbering them, it seems appropriate to assume that the vast majority of metabolites in our bodies are not of human origin and that microbes significantly alter the human microbiome.
Figure 1. The microbe-host interface.

Individual differences, including sex-specific differences between men and women (blue and red respectively), may control the microbiota or interact with the microbiota to influence immune function in several ways, including release of metabolites and direct microbial interactions. Much of this interface occurs in the gut (shown inset in the female figure), where the intestinal wall provides an interface between metabolites and immunological processes in the gut and the influence of these metabolites and immunological processes on the host systemically. In addition to normal metabolic function, specialized microbially-produced metabolic products (second inset), such as signaling molecules and antibiotics may play a key and specific role in driving microbial community composition, and subsequently the microbiome.
The number of potential gut bacterial metabolites is thus currently unknown, and include molecules of dietary, host and microbial origin. Furthermore, there is complex crosstalk between hosts, microbes, and the transformations that the metabolites undergo. These complex transformations often define the ultimate function of the molecules that are present (Martin et al., 2007). The tools currently available to harvest and visualize the diversity of microbial metabolites, including both primary and specialized metabolites, do not presently capture this complexity. Consequently, the state of knowledge for metabolites resembles the situation for microbes a decade ago: we know there are myriad metabolites of potential microbial origin or of microbial transformation, and we know that some are shared among people and some are unique. In a handful of examples that are clinically interesting, metabolites and their association with clinical phenotypes have been described using targeted techniques (Martin et al., 2007). For example, people are similar in some of the common metabolites they contain (such as amino acids or short-chain fatty acids in the gut), and that some of this variation correlates with disease. However, we lack a clear understanding of how many (and which) microbial metabolites influence host biology, and the overall nature of variation in the general population. This is due to the current limitations in technical approaches capable of building a comprehensive metabolomic “parts list” on a population-level scale. If we perform the analysis at the level of pathways rather than individual metabolites, they may appear to be more similar among individuals, but this apparent similarity may arise from limitations in the bioinformatics tools used. Just as we are starting to appreciate that our microbiomes augment our genome’s capabilities, and indeed can be more predictive for classifying people as healthy versus diseased than our human genome (Knights et al., 2011; Le Chatelier et al., 2013), we need to expand the concept of our metabolome to include the microbially-produced and microbially-modulated metabolites as factors contributing to pathogenesis.
Integrating immunology with metabolomics studies: a new frontier
Immunological responses by the host are also diverse, although our view of this diversity has been limited by the application of targeted approaches. Traditional research in immunology has focused on diverse immune responses to pathogenic infection. Viewed through this perspective, conventional wisdom in the field has guided studies to determine how the immune system attempts to control infectious agents, how pathogens subvert the arsenal of innate and adaptive immune mechanisms, and how individual mutations in the host may affect these processes. We now appreciate that host-microbial interactions at various body surfaces cannot be fully captured in this simple framework, but will likely need to involve complex and dynamic processes. These processes include not only immunity to pathogens, but also immune ignorance and/or tolerance mechanisms for symbiotic bacteria and opportunistic pathogens that are routinely found in microbiome studies. The functions of the immune system may even include promoting the growth of beneficial microbes, as well as limiting the growth of harmful microbes, as the same microbe may be harmful or beneficial in the context of different body sites, host physiological status, and so forth. Conceivably, individual members of the gut microbiota can have diverse effects on the immune system, with some indigenous organisms promoting pro-inflammatory responses while others promote anti-inflammatory reactions. A handful of microbes have been experimentally shown to directly impact infection (by other species, including bacteria, eukaryotes and viruses), autoimmunity and inflammation in animal models. These examples together with the genomic information indicate the existence of a vast constellation of microbes with bioactive properties waiting to be discovered. Consequently, the interplay between the gut microbiota and the immune response is likely far more diverse and dynamic than we currently appreciate, and the role that metabolites play in the evolutionary connection between symbiotic microbes and their hosts is a frontier of research in the field.
To date, observations connecting the microbiome, the metabolome, and immunological responses have been sporadic and incomplete. Investigators have made observations, like flashbulbs going off in the dark, but a systematic approach has not yet been applied in any complex biological system. Therefore, although we know that connections exist (as detailed below), only a small fraction of these connections are known at present. In part, this lack of a unified approach is due to “language barriers” among the practitioners of the different disciplines, and the lack of a unified research community making a concerted effort to bring their collective expertise to bear on these problems. Better ways of representing and visualizing the data, especially methods that translate across the various highly multivariate datasets collected by each discipline, and research practices and communities that allow translation of results among disciplines, will be critical going forward to uncover the diverse molecular interplay between gut microbial metabolites and the immune system. This perspective describes the current state of the art in our understanding of microbial-metabolite-immune connections, and provides a framework for the vast and exciting potential of developing an integrated program to decode the molecular conversation between mammals and their microbiomes. In doing so, we describe a roadmap for explaining current observations and discovering new interactions based on microbial metabolites in ways that will be useful for basic research, predictive modeling, patient stratification and potentially the development of transformative treatments for various diseases.
