

Abstract
Resistance arises quickly during chemotherapeutic selection and is particularly problematic during long-term treatment regimens such as those for tuberculosis, HIV infections, or cancer. Although drug combination therapy reduces the evolution of drug resistance, drug pairs vary in their ability to do so. Thus, predictive models are needed to rationally design resistance-limiting therapeutic regimens. Using adaptive evolution, we studied the resistance response of the common pathogen Escherichia coli to 5 different single antibiotics and all 10 different antibiotic drug pairs. By analyzing the genomes of all evolved E. coli lineages, we identified the mutational events that drive the differences in drug resistance levels and found that the degree of resistance development against drug combinations can be understood in terms of collateral sensitivity and resistance that occurred during adaptation to the component drugs. Then, using engineered E. coli strains, we confirmed that drug resistance mutations that imposed collateral sensitivity were suppressed in a drug pair growth environment. These results provide a framework for rationally selecting drug combinations that limit resistance evolution.
INTRODUCTION
Bacteria inevitably evolve antibiotic resistance in response to prolonged exposure to drugs; consequently, antibiotic resistance always follows the introduction of new antibiotics (1)—a trend that has led to the emergence of virtually untreatable multidrug-resistant bacteria such as extensively drug-resistant tuberculosis and carbapenem-resistant Escherichia coli and Klebsiella pneumoniae strains (2, 3). Drug resistance develops either through horizontal acquisition of resistance genes or through mutations in the bacterial genome. In the latter case, the length of treatment and patient compliance greatly influence resistance evolution, because prolonged subinhibitory drug concentrations select for resistant bacteria (4). A similar pattern applies to viral resistance, which evolves quickly because of a high abundance of viral particles in the host and the high error rate associated with viral replication (5).
One approach to counteract drug resistance development in bacteria and viruses is with combination drug therapy. This approach first proved its success in the late 1940s, when the combination of streptomycin and para-aminosalicylic acid was shown to markedly reduce evolution of resistant Mycobacterium tuberculosis compared to streptomycin monotherapy (6). Similarly, the use of combination therapy to treat HIV infections has been successful in reducing drug resistance, resulting in increased life expectancy of HIV patients (7). Furthermore, combination therapy is being used to combat drug resistance in cancer, which, like tuberculosis, requires months-long treatment with chemotherapeutic agents (8).
Other reasons for using drug combinations include synergistic therapeutic effects and increased spectrum of activity (9). Thus, the search for synergy has dominated the field of antimicrobial combination treatments for many decades, with successful examples including the sulfamethoxazole-trimethoprim combination and simultaneous treatment with a β-lactam antibiotic and a β-lactamase inhibitor (10-12). Synergy, however, can have two conflicting effects on resistance: it reduces evolution of resistance because it clears the infection faster, thereby limiting the time window available for resistant mutations to arise, but it also increases the selective advantage of single drug–resistant mutants (13). When competition for resources is strong, the latter effect can dominate (13); indeed, under these conditions, drug combinations that are antagonistic (that is, when the combination is less potent than the sum of its components) have been shown to limit evolution of resistance (14-16). The advantage of antagonistic combinations over synergistic combinations is the result of a reduced fitness gain, when bacteria develop resistance to antagonistic combinations compared to developing resistance to synergistic combinations (14-16).
In addition to drug interactions (synergy and antagonism), collateral sensitivity among the drugs is also believed to play a major role in driving the evolution of resistance against drug combinations. Collateral sensitivity and resistance, also known as cross-resistance, occurs when mutations conferring resistance to one drug increase or decrease sensitivity to another drug. The study of collateral susceptibility changes was pioneered by Szybalski and Bryson in the early 1950s and has since been reported for many different drugs and bacterial species, as well as for virus, cancer cell lines, and plants (17-28). We have recently shown that collateral sensitivity can be used to rationally design drug cycling regimens that limit the evolution of resistance (29).
Although the combined impact of both drug interactions and cross-resistance on selection in multidrug environments has been evaluated (16), it remains unclear which of these factors is more static over evolutionary times and thereby has a more dominant impact on long-term evolution of drug resistance. Here, we investigate how collateral resistance and interactions between component drugs affect the development of resistance to drug combinations over the long term. We show that resistance development is strongly reduced by collateral sensitivity. In contrast, the interactions between drugs changed with resistance development and thereby have a weaker effect on long-term adaptation. We further uncover the underlying genetic basis of adaptation and show examples of condition-specific selection for and against drug resistance mutations.
RESULTS
Resistance evolution
To investigate how combinations of antibiotics affect the evolution of resistance, we evolved E. coli in the presence of 10 antibiotic drug pair combinations and their single-drug components, totaling 15 drug conditions (Figs. 1 and 2, A to E, and fig. S1) (see Materials and Methods for details). Together, these drugs cover three major clinically relevant antibiotic targets in E. coli: DNA replication [ciprofloxacin (Cip)], mRNA translation [amikacin (Amk), tetracycline (Tet), and chloramphenicol (Chl)], and cell wall biosynthesis [piperacillin (Pip)]. In addition, the group includes both bactericidal drugs (Cip, Amk, and Pip) and bacteriostatic drugs (Tet and Chl) (Fig. 2, A to E). For each of the 15 drug conditions, three replicate bacterial lineages (A, B, and C) were evolved in parallel to evaluate the robustness of the evolutionary responses (Fig. 1).
Fig. 1. Experimental setup.
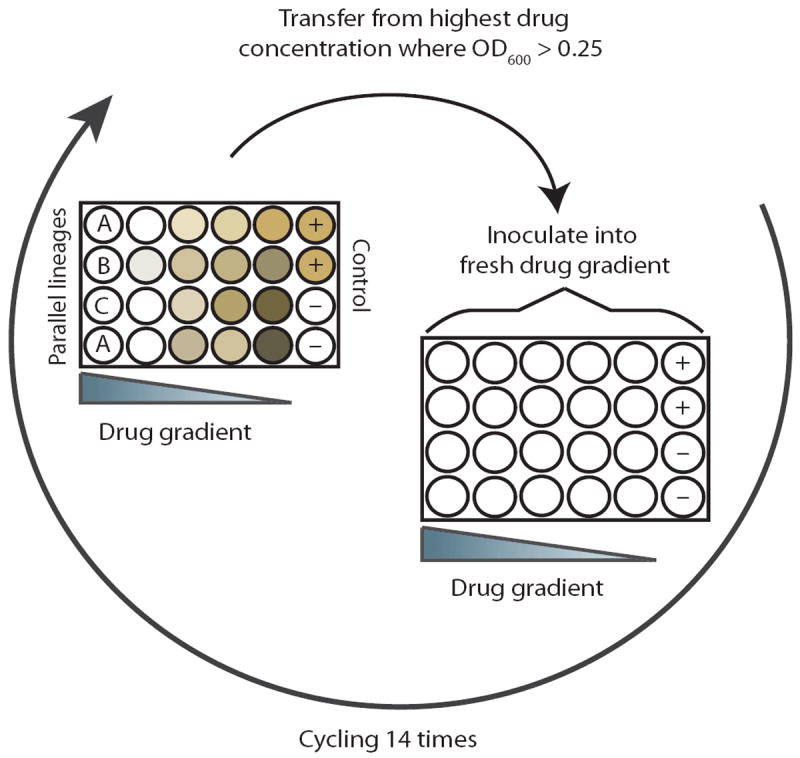
The in vitro evolution experiment was conducted in 24-well plates with drug gradients across the columns. The last column contained positive and negative controls. Every 20 hours, cells from the wells with the highest drug concentrations that displayed an OD600 >0.25 were diluted 1:40 into a freshly prepared drug gradient. Parallel-evolved lineages were inoculated across rows and labeled with the letters A, B, and C.
Fig. 2. Relative increases in IC90 values.
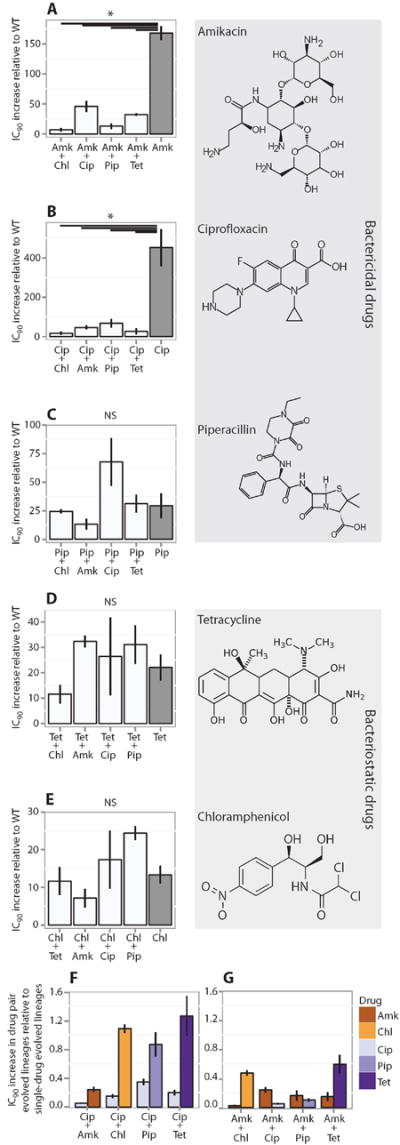
IC90 (means ± SEM, n = 3 biological replicates) of the evolved lineages relative to IC90 of the ancestral E. coli MG1655. (A to E) Data for lineages evolved to the single drugs Amk, Cip, Pip, Tet, and Chl, as well as lineages evolved to each of the 10 possible pairwise combinations. Only combinations containing either Amk (A) or Cip (B) showed significantly reduced relative increases in IC90 compared to the corresponding single-drug evolved lineages (*P < 0.05, ANOVA, followed by Tukey test). WT, wild type; NS, not significant. (F and G) Relative increase in IC90 for the individual components in Cip-containing combinations (F) and Amk-containing combinations (G) measured as relative increases in IC90 of the individual components in the drug pair evolved lineages divided by the relative IC90 increase in the single-drug evolved lineages (means ± SEM, n = 3 biological replicates). Only Amk-containing combinations limit resistance increase of both drugs in the drug pair.
