

Abstract
Autologous stem cells are a promising cell source for cartilage regeneration; however, cell replicative senescence and joint posttraumatic inflammation provide challenges to bring this treatment modality to fruition. In this study, we hypothesized that preconditioning with p38 MAPK inhibitor (sb203580) would recharge decellularized extracellular matrix (dECM) expanded human synovium-derived stem cell (hSDSC) chondrogenesis in an inflammatory environment. We found that preconditioning with sb203580 greatly enhanced dECM expanded hSDSC proliferation and chondrogenic potential while supplementation with sb203580 in an induction medium dramatically retarded hSDSC chondrogenic differentiation, even for dECM expanded cells. We also found that sb203580 preconditioning enhanced matrix-expanded hSDSC chondrogenic capacity even in an interleukin-1 (IL-1) induced inflammatory environment. Non-detectable expression of HLA-DR in the hSDSCs grown on allogeneic dECM indicates the feasibility of commercial preparation of these dECMs from healthy, young donors for patients who need autologous transplantation. Our study indicated that p38 MAPK inhibitor has a distinctive priming effect on dECM mediated stem cell cartilage regeneration. Combined rejuvenation with sb203580 and dECM expansion can precondition hSDSCs’ resurfacing capacity for osteoarthritic patients with cartilage defects.
Keywords: Decellularized extracellular matrix, p38 MAPK inhibitor, Mesenchymal stem cell, Chondrogenic potential, Interleukin-1 beta, Preconditioning
1. Introduction
Cartilage defects do not heal themselves due to a shortage of blood supply. Recently, adult stem cells have become a promising cell source for autologous cartilage regeneration. Adipose-derived stem cells (ASCs) have limited chondrogenic ability and bone marrow stromal cells (BMSCs) tend to undergo endochondral ossification upon chondrogenic induction [1]. By comparison, synovium-derived stem cells (SDSCs) are a tissue-specific stem cell for cartilage regeneration [2] and have been successfully used in cartilage regeneration studies [3–5]. In vitro expansion is a necessary step before in vivo application and represents a formidable challenge since stem cells are thought to exist in “niches” where extrinsic signals modulate the intrinsic signals that drive self-renewal and cell fate determination [6]. Outside of their niche, adult stem cells lose their developmental potential quickly and tend to either randomly differentiate or undergo apoptosis over time [7].
Decellularized extracellular matrix (dECM) could provide such an in vitro “niche”-like nanostructured microenvironment in which porcine SDSCs (pSDSCs) were greatly expanded with enhanced chondrogenic potential [8,9]. Despite success in using adult human SDSCs (hSDSCs) in this system [10], the rejuvenation effect was not as robust as that using young pSDSCs [8]. Different species and donor age might cause this discrepancy [7]. Furthermore, posttraumatic joint inflammation, normally accompanying cartilage defects [11,12], could perhaps lead to reduced efficiency of dECM on the rejuvenation of adult hSDSCs [13].
The p38 mitogen-activated protein kinase (MAPK) signaling cascade is known to be involved in various biological responses such as cell proliferation and differentiation [14]. Recently, p38 MAPK was also found to be activated by various pro-inflammatory and stressful stimuli [15]. There is increasing evidence showing that in vivo application of p38 MAPK inhibitors can decrease inflammation and related damage [16]; unfortunately, these inhibitors also arrest tissue regeneration [17–19].
To maximize advantages and minimize disadvantages, in this study, a p38 MAPK inhibitor was used to precondition hSDSCs during cell expansion on either plastic flasks (Plastic) or dECM followed by chondrogenic induction in a pellet culture system. In comparison, p38 MAPK inhibitor was supplemented in induction medium instead for the assessment of the direct effect on hSDSC chondrogenesis. Expanded hSDSCs were also evaluated for chondrogenic capacity in an interleukin-1 beta (IL-1β) induced inflammatory environment. Lastly, dECM deposited by allogeneic cells were evaluated for eliciting potential immune issues in hSDSCs after expansion. We hypothesized that preconditioning with p38 MAPK inhibitor would recharge dECM expanded hSDSC chondrogenesis in an inflammatory environment.