Microbial metabolism and inflammation
The microbiome produces a wide range of metabolites and has a pervasive effect on various host processes, although many of these effects are just starting to be explored in detail. It has long been known that bacteria in the gut are involved in metabolizing otherwise indigestible food components such as dietary fiber, but additional functions are being discovered at a rapid pace. For example, microbial production of vitamins and amino acids is important during human infant development (Yatsunenko et al., 2012), and has been implicated in malnutrition (Smith et al., 2013a), which in turn affects susceptibility to a variety of infectious diseases. Similarly, different bacteria in different people have variable effects on the metabolism of drugs ranging from the painkiller acetaminophen (Clayton et al., 2009) to the cardiac glycoside digoxin (Haiser et al., 2013) to the cancer drug cyclophosphamide (Viaud et al., 2013), and likely also have an effect on immunomodulatory drugs.
Microbial metabolites have long been implicated in metabolic functions both inside and outside the gut (Figure 2). Of these, short chain fatty acid (SCFAs), breakdown products of dietary fiber such as butyrate, are especially important as an energy source for intestinal epithelial cells. Recent studies have expanded the role of SCFAs from gut bacteria to reveal impacts on the immune system (for more detail, see the accompanying Thorburn et al. review). Briefly, in ulcerative colitis, an inflammatory bowel disease (IBD), SCFAs are reduced, and treatment with dietary fiber provides clinical benefits. Mice raised germ-free without any gut bacteria have lower SCFA levels than animals with a complex microbiota (Maslowski et al., 2009). This phenotype is attributed to increased intestinal inflammation in a mouse model of IBD, suggesting that SCFAs provide beneficial or immunosuppressive functions. Accordingly, deletion of GPR43, a sensor of SCFAs, leads to exacerbated intestinal disease in mice.
Figure 2. A simplified depiction of the effects of specific gut microbial or microbial-mammalian co-metabolites on different organ systems, including the immune system.
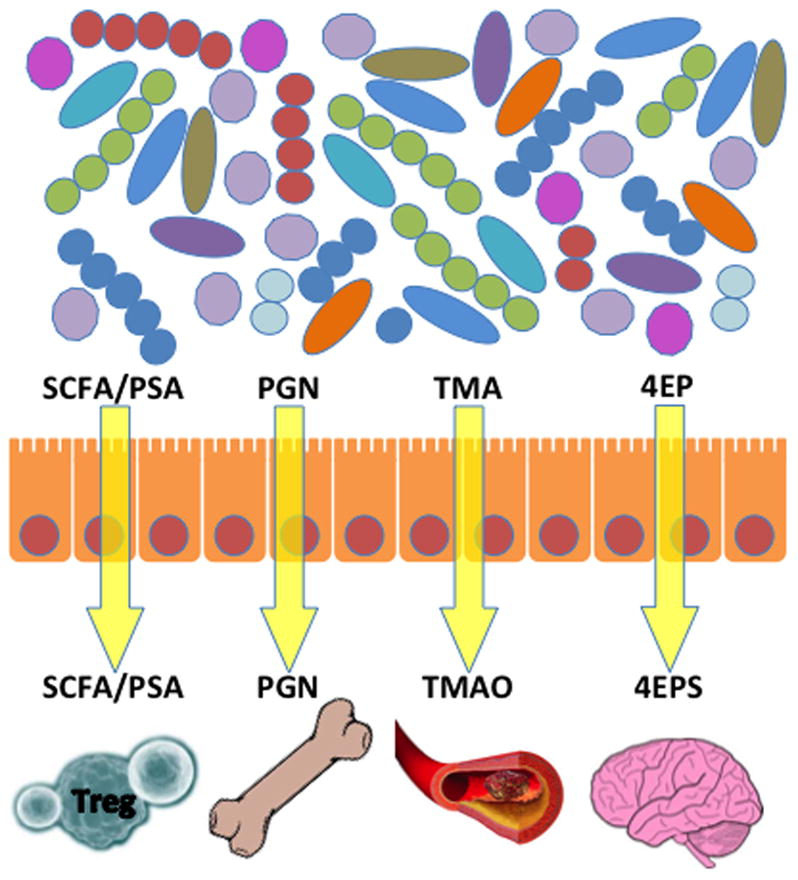
SCFA and PSA affect Treg cell development. PGN affects bone resorption. TMAO affects the vascular system and influences the risk of heart disease. 4EPS affects brain function and leads to anxiety-like behavioral defects similar to those observed in autism. Although some of these examples illustrate harmful effects, it is likely that many microbially produced metabolites in addition to SCFA and PSA produce beneficial effects on the host, although these tend to be less well studied. SCFA: short chain fatty acids; PSA: Polysaccharide A; PGN: peptidoglycan; TMA: Trimethylamine; TMAO: Trimethylamine N-oxide; 4EP: 4-ethylphenol; 4EPS: 4EP: 4-ethylphenol sulfate.