The experiment was carried out in a twofold dilution gradient in a 24-well plate. After 20 hours of incubation, the culture exposed to the highest drug concentration that had an optical density at 600 nm (OD600) greater than 0.25 (corresponding to 109 colony-forming units/ml) was diluted 40 times into a freshly prepared drug gradient (Fig. 1). As resistance increased, the drug gradients were scaled to maintain a selective pressure. In total, 14 consecutive passages were made, corresponding to ~75 generations or 1011 cumulative cell divisions per experiment assuming an average endpoint OD600 of 0.6 (30). The experiment was ended when all lineages that were evolved to a single drug (called single-drug evolved lineages) had reached their clinical breakpoint [as defined by the The European Committee on Antimicrobial Susceptibility Testing (EUCAST)] (see Materials and Methods). This approach ensured that all lineages were evolved throughout the same period of time, allowing us to investigate how the evolution of resistance against one drug is affected by the presence of another drug.
To measure the increase in drug resistance, we performed micro-broth dilution inhibition assays (Materials and Methods). We tested the 15 single-drug evolved lineages against all single drugs and the 30 drug pair evolved lineages against the drug combination they had been evolved to as well as its component drugs. All measurements were performed in triplicate. Using the normalized OD600 measurements, we plotted a dose-response curve and fitted a four-parameter (Hill equation) dose-response curve to the data points (Materials and Methods, fig. S2, and table S1). Traditional dose-response analyses, such as receptor-antagonist studies, normally use 50% inhibition levels [median inhibitory concentration (IC50)] to compare drug effects. However, because we want to investigate changes in the minimal drug concentration required to inhibit bacterial growth [minimum inhibitory concentration (MIC)], we measured the drug concentration at a 90% inhibition level (IC90), which resembles the MIC but can be more precisely determined through fitting of dose-response curves. To investigate if there were any major differences in calculations based on different levels of growth inhibition [that is, IC50, IC90, and MIC (defined as the drug concentration leading to 95% inhibition in our dose-response curves)], we compared calculations of the fold change in drug tolerance based on IC50, IC90, and MIC estimates (fig. S3). The data revealed a very strong linear correlation between the different IC levels, indicating that there are no major differences between calculations and conclusions based on IC50, IC90, and MIC.
The overall IC90 increase for the different drug pairs and their component drugs varied from 15- to more than 300-fold in the various evolved E. coli lineages relative to the wild-type strain (Fig. 2, A to E). For the single-drug evolved lineages, the greatest IC90 increase was seen in those evolved to Cip (Fig. 2B), for which two of the three lineages had more than a 300-fold IC90 increase compared to the wild type. This is consistent with previous findings showing that high-level Cip resistance only requires a few mutations in the specific target genes gyrA, parC, and parE (31). In contrast, lineages evolved to Chl (Fig. 2E) only increased their IC90 by 15-fold, underscoring the large differences in easily accessible high-level resistance mutations among the drugs used in the study.
For the drug pair evolved lineages, we observed IC90 increases between 2- and 100-fold. Notably, when comparing single-drug evolved lineages of Pip, Tet, and Chl to drug pair lineages containing one of these drugs (Fig. 2, C to E), the presence of another drug did not significantly influence the relative increase in IC90 [P > 0.05, analysis of variance (ANOVA), followed by Tukey test]. This illustrates that not all drug combinations reduce evolution of resistance relative to the component drugs. In contrast, for lineages evolved to drug pairs containing Cip or Amk, the presence of a second drug significantly reduced the relative IC90 increases against the drug pair compared to the relative IC90 increases of the lineages evolved to either Cip or Amk alone (Fig. 2, A and B). This reduction could either be the result of the second drug limiting an otherwise large increase in Cip or Amk resistance or a genuine reduction in the IC90 increases of both drugs in the combination.
To investigate these possible mechanisms, we compared the IC90 increases for the individual drugs in the drug pair evolved lineages to the IC90 increases of the same drugs in the single-drug evolved lineages (Fig. 2, F and G). In lineages evolved to drug pairs that contained Cip, the IC90 increases for Cip were lower compared to the Cip-only evolved lineages. In contrast, the IC90 increases for the other drugs in the Cip combinations were comparable to those of their corresponding single-drug evolved lineage (Fig. 2F). This reveals that the reduced evolution of resistance in the lineages evolved to drug pairs that contained Cip was the result of the non-Cip component preventing high-level Cip exposure and consequently high-level resistance. In contrast, lineages evolved to drug pairs that contained Amk showed a substantial reduction in the IC90 increase for both Amk and the other drug in the combination, highlighting the fact that in Amk-containing drug pairs, evolution of resistance was restricted for both drugs (Fig. 2G).
Next, we investigated whether the level of resistance evolution to a drug combination could be predicted from the level of resistance evolution to its component drugs. We compared the IC90 increases of the drug pairs to the increases of the slowest- and fastest-evolving components, as well as to the average IC90 increases of the component drugs. We observed no apparent correlation between the resistance increases in the drug pairs and those of the component drugs (Fig. 3, A to C), suggesting that when drugs are used in combination, other factors have an impact on the evolution of resistance toward drug combinations.
Fig. 3. Increase in IC90 for drug pairs compared to individual components.
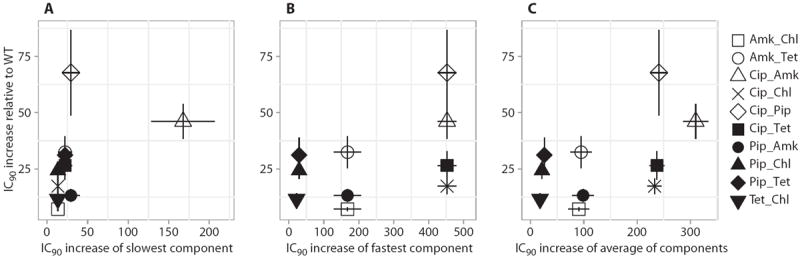
(A to C) Drug pair IC90 increases relative to the IC90 increases of the slowest-evolving component (A), the fastest-evolving component (B), and the average of the two (C). Error bars depict SEMs. The IC90 increases for the drug pairs are not correlated to the increases for the component drugs (P > 0.05, Bonferroni-corrected Spearman correlation; n = 10).
Lack of correlation between fractional inhibitory concentration indices and levels of resistance evolution
Drug interactions can be classified as being synergistic, additive, or antagonistic and can be assessed with various mathematical models (32). A commonly used model is the Loewe additivity model, which describes drug interactions as the sum of the inhibitory concentration of each drug in a combination compared to the drug alone and is reported at a given effect level [for example, 90% growth inhibition (IC90)]. The result is a fractional inhibitory concentration index (FICI) that describes the drug interactions (see Materials and Methods). A main advantage of the Loewe additivity model compared to other models, such as the Bliss independence model, is that it assumes that a drug cannot be synergistic with itself. This is particularly relevant when considering drug combinations that share the same target, for example, the ribosome. Consequently, in a thought experiment in which a drug is combined with itself, the FICI value would be 1, indicating that the interaction is additive (that is, that the effect of the drug combination is the sum of the effect of the individual drugs). In contrast, a combination of two drugs resulting in a FICI of less than 1 indicates that the drugs interact synergistically, meaning that the inhibitory effect of the combination is greater than the sum of the effects of the individual components. Conversely, an index above 1 indicates an antagonistic interaction in which the drug combination is less potent than the sum of the individual components.
Recently, it was shown that growth rate adaptation in a growth medium that contains binary combinations of antibiotics is accelerated for synergistic and reduced for antagonistic drug combinations (15). Therefore, we investigated whether the drug interaction profile also correlated with the increase in resistance during long-term evolution. We calculated the FICI for each drug pair and compared that to the degree of resistance evolution in the drug pair evolved lineages. To measure the degree of resistance evolution, we introduce the “evolvability index.” It compares the resistance increase of the individual drugs across single-drug and drug pair evolved lineages, thereby describing how the evolution of resistance against a drug is affected by the presence of the other drug. Calculation of the evolvability index is possible because all the lineages were evolved over the same period of time. Specifically, it is obtained by first calculating the IC90 increase for each drug in a drug pair evolved lineage and dividing it by the IC90 increase of the corresponding single-drug evolved lineage. Finally, the average of the ratios for each drug in the drug pair is calculated (Materials and Methods). Thus, an evolvability index of 1 implies that resistance against the component drugs evolves to the same extent in the drug combination as in the individual single-drug evolved lineages, whereas a value greater than 1 indicates that resistance evolves to a larger extent in the drug combination, whereas a value less than 1 indicates that resistance evolves to a lesser extent in the drug combination, relative to the individual drugs. The evolvability index allows direct comparison of the resistance-reducing capacity of different drug combinations in spite of differences in the absolute capacity to evolve resistance against the component drugs.