2. Materials and Methods
2.1. SDSC culture
Adult human synovial fibroblasts (4 donors, two male and two female, average 43 years old, all had no known joint disease), referred to as hSDSCs [10,20,21], were obtained from Asterand (North America Laboratories, Detroit, MI). Human SDSCs were plated and cultured in a growth medium [alpha minimum essential medium (αMEM) containing 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL fungizone (Invitrogen, Carlsbad, CA)] at 37°C in a humidified 5% CO2 and 21% O2 incubator. The medium was changed every three days.
2.2. dECM preparation
The preparation of dECM was described in our previous study [10,21,22]. Briefly, plastic flasks (Plastic) were precoated with 0.2% gelatin (Sigma-Aldrich, St. Louis, MO) at 37°C for 1 h and seeded with passage 3 (P3) SDSCs. After cells reached 90% confluence, 250 μM L-ascorbic acid phosphate (Wako Chemicals USA, Inc., Richmond, VA) was added for 8 days. The deposited matrix was incubated with 0.5% Triton X-100 containing 20 mM ammonium hydroxide at 37°C for 5 min and stored at 4°C in phosphate-buffered saline (PBS) containing 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL fungizone.
2.3. Morphological characterization of dECM with or without hSDSCs
Representative samples (n=3) were primarily fixed in 2.5% glutaraldehyde (Sigma-Aldrich) for 2 h, followed by secondary fixation in 2% osmium tetroxide (Sigma-Aldrich) for another 2 h. The samples were then dehydrated in a gradient ethanol series, in hexamethyldisilazane (HMDS, Sigma-Aldrich) at a ratio of 1:1 with ethanol twice for 1 h each time, in HMDS at a ratio of 1:2 with ethanol overnight, and in HMDS three times for 4 h each time. The samples were air-dried for 24 h and gold sputter was added. The images were recorded by a scanning electron microscope (SEM) (Hitachi, Model S 2400).
2.4. Preconditioning using p38 MAPK inhibitor during cell expansion
Passage 3 hSDSCs were cultured at 3000 cells/cm2 for one passage (8 days) on two substrates: dECM or Plastic. Ten μM of sb203580 (p38 MAPK inhibitor, LC Laboratories, Woburn, MA) were added 48 h after cell seeding throughout the culture. The groups without sb203580 preconditioning served as a control. There were four groups: Plastic expansion alone (Pcontrol), Plastic expansion plus sb203580 (Psb203580), dECM expansion alone (Econtrol), and dECM expansion plus sb203580 (Esb203580). The cell number in T175 flasks (n=6) from each group was counted using a hemocytometer. For proliferation index (PI), P3 hSDSCs (before expansion) were labeled with CellVue® Claret (Sigma-Aldrich) at 2×10−6 M for 5 min according to the manufacturer’s protocol. After passaging, expanded cells were collected and measured using a BD FACS Calibur™ flow cytometer (dual laser) (BD Biosciences, San Jose, CA). Twenty thousand events of each sample were collected using CellQuest Pro software (BD Biosciences) and PI was analyzed by ModFit LT™ version 3.1 (Verity Software House, Topsham, ME).
The following primary antibodies were used to detect expanded hSDSC surface immunophenotype profiles: Alexa Fluor 488 anti-human CD29 antibody (BioLegend, San Diego, CA), FITC anti-human CD90 antibody (Biolegend), FITC anti-human CD105 antibody (BioLegend), PE anti-human SSEA4 (the stage-specific embryonic antigen 4) antibody (BioLegend), and isotype-matched IgGs (Beckman Coulter, Fullerton, CA). Samples (n=3) of each 2×105 expanded cells were incubated on ice in cold PBS containing 0.1% Chrom-Pure Human IgG whole molecule (Jackson ImmunoResearch Laboratories, West Grove, PA) and 1% NaN3 (Sigma-Aldrich) for 30 min. The cells were then incubated in the dark in the primary antibody for 30 min. Fluorescence was analyzed by a FACS Calibur (BD Biosciences) using FCS Express software package (De Novo Software, Los Angeles, CA).