SCFAs directly affect development and function of anti-inflammatory regulatory T cells (Tregs), which restrain uncontrolled inflammation. SCFAs, and in particular propionate, increase both the proportion and the absolute count of Tregs in germ-free mice, and augment Treg function to promote suppression in colitis models (Smith et al., 2013b). Bacteria such as spore-forming Group XIVa Clostridia appear to produce butyrate that also promotes Treg differentiation both in vivo and in vitro (Furusawa et al., 2013). The effects of SCFAs extend beyond TREGS and the gut. For example, mice fed a high-fiber diet had both increased SCFA levels and protection from allergic inflammation in the lungs, attributed to effects on hematopoiesis of innate immune cells by propionate (Trompette et al., 2014). Collectively, these studies highlight how gut microbial metabolites, specifically, SCFAs, provide benefits to the host via enhancing the innate and adaptive immune systems in various animal models and contexts. Future work will be needed to determine whether this mechanism translates to humans. Dietary and microbial interventions for allergic, inflammatory and autoimmune diseases would be relatively safe and feasible approaches that could be rapidly validated in many populations and societies around the world.
Specific immune signals from microbially-produced metabolites
Many of the diseases that have been linked to the microbiome also have an immunological component. These conditions include IBD (Frank et al., 2011; Frank et al., 2007; Gevers et al., 2014), obesity (Cotillard et al., 2013; Le Chatelier et al., 2013; Ley et al., 2006; Ridaura et al., 2013; Turnbaugh et al., 2009), rheumatoid arthritis (Scher et al., 2013), food allergies (Ling et al., 2014), asthma (Fuchs and von Mutius, 2013), and animal models of multiple sclerosis (Berer et al., 2011; Lee et al., 2011), autism (Hsiao et al., 2013), resistance to infection (Abt et al., 2012; Ichinohe et al., 2011; Khosravi et al., 2014), and a wide range of other conditions. Examples of specific microbial triggers including lipopolysaccharide (LPS), double stranded RNA (dsRNA), quorum sensing molecules (homoserine lactones and their derivatives) are also well-known. We will summarize some of this literature, but these topics have been extensively reviewed elsewhere. Microbe-brain connections are also increasingly emerging as interesting and important (Cryan and Dinan, 2012). For example, neurotransmitters such as dopamine and serotonin are either produced by bacteria or production is stimulated in host cells by the presence of bacteria; viewing these neurotransmitters as metabolites that affect the immune system is an important emerging perspective (Baganz and Blakely, 2013).
Metabolites and pattern recognition by the immune system
Of particular interest is how molecules produced by bacteria, including metabolites, can act in pattern recognition by the immune system. The innate immune system has evolved pattern recognition receptors (PRRs) that recognize conserved microbial ligands. Binding to these ligands alerts the host to the presence of the microbes that produce them. This class of molecules, commonly termed pathogen-associated microbial patterns (PAMPs), has primarily been studied in the context of pathogenesis. Remarkably, some PAMPs produced by the gut microbiota can also positively impact the immune system and health. For example, in a model of intestinal injury and inflammation, more severe disease occurred in the absence of commensal microbes, but this effect could be ameliorated by adding LPS or lipotiechioc acid (LTA), two highly conserved products produced by microbes (Rakoff-Nahoum et al., 2004). The beneficial effects of microbial molecules extend beyond the gut. For example, peptidoglycan (a PAMP) from gut bacteria augments the function of innate immune cells that originate from the bone marrow, such as neutrophils, to help fight off systemic bacterial infection (Clarke et al., 2010). Depletion of gut bacteria also renders mice susceptible to influenza virus infection in the lungs, and to systemic LCMV infection (Abt et al., 2012; Ichinohe et al., 2011). The administration of PAMPs restored the host’s ability to control infection, demonstrating that microbial molecules previously studied in the context of infection can actually host-protective functions when produced at physiological amounts by gut bacteria. Finally, germ-free animals are defective in the differentiation of specific innate immune cell subsets that are critical for resistance to systemic bacterial infection (Khosravi et al., 2014). Oral administration of microbial ligands restored these defects, illustrating that microbial molecules regulate immune system development at its core -- during hematopoiesis.