The drug combinations used in our study cover all types of drug interactions. For instance, Amk + Tet interact synergistically, Cip + Pip interact additively, and Cip + Chl display an antagonistic interaction (Fig. 4A). In contrast, the evolvability index displays a bimodal distribution with a group evolving less resistance than their corresponding single-drug evolved lineages and another group evolving resistance to the same level as the lineages evolved to their component drugs (Fig. 4B).
Fig. 4. Drug interactions and evolution of resistance.
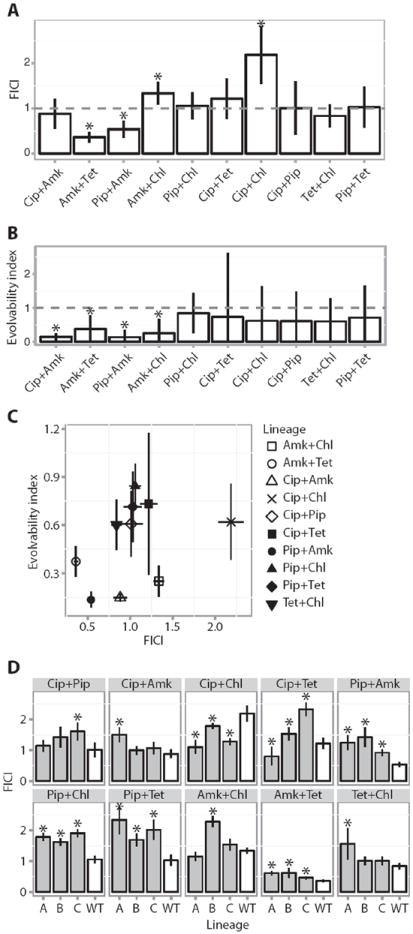
(A) Mean FICI of the various drug pairs tested against WT E. coli MG1655 (Materials and Methods). Error bars depict a 95% confidence interval (CI) (n = 3 biological replicates). Asterisks indicate a FICI significantly different from additivity (FICI = 1). (B) Evolvability index of the various drug pairs calculated as the average of the fold increase in IC90 for the individual drugs in a drug pair evolved lineage relative to the single-drug evolved lineages (Materials and Methods). Error bars depict 95% CI (n = 3 biological replicates). Asterisks indicate a significantly different level of resistance evolution relative to single-drug evolved lineages (evolvability index = 1). (C) Evolvability as a function of FICI. The plot shows that the evolvability index does not correlate with the FICI (P > 0.05, Spearman correlation; error bars depict SEMs). (D) Change in FICI during resistance evolution. Comparison of FICI for the WT MG1655 strain (white) and the three parallel evolved lineages (A, B, and C) for each drug pair (gray). Error bars depict SEMs. Asterisks indicate significant changes in FICI in the evolved lineages compared to the WT FICI (Student’s t test).
We find that the evolvability index of a drug combination was not correlated with the drug interaction profile (Fig. 4C) (P > 0.05, Spearman correlation). This suggests that during long-term evolution, drug interactions do not significantly influence evolution of resistance against drug combinations. To investigate whether other measures of resistance evolution correlated with drug interactions, we compared the FICI to resistance evolution quantified as the drug pair IC90 increase relative to the slowest and fastest components, and none of these were significant (P > 0.05, Bonferroni-corrected Spearman correlation) (fig. S4).
Modulation of drug interactions by evolution
In many cases, the nature of drug interactions can be specific to a particular organism and can vary substantially between organisms. Indeed, drug interactions do not result just from the properties of the drugs in a combination, but are a result of a complex interplay between the drug combination and the cellular network state of a particular organism (33). If the drug interactions observed in our wild-type E. coli strain are affected by the genotype, an outcome one would expect is that as the lineages mutate to become resistant, the drug interaction profile would change. This has been demonstrated previously for the synergistic combination Tet plus erythromycin (34). To explore whether this is a general principle, we compared the FICI values of the evolved lineages to the FICI values of the wild-type lineage (Fig. 4D). Inmost cases, there was a significant shift in the drug interaction profiles as the lineages evolved resistance (P > 0.05, Student’s t test). For example, Pip + Amk shifted from a synergistic interaction profile toward a more additive interaction profile, whereas Cip + Chl shifted from an antagonistic interaction profile toward a more additive profile. In all cases, except Cip + Tet, lineages evolved to the same drug pair exhibited a FICI shift in the same direction, highlighting that the changes in drug interaction profiles during resistance evolution are consistent (Fig. 4D). This dependency of drug interactions on genotype conditioned by resistance evolution highlights the complexity of assessing the factors involved in resistance evolution against drug combinations.
Limiting resistance evolution by collateral sensitivity
The evolvability index divided the drug combinations into two groups—one with drug pairs that limited evolution of resistance relative to their constituent drugs and one with drug pairs that did not (Fig. 4B). We showed recently that collateral sensitivity cycling can be used to counterselect a resistant subpopulation (29). We therefore speculated that the differences in the evolvability indices of the drug combinations could result from collateral resistance and collateral sensitivity occurring during adaptation to the different drugs. To investigate this hypothesis, we determined the IC90 values of the single-drug evolved lineages against all antibiotics to which they were not evolved (Fig. 5A). The results showed many examples of both collateral resistance and collateral sensitivity, all in agreement with previous findings (28, 29).
Fig. 5. Correlation between collateral IC90 change and evolvability.
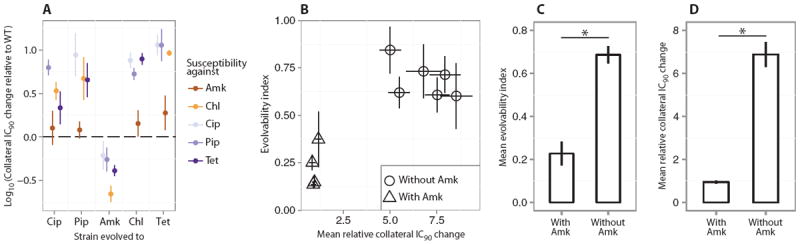
(A) Collateral resistance or sensitivity for the single-drug evolved lineages tested against the four drugs to which they had not been evolved. The figure shows the relative IC90 change calculated as IC90[drug A]drug B evolved/IC90[A]WT. Error bars depict SEMs (n = 3 biological replicates). For most lineages, collateral resistance is observed; however, the Amk-evolved lineages display a high degree of collateral sensitivity. (B) Correlation between the collateral IC90 changes in the single-drug evolved lineages and the evolvability indices of the drug pair evolved lineages. The mean collateral IC90 changes in the single-drug evolved lineages were calculated as {IC90[drug A]drug B evolved/IC90[drug A]WT + IC90[drug B]drug A evolved/IC90[drug B]WT}/2 (Materials and Methods). The evolvability index was calculated as {IC90[drug A]drug AB evolved/IC90[drug A]drug A evolved + IC90[drug B]drug AB evolved/IC90[drug B]drug B evolved}/2 (Materials and Methods). Triangles represent combinations that contained Amk, and circles represent combinations without Amk. Error bars depict SEMs (n = 3 biological replicates). (C) Mean evolvability indices of drug pairs containing Amk and drug pairs not containing Amk. Error bars represent SEMs across the various lineages. Amk-containing drug pairs evolve significantly less resistance than drug pairs without Amk, relative to their component drugs (*P < 0.05, Mann-Whitney test). (D) Average collateral IC90 changes for each pairwise combination of the single-drug evolved lineages, stratified by pairs with and without the Amk. Error bars represent SEMs (n = 3 biological replicates). The degree of collateral IC90 changes was significantly lower for combinations of single-drug evolved lineages that contained Amk (*P < 0.05, Mann-Whitney test).
The Amk-evolved lineages displayed a high degree of collateral sensitivity, corroborating recent work showing that mutations that led to Amk resistance conferred increased sensitivity to the other antibiotics (28). In addition, Amk collateral resistance was not present among the non–Amk-evolved lineages, thus revealing that the evolution of Amk resistance shares little overlap with evolution of resistance to the other drugs. Collateral resistance against all drugs except Amk was observed for all the remaining lineages. The Chl- and Tet-evolved lineages especially displayed an increased resistance to drugs to which they had not been evolved (apart from Amk), with an up to 20-fold collateral increase in IC90 (Fig. 5A).
To examine whether collateral resistance and collateral sensitivity affect the evolution of resistance, we compared the evolvability indices of the drug pair evolved lineages to the average collateral IC90 changes among the constituent single-drug evolved lineages (Fig. 5B). In agreement with the intuitive expectation that collateral-sensitivity interactions between component drugs would reduce evolution of resistance toward drug combinations, our data showed that low-average collateral IC90 changes within the single-drug evolved lineages were associated with reduced evolution of resistance in the drug pair evolved lineages. Notably, the Amk-containing combinations both displayed significantly lower degrees of collateral IC90 change and have significantly lower evolvability indices compared to the non–Amk-containing drug pairs (Fig. 5, C and D) (P < 0.05, Mann-Whitney test).