2.5. Chondrogenic induction of expanded hSDSCs with dECM and/or p38 MAPK inhibitor preconditioning
After in vitro expansion, 0.3×106 of hSDSCs from each group (Pcontrol, Psb203580, Econtrol, and E203580) were centrifuged at 500 g for 5 min in a 15-mL polypropylene tube to form a pellet. After overnight incubation, the pellets were cultured for 35 days in a serum-free chondrogenic medium consisting of high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM), 40 μg/mL proline, 100 nM dexamethasone, 100 U/mL penicillin, 100 μg/mL streptomycin, 0.1 mM ascorbic acid-2-phosphate, and 1×ITS™ Premix [6.25 μg/mL insulin, 6.25 μg/mL transferrin, 6.25 μg/mL selenous acid, 5.35 μg/mL linoleic acid, and 1.25 μg/mL bovine serum albumin (BSA), from BD Biosciences] with supplementation of 10 ng/mL transforming growth factor beta 3 (TGFβ3, PeproTech Inc., Rocky Hill, NJ). Chondrogenic differentiation was evaluated using histology, immunostaining, and biochemical analysis.
Representative pellets (n=3) were fixed in 4% paraformaldehyde at 4°C overnight, followed by dehydrating in a gradient ethanol series, clearing with xylene, and embedding in paraffin blocks. Five-μm thick sections were histochemically stained with Alcian blue (Sigma-Aldrich; counterstained with fast red) for sulfated glycosaminoglycans (GAGs). For immunohistochemical analysis, the sections were immunolabeled with primary antibody against type II collagen (Col II; II-II6B3; Developmental Studies Hybridoma Bank, Iowa City, IA) followed by the secondary antibody of biotinylated horse anti-mouse IgG (Vector, Burlingame, CA). Immunoactivity was detected using Vectastain ABC reagent (Vector) with 3,3′-diaminobenzidine as a substrate.
Representative pellets (n=4) were digested for 4 h at 60°C with 125 μg/mL papain in PBE buffer (100 mM phosphate and 10 mM ethylenediaminetetraacetic acid, pH 6.5) containing 10 mM cysteine, by using 200 μL of enzyme per sample. To quantify cell density, the amount of DNA in the papain digestion was measured using the Quant-iT™ PicoGreen® dsDNA assay kit (Invitrogen) with a CytoFluor® Series 4000 (Applied Biosystems, Foster City, CA). GAG was measured using dimethylmethylene blue dye and a Spectronic BioMate 3 Spectrophotometer (Thermo Scientific, Milford, MA) with bovine chondroitin sulfate as a standard.
2.6. Treatment using p38 MAPK inhibitor during chondrogenic induction
After in vitro expansion, the pellets from both the Plastic and dECM groups were cultured for 28 days in a serum-free chondrogenic medium supplemented with 10 μM of sb203580 throughout the culture. The groups without sb203580 treatment served as a control. Chondrogenic differentiation was evaluated using Alcian blue staining for sulfated GAGs, immunostaining for type II collagen, and biochemical analysis for DNA and GAG amounts in pellets.
2.7. Preconditioning strategy for hSDSC-based chondrogenesis in an inflammatory environment
After in vitro expansion, the pellets from both the Plastic and dECM groups were cultured for 24 days in a serum-free chondrogenic medium. Two dosages of IL-1β, 0.1 and 1 ng/mL, were applied either throughout the culture (day 0–24), at the early (day 0–15), or late stage (day 15–24) of chondrogenic induction. Chondrogenic differentiation was evaluated using Alcian blue staining for sulfated GAGs, immunostaining for type II collagen, biochemical analysis for DNA and GAG amounts in pellets, and TaqMan® real-time polymerase chain reaction (PCR) for chondrogenic markers including SOX9 [SRY (sex determining region Y)-box 9], ACAN (aggrecan), and COL2A1 (type II collagen).
After in vitro expansion with or without 10 μM of sb203580, the pellets from both the Plastic and dECM groups were cultured for 35 days in a serum-free chondrogenic medium with 1 ng/mL of IL-1β. Chondrogenic differentiation was evaluated using Alcian blue staining for sulfated GAGs, immunostaining for type II collagen, biochemical analysis for DNA and GAG amounts in pellets, and real-time PCR for chondrogenic markers in terms of ACAN and COL2A1 and inflammatory factors including PTGS2 (prostaglandin G/H synthase and cyclooxygenase), VEGFA (vascular endothelial growth factor A), and MMP13 (matrix metallopeptidase 13).