Another example of a PAMP that can act as a beneficial signal, not just as a pathogenic trigger, is polysaccharide A (PSA) from the human commensal Bacteroides fragilis (Mazmanian et al., 2005). PSA signals to the immune system through a specific PRR to induce development and function of Foxp3+ Tregs, which prevent inflammation in the gut and in the central nervous system through the effects of interleukin-10 (IL-10) (Ochoa-Reparaz et al., 2010; Round and Mazmanian, 2010). Therefore, not all interactions between microbial molecules and PRRs lead to inflammation and disease (Chu and Mazmanian, 2013). One can therefore view PRRs as sensors not of pathogens, but of microbes in general. Thus, PRRs can be viewed as the immune system’s “eyes” that observe the microbial world, with the potential for beneficial or harmful outcomes to the host dependent on the context of the interaction. This perspective suggests a reconsideration of term PAMP to a more broad terminology proposed by many advocating for the use of microbial associated molecular patterns, or MAMPs (Mackey and McFall, 2006). The situation is somewhat analogous to antibiotics, which are often used in lower concentrations as signaling molecules in natural microbial ecosystems (Linares et al., 2006).
Microbially derived metabolites affect the immune system
The human immune system is significantly impacted by many common microbially-derived molecules. LPS is perhaps the best-studied microbially-produced molecule that impacts the inflammatory status of humans or mice, with over 85,000 articles on this topic in PubMed at the time of this publication. However, other bacterial molecules are also important. Tryptophan, which is an essential amino acid in humans and which we obtain both from our diet and from tryptophan biosynthesis by our microbial symbionts, as well as its microbially-produced breakdown products, plays an important role in the immune system. For example, in mice fed unrestricted tryptophan diets, lactobacilli (typically thought of as anti-inflammatory, although the genus Lactobacillus contains considerable genetic and phenotypic diversity) produce indole-3-aldehyde, which upregulates IL-22 in the host and induces a mucosal response limiting colonization of the gut by the fungal pathogen Candida albicans (Zelante et al., 2013). Another interesting case is the common quorum-sensing molecule N-(3-oxo-dodecanoyl) homoserine lactone, which disrupts NF-κB signaling (Kravchenko et al., 2008). Although no homoserine lactones have yet been detected in the gut, the gut microbiome harbors many genes capable of producing this class of signaling molecules (Swearingen et al., 2013). Part of the challenge in detecting homoserine lactones is that they are rapidly degraded by microbes (Moroboshi et al., 2005; Tinh et al., 2007), and they are only produced under specific conditions even by bacteria that contain the relevant genes (Wang et al., 2006). Better methods for detecting transiently produced and rapidly degraded molecules may be required in order to pinpoint the role of such molecules in shaping the gut immune system.
Microbes also contribute to the alteration of arachidonic acid-derived lipids. Although mostly studied in the context of pathogenesis (Eberhard et al., 2002), many organisms that can metabolize these lipids are found in the normal human microbiota, and alter the amount of arachidonic acid and its downstream metabolites such as prostaglandins and leukotrienes. For example, colonizing germ-free mice with Bacteroides thetaiotaomicron (B. theta) or colonizing them with B. theta and Bifidobacterium longum together increased prostaglandin E2 production (Rath et al., 2012). If insufficient arachidonic acid is present in the diet, it can also be obtained from hydrolysis of membrane lipids, which store arachidonic acid. Arachidonic acid release can be mediated by several mechanisms. Although arachidonic acid release has not yet been correlated with or attributed to the gut microbiota, many microbes, including many members of the normal gut microbiota, have the necessary hydrolases to produce arachidonic acid, and likely perform similar roles as phospholipase A2.
Unknown functions of specialized gene clusters in the microbiome: new natural products?
Finally, many specialized metabolite-producing gene clusters (for example natural products including polyketides, sterols, isoprenoids and non-ribosomally synthesized peptides) are found within the gut microbiome. The functions of these metabolites are almost completely all unknown. Given that many specialized metabolites isolated from other microbes, such as rapamycin, are now in clinical use to control immune-mediated diseases, it is likely that the microbiome has co-evolved numerous gene clusters whose products interact with the immune system directly or indirectly, perhaps in concert with other microbial inhabitants of the gut. Many microbes produce the classes of molecules we have described, although understanding the full phylogenetic spectrum of microbes capable of their production, and the conditions under which they are produced, remain largely uncharacterized especially in the context of the community.