Our data suggest that the collateral IC90 change between two single-drug evolved lineages influences the evolution of resistance against drug combinations. Consequently, knowledge about the evolutionary changes leading to resistance and sensitivity against individual drugs can be used to design drug combinations that limit the evolution of resistance.
Genetic determinants underlying evolution of resistance
To explore the evolutionary events underlying resistance evolution, we sequenced the genomes of isolates from all evolved lineages as well as wild-type E. coli MG1655 (table S2). In total, 46 strains were sequenced. By comparing the genome of the wild-type strain to the evolved lineages, we identified single-nucleotide polymorphisms (SNPs) and insertions and deletions (INDELs) that characterized the evolutionary endpoint (Fig. 6, A to C, and table S3) (Materials and Methods).
Fig. 6. Sequencing the evolved lineages.
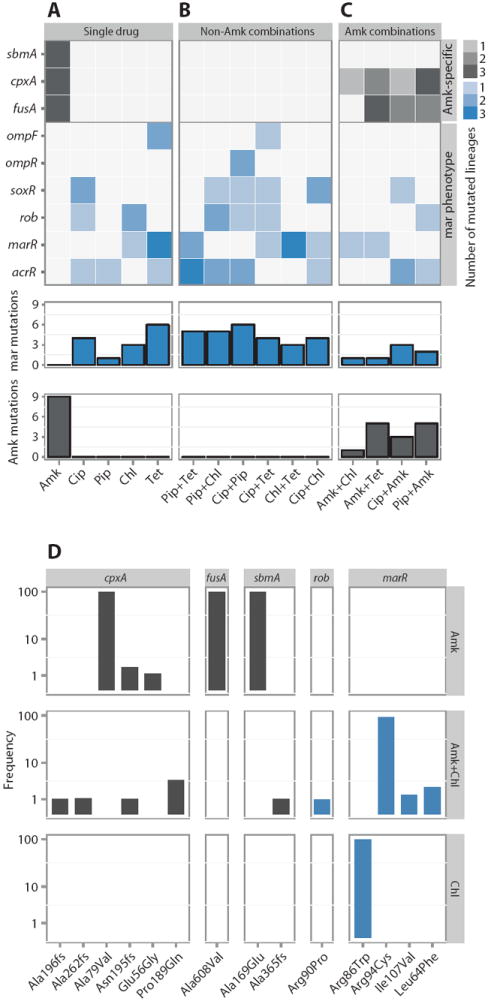
(A to C) Heat map depicting mutations (SNPs and INDELs) in the 45-endpoint sequenced lineages known to be involved in Amk and mar resistance. Mutations in the single-drug evolved lineages (A), mutations found in lineages evolved to drug pairs without Amk (B), and lineages evolved to drug pairs with Amk (C). The three parallel-evolved lineages are collapsed by drug condition. The gene targets are grouped by the phenotypic characteristics (gray, Amk-specific mutations; blue, mutations known to confer the mar phenotype). The legend indicates the number of parallel lineages that contained a mutation in the specific target. The bars below each drug condition summarizes the mar- and Amk-specific mutations, respectively. (D) Population frequency sequencing. Total DNA from the populations evolved to Amk, Chl, and Amk + Chl (drug conditions are noted in the vertical strip text) was sequenced, and frequencies of the individual SNPs/INDELs were calculated (loci are noted in the horizontal strip text). SNPs/INDELs in genes linked to Amk resistance (fusA, sbmA, and cpxA) were fixed in the Amk-evolved population but could not be fixed when Chl was present. In contrast, SNPs in marR were observed in both the Chl and Amk + Chl evolved lineages. These findings corroborate the single-isolate sequencing results.
We identified similar mutational profiles in the lineages evolved to the drugs Pip, Chl, Tet, and Cip (Fig. 6A). These drugs all selected for mutations in acrR or ompF/R and the marR, soxR, and rob genes. Mutations in these genes occurred at high frequencies, with 76% of all lineages having a mutation in at least one of these genes (Fig. 6A). All of these genes are implicated in adaptive antibiotic resistance and have been found to contribute to resistance in clinical isolates (35-43). The AcrR protein regulates expression of the gene encoding the AcrAB multidrug efflux pump, which exports a wide range of antibiotics, including Tet, Cip, ampicillin, and Chl (39). In contrast, the gene products of marR, soxR, and rob each regulate a wide range of genes (44-46). They share a common regulon core that includes genes known to result in the multiple antibiotic resistance (mar) phenotype, which is characterized by up-regulation of AcrAB and simultaneous down-regulation of the outer-membrane porin OmpF, a protein that increases the membrane permeability. Together, these modulations result in broad-spectrum antibiotic resistance (47). Mutations in marR and deletion of ompF and ompC, or mutations in the gene that encodes their common regulator ompR, can cause carbapenem resistance in non–carbapenemaseproducing clinical isolates of E. coli, either directly or in synergy with extended-spectrum β-lactamases, highlighting the clinical relevance of this mutational resistance (48, 49). It should be noted that AcrAB does not export Amk, which provides an explanation both for the low level of collateral resistance to Amk and for why no mutations in the Amk-evolved lineages were involved in acrAB regulation (Figs. 5A and 6A) (50).
The Amk-evolved lineages had a mutational profile that was distinct from those of the other antibiotics included in the study (Fig. 6A); all had mutations in the fusA, cpxA, and sbmA genes (Fig. 6A). fusA encodes elongation factor G and is named for its involvement in fusidic acid resistance. However, mutations in this gene have also been reported to confer aminoglycoside resistance (34, 51, 52). In addition, a fusA mutant has also been shown to result in low-level collateral sensitivity to other antibiotics, including β-lactam drugs and Chl (53). CpxA is a part of the CpxRA two-component system involved in sensing and responding to envelope stress (54). CpxA works via CpxR to activate genes whose products participate in protein folding and degradation and responds to aminoglycoside-induced stress, which is known to produce mistranslated membrane proteins (34, 55, 56). SbmA is a less well-characterized inner membrane protein reported to be involved in resistance to peptide antibiotics (57). However, SbmA is also involved in hypersensitivity to Tet, supporting the notion that mutations in this gene, while imparting Amk resistance, also cause collateral sensitivity (58).
In the phenotypic characterization, we observed that drug combinations that exhibited collateral sensitivity evolved less resistance compared to combinations of drugs with no collateral-sensitivity interactions. This correlation was also reflected in the accumulation of resistance mutations in the drug pair evolved lineages. In the lineages evolved to drug combinations wherein both of the component drugs select for the mar phenotype, we found that mutations involved in the establishment of this phenotype were highly abundant (Fig. 6B). In contrast, in lineages evolved to drug pairs containing Amk, which displayed a high degree of collateral sensitivity, mutations that were otherwise commonly found in the single-drug evolved lineages were less frequent or even completely absent. For instance, none of the lineages evolved to the Amk-containing drug pairs had the otherwise ubiquitous sbmA mutation. Similarly, mutations in fusA and cpxA, which were also consistently found in all of the Amk-evolved lineages, were found in only 7 of the 12 (58%) lineages evolved to Amk-containing combinations (Fig. 6C). Likewise, the mar mutations, which were, on average, present with 1.5 mutations per non-Amk drug pair evolved lineage, were found in only 7 of 12 (58%) of the Amk-containing drug pair evolved lineages (Fig. 6, B and C). These findings suggest that collateral sensitivity prevents the simultaneous acquisition of the ubiquitous Amk mutations and mar mutations.
To investigate how accurately the sequenced isolates represented the evolved populations, we performed whole-population sequencing on DNA extracted from lineages evolved to Amk, Chl, and Amk + Chl. We found that for the Amk-evolved lineages, mutations in cpxA, fusA, and sbmA were completely fixed in the population (Fig. 6D). This observation agrees with the single-isolate sequencing (Fig. 6A). Likewise, for the Chl-evolved lineage, a marR mutation was completely fixed in the population, again in agreement with the single-isolate sequencing results (Fig. 6, A and D, and table S3). For the Amk + Chl–evolved lineage, a marR mutation was completely fixed in the population, which was also the case for the sequenced single isolate (Fig. 6, C and D, and table S3). In contrast, the canonical Amk mutations were absent or present only at low frequencies in the drug pair evolved lineages. The most abundant Amk-specific mutation was one present in cpxA at just less than 4%; similarly, mutations in sbmA were found at only 1%, whereas no fusA mutations were detected (Fig. 6D). Overall, these population-sequencing results show that the general mutational patterns of the single-isolate sequencing fits with the population frequencies.
Condition-specific selection and counter-selection of resistant mutants
To explore whether the mutational patterns observed in our evolved populations were a result of selection and counterselection by different drug treatments, we constructed a set of mutant strains that harbored the individual mutations identified in our evolution experiment. We constructed single-mutant strains containing each of themutated alleles of cpxA, fusA, and sbmA identified in the Amk-evolved lineage B (Materials and Methods). These alleles all represent mutations that appeared incompatible with the Amk + Chl combination. We also constructed strains containing the gyrA and marR mutations found in the Cip B– and Tet A–evolved lineages, respectively; these lineages represented mutations that appeared compatible with both mono and combination treatment with Cip and Tet. Next, we conducted a series of competition experiments using a ΔlacZ wild-type strain and engineered mutant strains.
The wild-type and mutant strains were mixed 1:1 and grown overnight in subinhibitory concentrations of antibiotics and no antibiotics. Subsequently, quantitative polymerase chain reaction (qPCR) was used to measure the ratio between wild-type and mutant alleles, and changes relative to growth without antibiotics were reported (Fig. 7, A to E). The results showed that the three mutations conferring resistance to Amk (in cpxA, fusA, and sbmA) were counterselected by Chl (Fig. 7, D to F). This effect was also seen when the cell mixture was grown in a combination of the two drugs, illustrating how collateral sensitivity can effectively reduce the evolution of resistance. In contrast, the gyrA mutation was strongly enriched by Cip alone as well as by Cip + Amk, but not in Amk alone. This shows that without collateral sensitivity, drug combinations cannot prevent selection of resistance mutations (Fig. 7G). The marR mutation was positively selected in Tet and Chl alone, as well as when the two drugs were combined, showing that drug combinations do not necessarily reduce the evolution of resistance (Fig. 7H). Our competition results were confirmed through plating the overnight cultures on LB + IPTG (isopropyl-β-D-thiogalactopyranoside) + Xgal (fig. S5).
Fig. 7. Competition experiment.
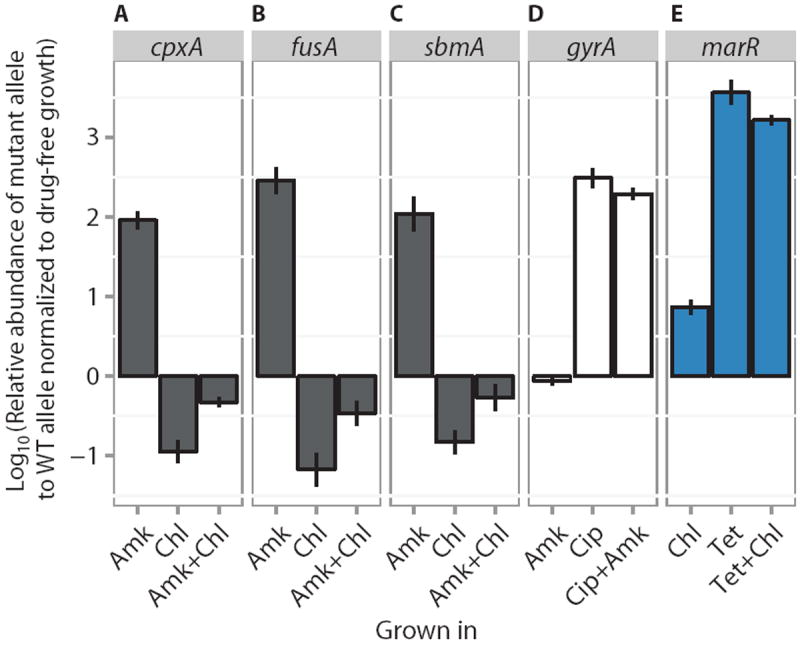
(A to E) Competition assays between mutant and WT alleles. For each competition experiment, the ratios of mutant to WT alleles after competitive growth in subinhibitory concentrations of either single drugs or drug pairs (listed below each panel) are reported relative to the ratio in antibiotic-free growth medium (means ± SEM, n = 3 biological replicates). Alleles are cpxA, fusA, sbmA, gyrA, and marR, for (A) to (E), respectively. Gray fill indicates mutations found to give Amk resistance. Blue fill indicates mutations known to confer the mar phenotype. White fill indicates mutations that confer Cip resistance.
Together with the in vitro evolution experiments and population sequencing, the competition experiments revealed how combinations of drugs that display collateral sensitivity can be used to create an “evolutionary tension” that works against fixation of resistance alleles that also confer collateral sensitivity.
DISCUSSION
Combination therapy is often used to counteract the evolution of resistance. Here, we show that the degree to which a combination can reduce the evolution of resistance depends on the degree of collateral resistance and collateral sensitivity between its component drugs. If resistance to one drug gives collateral resistance to another drug, a combination of the two drugs will not be very effective in reducing the evolution of resistance (Fig. 8A). This is the case for drug pairs of Cip, Pip, Tet, and Chl. Conversely, if resistance to one drug confers collateral sensitivity to another drug, resistance to a combination of these drugs will be significantly limited (Fig. 8B). These findings, together with studies of drug interactions, illustrate that a complex interplay between collateral susceptibility changes and drug interactions governs the evolution of resistance against drug combinations (28, 59).
Fig. 8. Impact of collateral IC90 changes on the evolution of drug resistance.
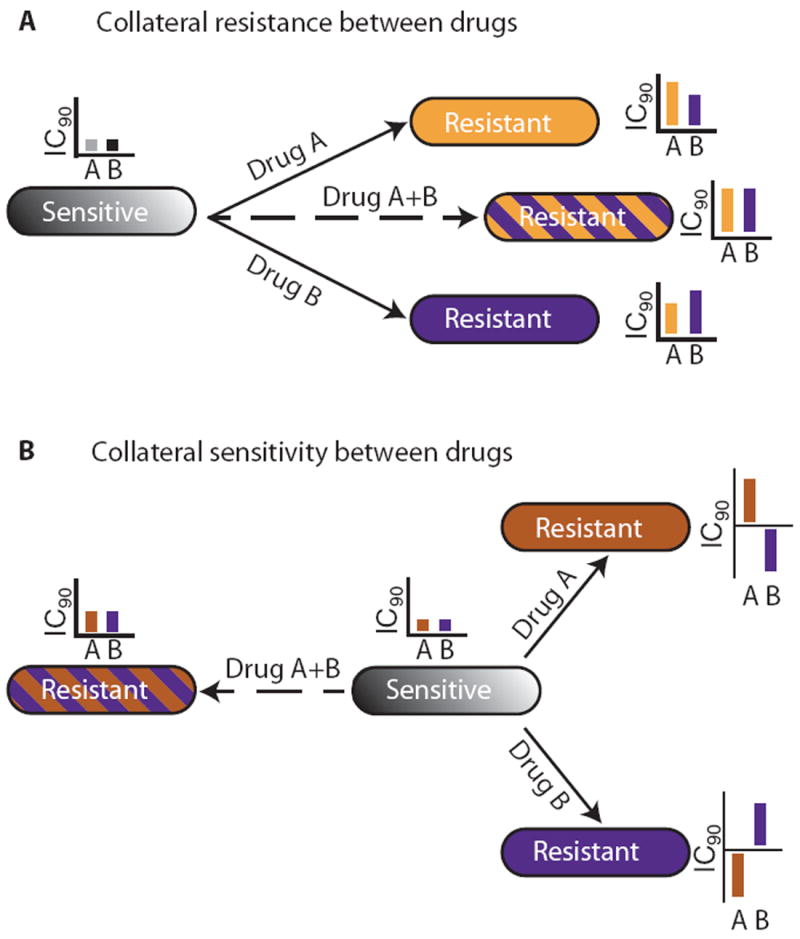
(A) If two drugs display collateral resistance (such as Tet and Chl), a combination of the two drugs will not effectively reduce resistance development, because they both will select for the same mutational profile. (B) In contrast, if the two drugs display collateral sensitivity (such as Amk and Chl), a combination of the two will be effective at reducing evolution of resistance because of suppressed fixation of resistance mutations in the population.
To identify the genetic changes underlying the observed collateral sensitivity and resistance, we sequenced the evolved lineages and found that Cip, Pip, Tet, and Chl selected for mutations that lead to an increased efflux and decreased membrane permeability, whereas Amk selected for an entirely different set of mutations specifically involved in aminoglycoside-induced membrane stress. These findings explain how the drugs in the drug pairs containing Cip, Pip, Tet, or Chl effectively work together to select for the same genotype, whereas the drugs in the drug pairs containing Amk select for two subsets of mutations, where one of them counteracts the effect of the other.
Furthermore, we used a competition assay to show that Amk-resistant mutants are counterselected when grown in the presence of Chl. This example illustrates that for drug pairs that display collateral sensitivity, the balance between resistance and sensitivity is difficult to maintain in a drug pair environment, resulting in decreased evolution of resistance. Our findings were corroborated by population sequencing showing that mutations that were fixed in single-drug evolved populations could not be fixed in populations evolved in a combination of drugs that displayed collateral sensitivity. The genes mutated in our study are broadly conserved across E. coli genomes, and mutations in these genes have been linked to antibiotic resistance in clinical isolates of E. coli, underscoring the clinical relevance of our findings (48, 49).