Defining a research program to integrate microbiota, metabolism and immunity
As noted above, many connections have been made between microbes and metabolism, between microbes and immunity, and between metabolites and immunity. However, connections that link all three are scarce at present. An integrated, comprehensive, discovery and hypothesis-driven research program could yield immense dividends in terms of insights into all three fields (microbes, metabolism, immunity) and their fascinating connections. We therefore define approaches that can systematically identify these connections. In particular, these approaches will identify small-molecule phenocopies of what are currently thought to be host immune issues: these issues may or may not include known bacterial components. We should aim to design a pipeline that transcends descriptive cataloging studies (microbiomes, metabolomes, etc), and instead gets to underlying mechanisms that represent the molecular foundations host-microbial symbiosis.
A paradigm for studies linking metabolites, microbes, and the immune system
An example of what this research program might look like is provided by a recent study examining the links between the immune system, the microbiota, and metabolism in a mouse model of autism (Hsiao et al., 2013). Briefly, mice born to mothers subject to maternal immune activation, which simulates viral infection, develop symptoms that resemble autism spectrum disorders (ASD), including lack of social interaction, repetitive behavior, deficits in communication, gut barrier dysfunction, immunological changes and dysbiosis of the microbiome. This combination of symptoms has been reported in ASD.
Importantly, systematic changes in the serum metabolome are observed, including overproduction of a metabolite, 4-ethyl phenyl sulfate (4-EPS), that when individually administered to normal mice recapitulates some of the same phenotypes. Introduction of a probiotic strain of B. fragilis results in lowered 4-EPS production and also ameliorates intestinal and behavioral abnormalities. This combination of immunological manipulation, generation of lead microbes and metabolites through untargeted microbiome and metabolite profiling, and tests in gnotobiotic mice (initially germ-free mice colonized with defined communities of microbes) provides a paradigm for identifying connections -- although we emphasize that studies like this that have been performed to date only scratch the surface of the full range of microbial and metabolic components of the altered response.
Towards an integrative pipeline
A pipeline for identifying novel connections in higher throughput might include a program to screen small-molecule compound libraries for effects on immune cells in tissue culture, identifying metabolites produced by bacteria or by bacterial communities that have been linked to diseases and testing candidate microbes and metabolites, alone or in combination, in gnotobiotic mice. Additionally, screening bacteria in high-throughput in organoid systems with reporter gene assays to identify immunomodulatory effects may be more facile than use of live animals. Essentially, the pipeline needs to include case-control studies to establish that there are microbial or metabolic differences to explain in the first place, spatial mapping and multivalent characterization (microbiome, metabolome, immune repertoire) to generate hypotheses about possible connections, prospective longitudinal studies in humans and preclinical experimental manipulation studies in mice or other model systems to establish causality. This approach may ultimately lead to drug candidates for clinical trials in humans.
This pipeline will be complicated by the bi-directional connections between the microbiome and the immune system. For example, genetic deletion of TLR5, a component of the innate immune system that recognizes bacterial flagellin, results in a substantially altered bacterial community, which in turn (depending on the microbial background) can lead to phenotypes ranging from metabolic syndrome (Vijay-Kumar et al., 2010) to colitis (Carvalho et al., 2012a; Carvalho et al., 2012b), the latter stemming from an inability to regulate pro-inflammatory proteobacteria. Additional research has shown that, rather than producing a single altered microbiome, TLR5-deficient mice produce different microbiota coupled to different phenotypes that can be transmitted vertically within a family (facilitated by the fact that mice are coprophagous) (Ubeda et al., 2012). Microbiome profiling and specific cytokine assays have been performed in these animals, and some of the phenotypes have been transferred to previously germ-free mice by transmitting the altered microbiota. Such phenotypes include, fascinatingly, the behavioral phenotype that causes the TLR5-deficient mice to overeat and thereby develop metabolic syndrome. Unfortunately, however, the combination of microbiome, metabolite and immunological profiling in a longitudinal study design that would be ideal for identifying lead microbes and metabolites has not yet been performed.
Moving beyond the gut
In our discussions thus far, we have mainly focused on the gut and on systemic effects of gut microbes at distal sites, because these have been best studied to date. However, there is intriguing although preliminary evidence that there may be microbes at sites previously thought to be sterile in healthy adults, including the lungs, the adipose tissue, the pancreas, the liver, the amniotic fluid, and even the brain. Studying whether microbes inhabit these sites in gnotobiotic mice, and the immunological and host responses at distal sites, together with the metabolites produced by the host, the bacteria, or the combination of the two, has substantial potential for uncovering fundamentally new mechanisms of disease. Microbial metabolites are dramatically understudied in the context of the human nervous system, immune system, and metabolism. We propose that future studies should focus on interactions between microbial metabolites and the host in various contexts, conditions and diseases.