Central to the phenomena of collateral sensitivity is the fact that it is a property present in single-drug evolved lineages. Accordingly, it should be possible to predict drug combinations with low rates of resistance evolution by studying the collateral responses to individual drugs. In turn, this affects screening programs seeking to identify resistance-limiting combinations because the number of combinations required to screen to find a successful candidate can be markedly lowered. This is exemplified in a recent study in which we analyzed the collateral sensitivity network of a laboratory E. coli strain as well as two clinical isolates and identified 20 potential antiresistance drug combinations and more than 30 drug combinations that should be avoided in clinical practice because of collateral-resistance interactions (28).
Our results have been obtained under ideal in vitro conditions. To more generally translate our results to in vivo settings, quantitative modeling of the pharmacokinetic and pharmacodynamic properties of the constituent drugs would be beneficial. Such pharmacokinetic and pharmacodynamic modeling has proven valuable for optimization of single-drug treatment regimens (60, 61), and recent work has highlighted the use of pharmacokinetic and pharmacodynamic modeling for drug combinations to explore the dynamics of resistance development (62).
In addition to treating bacterial infections, combination therapy is also frequently used in the management of tuberculosis, HIV infections, and cancer, where it has proven indispensable in the fight against resistance. Although our study has focused on adaptive resistance in a bacterial system, E. coli, collateral resistance and collateral sensitivity have been described in viruses, cancer cell lines, and plants (17, 23-25). Hence, we expect that the general concept of predicting resistance evolution of drug combinations based on the collateral sensitivity profiles of the component drugs should be extendable to a broad range of diseases.
MATERIALS AND METHODS
Study design
E. coli MG1655 was evolved to the five antibiotics: Cip hydrochloride (AppliChem), Tet hydrochloride (Sigma), Amk sulfate (Sigma), Chl (Sigma), and Pip sulfate (Sigma), as well as all pairwise combinations thereof. These drugs were chosen to represent a broad range of chemical classes and clinical relevant targets.
The evolution experiments were performed in triplicate for each drug condition using LB medium as the growth medium. Selection was carried out in twofold dilution steps of antibiotics in 24-well plates using 1-ml total volume. Each plate contained two medium control wells; none of these showed growth during the experiment. After 20 hours of incubation at 37°C with shaking, OD600 was read, and 25 μl from the lowest antibiotic concentration with OD600 greater than 0.25 was diluted into a new antibiotic gradient. In total, 14 passages were performed. All lineages underwent the same number of passages to ensure a comparable number of generations.
The drug pairs were evolved to a 1:1 mixture of the component drug’s IC90; the resulting molar ratios were the following: Cip/Pip, 0.012; Cip/Amk, 0.015; Cip/Chl, 0.004; Cip/Tet, 0.021; Pip/Amk, 0.559; Pip/Chl, 0.230; Pip/Tet, 0.641; Amk/Chl, 0.191; Amk/Tet, 0.585; and Tet/Chl, 0.144. The evolution experiment was ended when the single-drug evolved lineages had reached the clinical breakpoint for the antibiotic. Clinical breakpoints were defined according to The EUCAST:Cip, 1 μg/ml; Pip, 16 μg/ml; Amk, 16 μg/ml; Chl, 8 μg/ml; and Tet, 16 μg/ml. On the last day, all lineages were streaked on LB agar plates to be used for IC90 determination.
IC90 determination
IC90 determination was performed in 96-well microtiter plates prepared using a Hamilton STAR pipetting robot. Each drug gradient consisted of 11 points in a twofold dilution series prepared in MHBII (Mueller-Hinton broth 2) (Sigma) medium with a total of 150 μl in each well. For the single-drug evolved lineages, the IC90 was determined for all single drugs as well as all combinations containing the evolved-to drug, and the experiments were carried out in triplicate and quadruplicate, respectively. For the drug pair evolved lineages, the IC90 was determined for the two drugs in the combination as well as the drug combination, and experiments were done in five replicates. For every IC90 test, the wild-type strain was included to determine the IC90 reference point. The IC90 plates were inoculated with about 105 cells per well using a 96-pin replicator. The plates were incubated at 37°C with shaking for 18 to 20 hours, and OD600 was read on a BioTek H1 plate reader.
Data analysis
The OD600 data files were analyzed using R (63). In brief, control wells were analyzed (one contamination of 616 blanks and growth in all positive controls). To obtain inhibition curves, the OD600 values for the dose-response series were converted into values of percent inhibition calculated as 1 − [OD600(growth in drug) − OD600(negative control)]/[OD600(positive control) − OD600(negative control)] and plotted against the molar concentration of the antibiotic, and a dose-response curve was fitted using the drc package with the default four-variable logistic model, where x is the molar drug concentration:
(fig. S2) (64). IC90 was calculated via the inverse function of the fit. Graphs were made in R with the packages plotrix and ggplot2 (65, 66).
FICI was calculated as:
where ω signifies the molar fraction of drug A in the drug combination AB.
Calculation of evolvability index
The evolvability index is used to assess how the development of resistance against a drug is affected by the presence of another drug. By summing the effect from each component drug in a drug pair, it gives an overall value to describe the degree of resistance development in drug pair evolved lineages relative to the resistance development in single-drug evolved lineages. Specifically, the evolvability index is calculated by taking the average of the relative change in resistance development for each component drug of a drug pair evolved lineage and divided it by the relative change in resistance development in the single-drug evolved lineages:
where IC90[A]AB signifies the IC90 of the AB evolved strain tested against drug A.
Collateral IC90 change is the average collateral IC90 change between two single-drug evolved lineages calculated as:
where IC90[A]B signifies the IC90 of the B evolved lineage tested against drug A.
SOLiD sequencing
A single colony from each evolution experiment was grown up in LB, and DNA was extracted using the DNeasy kit (Qiagen). The DNA was sheared into 200-bp fragments using Covaris E210, and barcoded libraries were made for SOLiD sequencing. SOLiD reads were aligned to E. coli MG1655 reference genome (NC_000913) using Bowtie 2 (67). Each sample had at least 98% of the genome covered with three times coverage or greater, and the mean percentage with at least three times coverage was 99.71% [table S2, (68)]. The alignments were further tuned by GATK (Genome Analysis Toolkit) (69) by re-aligning identified possible INDEL sites to discriminate between SNP and INDEL sites (70). Variant calling for SNPs and INDELs was done using SAMtools (71), with INDELs verified by aligning constructed contigs around INDEL sites to the reference genome (72, 73). Further analysis was done by custom-written scripts using Biopython (74).
Population sequencing
Total DNA was extracted from the populations of lineage B evolved to Amk, Chl, and Amk + Chl using the DNeasy kit (Qiagen). Sequencing libraries for the Illumina MiSeq platform were prepared using the Illumina Nextera XT kit. The average coverage was 167 for the Amk population, 143 for the Chl populations, and 156 for the Amk + Chl population. SNPs and INDELs were identified using CLC Genomics Workbench (Qiagen) by mapping reads onto the reference E. coli MG1655 genome (NC_000913).
Competitive growth selection
The five single mutants cpxA, fusA, sbmA, gyrA, and marR were engineered in EcHW24 using the MAGE (multiplex automated genomic engineering) technique with relevant SNP oligos (table S3) (75). A wild-type lacZ mutant was engineered in a similar fashion. Each of the five single mutants was mixed 1:1 with the wild-type lacZ mutant at OD600 equivalent to 0.1. This mixture (1 μl) was inoculated into subinhibitory concentrations of antibiotics in the following order: cpxA, fusA, and sbmA were inoculated into Amk, Chl, Amk + Chl, and LB; gyrA was inoculated into Cip, Amk, Cip + Amk, and LB; marR was inoculated into Tet, Chl, Tet + Chl, and LB. All were grown overnight at 30°C. To confirm that the antibiotic concentration was indeed subinhibitory, the wild-type lacZ was inoculated into all antibiotic solutions, and the growth rate was measured. The competitive growth selection experiments were performed in triplicates.
qPCR assay
For each of the five mutant alleles in the competitive growth selection experiment, a wild-type primer pair and a mutant primer pair were designed (table S4). The optimal annealing temperature was identified in a temperature gradient, and for each growth condition, a separate qPCR was performed with the wild-type primer pair and the mutant primer pair using 1 μl of a 100-fold dilution of the overnight competitive selection culture as template in a SYBR Green qPCR (SSO, Bio-Rad). The ΔCT for the wild-type and mutant primer pair was calculated and normalized to the ΔCT for the LB growth, resulting in a ΔΔCT value.
Plating validation of the competitive growth selection
After overnight, competitive growth selection cultures were diluted and plated on LB with IPTG and Xgal. Representative pictures were taken and used in fig. S5.
Statistical analyses
Growth inhibition curves were analyzed as described in the data analysis section. For each lineage and drug condition, the average IC90 and SD were calculated (n = 3 technical replicates for single drugs, and n = 4 for drug pairs), and these values were normalized to the average IC90 values for the wild-type E. coli strain, taking into account the SD of both the numerator and the denominator. The averages of the biological replicates for each drug condition (n = 3) were used in the calculations of the evolvability index and the collateral IC90 change. All analyses were performed with software R (63).
Throughout the manuscript, a significance level of 0.05 is used. ANOVA, followed by Tukey test, was used to identify differences in the relative increase in resistance between the single-drug and the drug pair evolved lineages (Fig. 1). Spearman rank correlation was used to assess correlation between evolvability index and FICI (Fig. 4C), and in case of multiple hypothesis testing, the significance level was Bonferroni-corrected (fig. S4). Student’s t test was used to assess changes in FICI (Fig. 4D). Mann-Whitney U-test was used to compare evolvability index and collateral IC90 change in drug pairs with Amk to those without Amk (Fig. 5, C and D).
Supplementary Material
www.sciencetranslationalmedicine.org/cgi/content/full/6/262/262ra156/DC1
Fig. S1. Fold increase in drug tolerance during evolution experiment.
Fig. S2. IC90 dose-response curves.
Fig. S3. Correlation between IC50 or MIC, and IC90-based measurements.
Fig. S4. Alternative correlation between FICI and evolution of resistance.
Fig. S5. Competition between wild-type ΔlacZ and mutant.
Table S1. Raw OD600 readings.
Table S2. Sequencing coverage.
Table S3. Individual SNPs and INDELs.
Table S4. Oligos for MAGE and qPCR primers.
Reference (76)
Acknowledgments
We thank T. Thomsen for assisting with the robotics and R. Kishony for helpful comments on the manuscript. Funding: This research was funded by the Lundbeck Foundation and the Danish Free Research Councils. M.O.A.S. acknowledges additional funding from the Novo Nordisk Foundation and the European Union Seventh Framework Programme–Health Program Evolution and Transfer of Antibiotic Resistance (grant 282004). H.H.W. acknowledges funding by the U.S. NIH Director’s Early Independence Award (grant 1DP5OD009172-01).
Footnotes
Author contributions: C.M., M.O.A.S., and H.H.W. designed the experiments; C.M. and A.I.N.W. performed all the experiments; C.M., M.O.A.S., and H.K.G. analyzed the data; and C.M. and M.O.A.S. wrote the paper.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: Raw sequence data are available at National Center for Biotechnology Information under project no. PRJNA260999.
REFERENCES AND NOTES
- 1.Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. 2010;74:417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nordmann P, Cuzon G, Naas T. The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. Lancet Infect Dis. 2009;9:228–236. doi: 10.1016/S1473-3099(09)70054-4. [DOI] [PubMed] [Google Scholar]
- 3.Donald PR, van Helden PD. The global burden of tuberculosis—Combating drug resistance in difficult times. N Engl J Med. 2009;360:2393–2395. doi: 10.1056/NEJMp0903806. [DOI] [PubMed] [Google Scholar]
- 4.Mitchison DA. How drug resistance emerges as a result of poor compliance during short course chemotherapy for tuberculosis. Int J Tuberc Lung Dis. 1998;2:10–15. [PubMed] [Google Scholar]
- 5.Clavel F, Hance AJ. HIV drug resistance. N Engl J Med. 2004;350:1023–1035. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- 6.Dunner E, Brown WB, Wallace J. The effect of streptomycin with para-amino salicylic acid on the emergence of resistant strains of tubercle bacilli. Dis Chest. 1949;16:661–666. doi: 10.1378/chest.16.6.661. [DOI] [PubMed] [Google Scholar]
- 7.Palella FJ, Jr, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N Engl J Med. 1998;338:853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 8.Glickman MS, Sawyers CL. Converting cancer therapies into cures: Lessons from infectious diseases. Cell. 2012;148:1089–1098. doi: 10.1016/j.cell.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamma PD, Cosgrove SE, Maragakis LL. Combination therapy for treatment of infections with Gram-negative bacteria. Clin Microbiol Rev. 2012;25:450–470. doi: 10.1128/CMR.05041-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bushby SR. Trimethoprim-sulfamethoxazole: In vitro microbiological aspects. J Infect Dis. 1973;128:S442–S462. doi: 10.1093/infdis/128.supplement_3.s442. [DOI] [PubMed] [Google Scholar]
- 11.Cottarel G, Wierzbowski J. Combination drugs, an emerging option for antibacterial therapy. Trends Biotechnol. 2007;25:547–555. doi: 10.1016/j.tibtech.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 12.Shahid M, Sobia F, Singh A, Malik A, Khan HM, Jonas D, Hawkey PM. Beta-lactams and beta-lactamase-inhibitors in current- or potential-clinical practice: A comprehensive update. Crit Rev Microbiol. 2009;35:81–108. doi: 10.1080/10408410902733979. [DOI] [PubMed] [Google Scholar]
- 13.Torella JP, Chait R, Kishony R. Optimal drug synergy in antimicrobial treatments. PLOS Comput Biol. 2010;6:e1000796. doi: 10.1371/journal.pcbi.1000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chait R, Craney A, Kishony R. Antibiotic interactions that select against resistance. Nature. 2007;446:668–671. doi: 10.1038/nature05685. [DOI] [PubMed] [Google Scholar]
- 15.Hegreness M, Shoresh N, Damian D, Hartl D, Kishony R. Accelerated evolution of resistance in multidrug environments. Proc Natl Acad Sci U S A. 2008;105:13977–13981. doi: 10.1073/pnas.0805965105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michel JB, Yeh PJ, Chait R, Moellering RC, Jr, Kishony R. Drug interactions modulate the potential for evolution of resistance. Proc Natl Acad Sci U S A. 2008;105:14918–14923. doi: 10.1073/pnas.0800944105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gadamski G, Ciarka D, Gressel J, Gawronski SW. Negative cross-resistance in triazine-resistant biotypes of Echinochloa crus-galli and Conyza canadensis. Weed Science. 2000;48:176–180. [Google Scholar]
- 18.Dragosits M, Mozhayskiy V, Quinones-Soto S, Park J, Tagkopoulos I. Evolutionary potential, cross-stress behavior and the genetic basis of acquired stress resistance in Escherichia coli. Mol Syst Biol. 2013;9:643. doi: 10.1038/msb.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szybalski W, Bryson V. Genetic studies on microbial cross resistance to toxic agents. I. Cross resistance of Escherichia coli to fifteen antibiotics. J Bacteriol. 1952;64:489–499. doi: 10.1128/jb.64.4.489-499.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chao L. An unusual interaction between the target of nalidixic acid and novobiocin. Nature. 1978;271:385–386. doi: 10.1038/271385a0. [DOI] [PubMed] [Google Scholar]
- 21.Sanders CC, Sanders WE, Jr, Goering RV, Werner V. Selection of multiple antibiotic resistance by quinolones, β-lactams, and aminoglycosides with special reference to cross-resistance between unrelated drug classes. Antimicrob. Agents Chemother. 1984;26:797–801. doi: 10.1128/aac.26.6.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernández L, Hancock RE. Adaptive and mutational resistance: Role of porins and efflux pumps in drug resistance. Clin Microbiol Rev. 2012;25:661–681. doi: 10.1128/CMR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, Eisenson D, Narurkar R, Deng P, Nezi L, Lee MA, Hu B, Hu J, Sahin E, Ong D, Fletcher-Sananikone E, Ho D, Kwong L, Brennan C, Wang YA, Chin L, DePinho RA. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature. 2012;488:337–342. doi: 10.1038/nature11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pluchino KM, Hall MD, Goldsborough AS, Callaghan R, Gottesman MM. Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist Updat. 2012;15:98–105. doi: 10.1016/j.drup.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romano KP, Ali A, Aydin C, Soumana D, Ozen A, Deveau LM, Silver C, Cao H, Newton A, Petropoulos CJ, Huang W, Schiffer CA. The molecular basis of drug resistance against hepatitis C virus NS3/4A protease inhibitors. PLOS Pathog. 2012;8:e1002832. doi: 10.1371/journal.ppat.1002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gressel J, Segel LA. Negative cross resistance; a possible key to atrazine resistance management: A call for whole plant data. Z Naturforsch. 1990;45c:470–473. [Google Scholar]
- 27.Deeks SG. Treatment of antiretroviral-drug-resistant HIV-1 infection. Lancet. 2003;362:2002–2011. doi: 10.1016/S0140-6736(03)15022-2. [DOI] [PubMed] [Google Scholar]
- 28.Lázár V, Pal Singh G, Spohn R, Nagy I, Horváth B, Hrtyan M, Busa-Fekete R, Bogos B, Méhi O, Csörgő B, Pósfai G, Fekete G, Szappanos B, Kégl B, Papp B, Pál C. Bacterial evolution of antibiotic hypersensitivity. Mol Syst Biol. 2013;9:700. doi: 10.1038/msb.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Imamovic L, Sommer MO. Use of collateral sensitivity networks to design drug cycling protocols that avoid resistance development. Sci Transl Med. 2013;5:204ra132. doi: 10.1126/scitranslmed.3006609. [DOI] [PubMed] [Google Scholar]
- 30.Lee DH, Feist AM, Barrett CL, Palsson BØ. Cumulative number of cell divisions as a meaningful timescale for adaptive laboratory evolution of Escherichia coli. PLOS One. 2011;6:e26172. doi: 10.1371/journal.pone.0026172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hooper DC, Wolfson JS, Ng EY, Swartz MN. Mechanisms of action of and resistance to ciprofloxacin. Am J Med. 1987;82:12–20. [PubMed] [Google Scholar]
- 32.Greco WR, Bravo G, Parsons JC. The search for synergy: A critical review from a response surface perspective. Pharmacol Rev. 1995;47:331–385. [PubMed] [Google Scholar]