Finally, most studies to date have treated a given body compartment, such as the skin or the gut, as a homogeneous assemblage of microbes-- yet the spatial and dynamic associations of particular microbes, their metabolites, and components of the immune response are also critical for understanding function at these sites. For example, knowing which bacteria colonize which crypts in the gut (Lee et al., 2013; Pedron et al., 2012), and how they stimulate stem cell proliferation and other factors linked to IBD or cancer, may provide important information not accessible in readouts of the microbiome and/or metabolome through stool samples. In the context of IBD, for example, biomarkers in treatment-naive patients can be more clearly assessed in mucosal biopsies than in the stool (Gevers et al., 2014), the latter being frequently used for microbiome assessments due to its accessibility via non-invasive sampling. Especially given the success of imaging mass spectrometry in understanding how bacteria interact with one another to produce metabolites that they would not produce in isolation in pure culture (Traxler et al., 2013; Yang et al., 2009), as well as interkingdom interactions between bacteria and fungi (Moree et al., 2012), the potential for extending these spatially defined studies into the human body is immense. Furthermore, understanding how microbes and metabolites change over time in various contexts may lead to predictive models for disease diagnosis and patient stratification. The pipeline we propose should reveal how microbes are located spatially: correlating specific metabolite features with the microbiome distribution, then testing whether the molecules that co-localize impact the immune system, will provide an especially powerful mechanism for generating lead compounds for the downstream studies that may extend into the clinic. Unfortunately, this sampling is destructive using current methods; discovery of nondestructive readouts of the microbiome and metabolome would permit longitudinal within-subjects study designs, which, given high variability among individuals, might be important for clinical translation of these discoveries to improve human health.
Conclusion
A number of specific interactions between microbes, metabolites and the immune system have now been discovered: although many of these examples were identified in the context of specific diseases, the broader network of these connections are likely critical for a wide range of normal biological processes in the host. Development of a pipeline that allows far larger-scale discovery of these connections in both health and disease, in humans and animal models, is likely within our reach using technologies that exist today or are in current development. In particular, in the same way that microbe and gene catalogs are now saturating in many populations, saturating the metabolite repertoire and building reference databases that allow matching to known standards will accelerate metabolomic discoveries considerably. Similarly, ‘multi-omics’ approaches in which the microbiome, the metabolome, and the immune repertoire are assessed simultaneously in the same biological specimens will provide considerable advances over current approaches. The prospect of a fundamental advance in our understanding of biology, not just of the parts list but of the interactions among these parts that lead to a healthy supraorganism, and an engineering-level basis for developing technologies to arrest or reverse processes that lead to disease, is exciting and feasible. Overcoming the barriers in communication and philosophies among diverse disciplines, and among different experimental and computational technologies, may catalyze a revolution in understanding the microbe-metabolite-immune connection in numerous ways that will benefit mankind.
Recent research has developed specific connections between particular microbes, metabolites, and host immune processes, but a generalized map of this interaction network is still lacking. Dorrestein et al. describe these connections, and outline how we could move towards a global view.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity. 2012;37:158–170. doi: 10.1016/j.immuni.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, Rudensky AY. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baganz NL, Blakely RD. A dialogue between the immune system and brain, spoken in the language of serotonin. ACS chemical neuroscience. 2013;4:48–63. doi: 10.1021/cn300186b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, Wekerle H, Krishnamoorthy G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479:538–541. doi: 10.1038/nature10554. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J, Knights D, Gajer P, Ravel J, Fierer N, et al. Moving pictures of the human microbiome. Genome biology. 2011;12:R50. doi: 10.1186/gb-2011-12-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho FA, Koren O, Goodrich JK, Johansson ME, Nalbantoglu I, Aitken JD, Su Y, Chassaing B, Walters WA, Gonzalez A, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell host & microbe. 2012a;12:139–152. doi: 10.1016/j.chom.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho FA, Nalbantoglu I, Ortega-Fernandez S, Aitken JD, Su Y, Koren O, Walters WA, Knight R, Ley RE, Vijay-Kumar M, Gewirtz AT. Interleukin-1beta (IL-1beta) promotes susceptibility of Toll-like receptor 5 (TLR5) deficient mice to colitis. Gut. 2012b;61:373–384. doi: 10.1136/gut.2011.240556. [DOI] [PubMed] [Google Scholar]