- 33.Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, Yaffe MB. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149:780–794. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pena-Miller R, Laehnemann D, Jansen G, Fuentes-Hernandez A, Rosenstiel P, Schulenburg H, Beardmore R. When the most potent combination of antibiotics selects for the greatest bacterial load: The smile-frown transition. PLOS Biol. 2013;11:e1001540. doi: 10.1371/journal.pbio.1001540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ariza RR, Li Z, Ringstad N, Demple B. Activation of multiple antibiotic resistance and binding of stress-inducible promoters by Escherichia coli Rob protein. J Bacteriol. 1995;177:1655–1661. doi: 10.1128/jb.177.7.1655-1661.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.George AM, Levy SB. Gene in the major cotransduction gap of the Escherichia coli K-12 linkage map required for the expression of chromosomal resistance to tetracycline and other antibiotics. J Bacteriol. 1983;155:541–548. doi: 10.1128/jb.155.2.541-548.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma D, Alberti M, Lynch C, Nikaido H, Hearst JE. The local repressor AcrR plays a modulating role in the regulation of acrAB genes of Escherichia coli by global stress signals. Mol Microbiol. 1996;19:101–112. doi: 10.1046/j.1365-2958.1996.357881.x. [DOI] [PubMed] [Google Scholar]
- 38.Miller PF, Gambino LF, Sulavik MC, Gracheck SJ. Genetic relationship between soxRS and mar loci in promoting multiple antibiotic resistance in Escherichia coli. Antimicrob Agents Chemother. 1994;38:1773–1779. doi: 10.1128/aac.38.8.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okusu H, Ma D, Nikaido H. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J Bacteriol. 1996;178:306–308. doi: 10.1128/jb.178.1.306-308.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toprak E, Veres A, Michel JB, Chait R, Hartl DL, Kishony R. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat Genet. 2011;44:101–105. doi: 10.1038/ng.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oethinger M, Podglajen I, Kern WV, Levy SB. Overexpression of the marA or soxS regulatory gene in clinical topoisomerase mutants of Escherichia coli. Antimicrob Agents Chemother. 1998;42:2089–2094. doi: 10.1128/aac.42.8.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maneewannakul K, Levy SB. Identification for mar mutants among quinolone-resistant clinical isolates of Escherichia coli. Antimicrob Agents Chemother. 1996;40:1695–1698. doi: 10.1128/aac.40.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang H, Dzink-Fox JL, Chen M, Levy SB. Genetic characterization of highly fluoroquinolone-resistant clinical Escherichia coli strains from China: Role of acrR mutations. Antimicrob Agents Chemother. 2001;45:1515–1521. doi: 10.1128/AAC.45.5.1515-1521.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barbosa TM, Levy SB. Differential expression of over 60 chromosomal genes in Escherichia coli by constitutive expression of MarA. J Bacteriol. 2000;182:3467–3474. doi: 10.1128/jb.182.12.3467-3474.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bennik MH, Pomposiello PJ, Thorne DF, Demple B. Defining a rob Regulon in Escherichia coli by using transposon mutagenesis. J Bacteriol. 2000;182:3794–3801. doi: 10.1128/jb.182.13.3794-3801.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pomposiello PJ, Bennik MH, Demple B. Genome-wide transcriptional profiling of the Escherichia coli responses to superoxide stress and sodium salicylate. J Bacteriol. 2001;183:3890–3902. doi: 10.1128/JB.183.13.3890-3902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin RG, Rosner JL. Genomics of the marA/soxS/rob regulon of Escherichia coli: Identification of directly activated promoters by application of molecular genetics and informatics to microarray data. Mol Microbiol. 2002;44:1611–1624. doi: 10.1046/j.1365-2958.2002.02985.x. [DOI] [PubMed] [Google Scholar]
- 48.Warner DM, Yang Q, Duval V, Chen M, Xu Y, Levy SB. Involvement of MarR and YedS in carbapenem resistance in a clinical isolate of Escherichia coli from China. Antimicrob Agents Chemother. 2013;57:1935–1937. doi: 10.1128/AAC.02445-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martínez-Martínez L. Extended-spectrum beta-lactamases and the permeability barrier. Clin Microbiol Infect. 2008;14(Suppl 1):82–89. doi: 10.1111/j.1469-0691.2007.01860.x. [DOI] [PubMed] [Google Scholar]
- 50.Vargiu AV, Nikaido H. Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proc Natl Acad Sci U S A. 2012;109:20637–20642. doi: 10.1073/pnas.1218348109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Norström T, Lannergård J, Hughes D. Genetic and phenotypic identification of fusidic acid-resistant mutants with the small-colony-variant phenotype in Staphylococcus aureus. Antimicrob Agents Chemother. 2007;51:4438–4446. doi: 10.1128/AAC.00328-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johanson U, Hughes D. Fusidic acid-resistant mutants define three regions in elongation factor G of Salmonella typhimurium. Gene. 1994;143:55–59. doi: 10.1016/0378-1119(94)90604-1. [DOI] [PubMed] [Google Scholar]
- 53.Macvanin M, Hughes D. Hyper-susceptibility of a fusidic acid-resistant mutant of Salmonella to different classes of antibiotics. FEMS Microbiol Lett. 2005;247:215–220. doi: 10.1016/j.femsle.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 54.Cosma CL, Danese PN, Carlson JH, Silhavy TJ, Snyder WB. Mutational activation of the Cpx signal transduction pathway of Escherichia coli suppresses the toxicity conferred by certain envelope-associated stresses. Mol Microbiol. 1995;18:491–505. doi: 10.1111/j.1365-2958.1995.mmi_18030491.x. [DOI] [PubMed] [Google Scholar]
- 55.Busse HJ, Wöstmann C, Bakker EP. The bactericidal action of streptomycin: Membrane permeabilization caused by the insertion of mistranslated proteins into the cytoplasmic membrane of Escherichia coli and subsequent caging of the antibiotic inside the cells due to degradation of these proteins. J Gen Microbiol. 1992;138:551–561. doi: 10.1099/00221287-138-3-551. [DOI] [PubMed] [Google Scholar]
- 56.Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell. 2008;135:679–690. doi: 10.1016/j.cell.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laviña M, Pugsley AP, Moreno F. Identification, mapping, cloning and characterization of a gene (sbmA) required for microcin B17 action on Escherichia coli K12. J Gen Microbiol. 1986;132:1685–1693. doi: 10.1099/00221287-132-6-1685. [DOI] [PubMed] [Google Scholar]
- 58.de Cristóbal RE, Vincent PA, Salomón RA. A combination of sbmA and tolC mutations in Escherichia coli K-12 Tn10-carrying strains results in hypersusceptibility to tetracycline. J Bacteriol. 2008;190:1491–1494. doi: 10.1128/JB.01844-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yeh PJ, Hegreness MJ, Aiden AP, Kishony R. Drug interactions and the evolution of antibiotic resistance. Nat Rev Microbiol. 2009;7:460–466. doi: 10.1038/nrmicro2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frimodt-Møller N. How predictive is PK/PD for antibacterial agents? Int J Antimicrob Agents. 2002;19:333–339. doi: 10.1016/s0924-8579(02)00029-8. [DOI] [PubMed] [Google Scholar]
- 61.Rybak MJ. Pharmacodynamics: Relation to antimicrobial resistance. Am J Infect Control. 2006;34:S38–S45. doi: 10.1016/j.ajic.2006.05.227. [DOI] [PubMed] [Google Scholar]
- 62.Ankomah P, Johnson PJ, Levin BR. The pharmaco –, population and evolutionary dynamics of multi-drug therapy: Experiments with S. aureus and E. coli and computer simulations. PLOS Pathog. 2013;9:e1003300. doi: 10.1371/journal.ppat.1003300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.R. R Core Team. R: A Language and Environment for Statistical Computing. available at http://www.R-project.org/)
- 64.Streibig JC, Ritz C. Bioassay analysis using R. J Stat Softw. 2005;12:1–22. available at http://econpapers.repec.org/RePEc:jss:jstsof:12:i05. [Google Scholar]
- 65.Lemon J. Plotrix: A package in the red light district of R. R-News. 2006;6:8–12. [Google Scholar]
- 66.Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer Publishing Company Incorporated; New York, NY: 2009. http://dl.acm.org/citation.cfm?id=1795559. [Google Scholar]
- 67.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Quinlan AR, Hall IM. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, de Hoon MJ. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang HH, Church GM. Multiplexed genome engineering and genotyping methods applications for synthetic biology and metabolic engineering. Methods Enzymol. 2011;498:409–426. doi: 10.1016/B978-0-12-385120-8.00018-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
www.sciencetranslationalmedicine.org/cgi/content/full/6/262/262ra156/DC1
Fig. S1. Fold increase in drug tolerance during evolution experiment.
Fig. S2. IC90 dose-response curves.
Fig. S3. Correlation between IC50 or MIC, and IC90-based measurements.
Fig. S4. Alternative correlation between FICI and evolution of resistance.
Fig. S5. Competition between wild-type ΔlacZ and mutant.
Table S1. Raw OD600 readings.
Table S2. Sequencing coverage.
Table S3. Individual SNPs and INDELs.
Table S4. Oligos for MAGE and qPCR primers.
Reference (76)