- Chu H, Mazmanian SK. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nature immunology. 2013;14:668–675. doi: 10.1038/ni.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nature medicine. 2010;16:228–231. doi: 10.1038/nm.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14728–14733. doi: 10.1073/pnas.0904489106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E, Almeida M, Quinquis B, Levenez F, Galleron N, et al. Dietary intervention impact on gut microbial gene richness. Nature. 2013;500:585–588. doi: 10.1038/nature12480. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nature reviews Neuroscience. 2012;13:701–712. doi: 10.1038/nrn3346. [DOI] [PubMed] [Google Scholar]
- Eberhard J, Jepsen S, Pohl L, Albers HK, Acil Y. Bacterial challenge stimulates formation of arachidonic acid metabolites by human keratinocytes and neutrophils in vitro. Clinical and diagnostic laboratory immunology. 2002;9:132–137. doi: 10.1128/CDLI.9.1.132-137.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, et al. The long-term stability of the human gut microbiota. Science. 2013;341:1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N, Lauber CL, Zhou N, McDonald D, Costello EK, Knight R. Forensic identification using skin bacterial communities. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:6477–6481. doi: 10.1073/pnas.1000162107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, Schoenfeld D, Nomicos E, Park M, Program NIHISCCS, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;498:367–370. doi: 10.1038/nature12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, Zhu W, Sartor RB, Boedeker EC, Harpaz N, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflammatory bowel diseases. 2011;17:179–184. doi: 10.1002/ibd.21339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs O, von Mutius E. Prenatal and childhood infections: implications for the development and treatment of childhood asthma. The lancet Respiratory medicine. 2013;1:743–754. doi: 10.1016/S2213-2600(13)70145-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell host & microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Program NCS, Bouffard GG, Blakesley RW, Murray PR, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Group NHW, Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, et al. The NIH Human Microbiome Project. Genome research. 2009;19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science. 2013;341:295–298. doi: 10.1126/science.1235872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamady M, Knight R. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome research. 2009;19:1141–1152. doi: 10.1101/gr.085464.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155:1451–1463. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project C. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, Iwasaki A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi A, Yanez A, Price JG, Chow A, Merad M, Goodridge HS, Mazmanian SK. Gut microbiota promote hematopoiesis to control bacterial infection. Cell host & microbe. 2014;15:374–381. doi: 10.1016/j.chom.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knights D, Parfrey LW, Zaneveld J, Lozupone C, Knight R. Human-associated microbial signatures: examining their predictive value. Cell host & microbe. 2011;10:292–296. doi: 10.1016/j.chom.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Grauer DC, Lehmann M, Meijler MM, Janda KD, Ulevitch RJ. Modulation of gene expression via disruption of NF-kappaB signaling by a bacterial small molecule. Science. 2008;321:259–263. doi: 10.1126/science.1156499. [DOI] [PubMed] [Google Scholar]
- Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature biotechnology. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- Lee SM, Donaldson GP, Mikulski Z, Boyajian S, Ley K, Mazmanian SK. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature. 2013;501:426–429. doi: 10.1038/nature12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(Suppl 1):4615–4622. doi: 10.1073/pnas.1000082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Linares JF, Gustafsson I, Baquero F, Martinez JL. Antibiotics as intermicrobial signaling agents instead of weapons. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:19484–19489. doi: 10.1073/pnas.0608949103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Z, Li Z, Liu X, Cheng Y, Luo Y, Tong X, Yuan L, Wang Y, Sun J, Li L, Xiang C. Altered fecal microbiota composition associated with food allergy in infants. Applied and environmental microbiology. 2014;80:2546–2554. doi: 10.1128/AEM.00003-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey D, McFall AJ. MAMPs and MIMPs: proposed classifications for inducers of innate immunity. Molecular microbiology. 2006;61:1365–1371. doi: 10.1111/j.1365-2958.2006.05311.x. [DOI] [PubMed] [Google Scholar]
- Martin FP, Dumas ME, Wang Y, Legido-Quigley C, Yap IK, Tang H, Zirah S, Murphy GM, Cloarec O, Lindon JC, et al. A top-down systems biology view of microbiome-mammalian metabolic interactions in a mouse model. Molecular systems biology. 2007;3:112. doi: 10.1038/msb4100153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122:107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Moree WJ, Phelan VV, Wu CH, Bandeira N, Cornett DS, Duggan BM, Dorrestein PC. Interkingdom metabolic transformations captured by microbial imaging mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:13811–13816. doi: 10.1073/pnas.1206855109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroboshi T, Ebata A, Nakazawa S, Kato N, Ikeda T. N-acyl Homoserine Lactone-Producing or -Degrading Bacteria Isolated from the Intestinal Flora of Ayu Fish (Plecoglossus altivelis) Microbes and Environments. 2005;4:264–268. [Google Scholar]
- Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L, Henrissat B, Knight R, Gordon JI. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332:970–974. doi: 10.1126/science.1198719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson JK, Holmes E, Wilson ID. Gut microorganisms, mammalian metabolism and personalized health care. Nature reviews Microbiology. 2005;3:431–438. doi: 10.1038/nrmicro1152. [DOI] [PubMed] [Google Scholar]
- Ochoa-Reparaz J, Mielcarz DW, Wang Y, Begum-Haque S, Dasgupta S, Kasper DL, Kasper LH. A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal immunology. 2010;3:487–495. doi: 10.1038/mi.2010.29. [DOI] [PubMed] [Google Scholar]
- Pedron T, Mulet C, Dauga C, Frangeul L, Chervaux C, Grompone G, Sansonetti PJ. A crypt-specific core microbiota resides in the mouse colon. mBio. 2012;3 doi: 10.1128/mBio.00116-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- Quehenberger O, Armando AM, Brown AH, Milne SB, Myers DS, Merrill AH, Bandyopadhyay S, Jones KN, Kelly S, Shaner RL, et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. Journal of lipid research. 2010;51:3299–3305. doi: 10.1194/jlr.M009449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Rath CM, Alexandrov T, Higginbottom SK, Song J, Milla ME, Fischbach MA, Sonnenburg JL, Dorrestein PC. Molecular analysis of model gut microbiotas by imaging mass spectrometry and nanodesorption electrospray ionization reveals dietary metabolite transformations. Analytical chemistry. 2012;84:9259–9267. doi: 10.1021/ac302039u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12204–12209. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife. 2013;2:e01202. doi: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloissnig S, Arumugam M, Sunagawa S, Mitreva M, Tap J, Zhu A, Waller A, Mende DR, Kultima JR, Martin J, et al. Genomic variation landscape of the human gut microbiome. Nature. 2013;493:45–50. doi: 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Simons K. Lipidomics: coming to grips with lipid diversity. Nature reviews Molecular cell biology. 2010;11:593–598. doi: 10.1038/nrm2934. [DOI] [PubMed] [Google Scholar]
- Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J, Kau AL, Rich SS, Concannon P, Mychaleckyj JC, et al. Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science. 2013a;339:548–554. doi: 10.1126/science.1229000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013b;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swearingen MC, Sabag-Daigle A, Ahmer BM. Are there acyl-homoserine lactones within mammalian intestines? Journal of bacteriology. 2013;195:173–179. doi: 10.1128/JB.01341-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinh NT, Asanka Gunasekara RA, Boon N, Dierckens K, Sorgeloos P, Bossier P. N-acyl homoserine lactone-degrading microbial enrichment cultures isolated from Penaeus vannamei shrimp gut and their probiotic properties in Brachionus plicatilis cultures. FEMS microbiology ecology. 2007;62:45–53. doi: 10.1111/j.1574-6941.2007.00378.x. [DOI] [PubMed] [Google Scholar]
- Traxler MF, Watrous JD, Alexandrov T, Dorrestein PC, Kolter R. Interspecies interactions stimulate diversification of the Streptomyces coelicolor secreted metabolome. mBio. 2013;4 doi: 10.1128/mBio.00459-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, Blanchard C, Junt T, Nicod LP, Harris NL, Marsland BJ. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nature medicine. 2014;20:159–166. doi: 10.1038/nm.3444. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubeda C, Lipuma L, Gobourne A, Viale A, Leiner I, Equinda M, Khanin R, Pamer EG. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. The Journal of experimental medicine. 2012;209:1445–1456. doi: 10.1084/jem.20120504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342:971–976. doi: 10.1126/science.1240537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Cai T, Weng M, Zhou J, Cao H, Zhong Z, Zhu J. Conditional production of acyl-homoserine lactone-type quorum-sensing signals in clinical isolates of enterobacteria. Journal of medical microbiology. 2006;55:1751–1753. doi: 10.1099/jmm.0.46756-0. [DOI] [PubMed] [Google Scholar]
- Yang YL, Xu Y, Straight P, Dorrestein PC. Translating metabolic exchange with imaging mass spectrometry. Nature chemical biology. 2009;5:885–887. doi: 10.1038/nchembio.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D’Angelo C, Massi-Benedetti C, Fallarino F, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39:372–385. doi: 10.1016/j.immuni.2013.08.003. [DOI] [PubMed] [Google Scholar]