Total RNA was extracted from pellets (n=4) using an RNase-free pestle in TRIzol® (Invitrogen). About 1 μg of mRNA was used for reverse transcription with a High-Capacity cDNA Archive Kit (Applied Biosystems) at 37°C for 120 min. Chondrogenic marker genes [COL2A1 (assay ID: Hs00156568_m1), ACAN (assay ID: Hs00153935_m1), and SOX9 (assay ID: Hs00165814_m1)] and inflammatory genes [PTGS2 (assay ID: Hs00153133_m1), VEGFA (assay ID: Hs00900055_m1), and MMP13 (assay ID: Hs00233992_m1)] were customized by Applied Biosystems as part of the Custom TaqMan® Gene Expression Assays. Eukaryotic 18S RNA (assay ID: Hs99999901_s1) was carried out as the endogenous control gene. Real-time PCR was performed with the iCycler iQ™ Multi-Color Real Time PCR Detection (Perkin-Elmer, Waltham, MA). Relative transcript levels were calculated as χ =2−ΔΔCt, in which ΔΔCt= ΔE−ΔC, ΔE=Ctexp−Ct18s, and ΔC=Ctct1−Ct18s.
2.8. Potential immune issues in hSDSCs after expansion on dECM deposited by either allogeneic or xenogeneic cells
Human SDSCs were expanded on either Plastic or dECM deposited by either hSDSCs (HECM) or pSDSCs (PECM) followed by the evaluation of surface marker expression for CD45 using anti-CD45 APC (BD Biosciences) and HLA-DR [major histocompatibility complex (MHC), class II, DR] using anti-human HLA-DR FITC (eBioscience, San Diego, CA), and isotype-matched IgGs (Beckman Coulter). Samples (n=5) of each 3×105 expanded cells were incubated on ice in cold PBS containing 0.1% Chrom-Pure Human IgG whole molecule (Jackson ImmunoResearch Laboratories) and 1% NaN3 (Sigma-Aldrich) for 30 min. The cells were then incubated in the dark in the primary antibodies (both HLA-DR and CD45 simultaneously) for 30 min. The fluorescence data were analyzed by a BD LSRFortessa™ (BD Biosciences), collected using BD FACSDiva 8.0 Software (BD Biosciences) at 10,000 events/sample. The results were analyzed using FCS Express 4 software (De Novo Software).
2.9. Statistics
Numerical data are presented as the mean and the standard error of the mean. A Mann-Whitney U test and a linear model with contrast analysis were used for pairwise comparison in biochemistry and real-time PCR data analysis. All statistical analyses were performed with SPSS 13.0 statistical software (SPSS Inc., Chicago, IL). A p value less than 0.05 was considered statistically significant.
3. Results
3.1. Preconditioning using sb203580 further promoted proliferation of dECM expanded cells
SEM data (Fig. 1A) showed that dECM deposited by hSDSCs was composed of randomly arranged fine matrix fibers and expansion on dECM yielded hSDSCs with a tiny fibroblast-like shape compared to a flattened and broad shape when grown on Plastic. Flow cytometry data (Fig. 1B) indicated that, despite a slight increase of proliferation index in hSDSCs when grown on Plastic, preconditioning using sb203580 presented a 2.62-fold increase in dECM expanded hSDSCs compared to a 1.65-fold increase in dECM expansion alone. Proliferation index data were confirmed by cell number change (Fig. 1C), in which preconditioning using sb203580 presented a 2.00-fold increase in dECM expanded hSDSCs compared to a 1.41-fold increase in dECM expansion alone. Flow cytometry data (Fig. 1D) also showed that mesenchymal stem cell (MSC) surface markers (CD29, CD90, and CD105) were down-regulated at the median in dECM expanded hSDSCs; when combined with sb203580 preconditioning, the expression of the above markers presented a slight increase. Interestingly, sb203580 preconditioning increased SSEA4 expression in both percentage and median of Plastic expanded hSDSCs followed by dECM expansion with the co-preconditioning of sb203580 and dECM expansion having the largest.
Fig. 1.

Preconditioning using sb203580 enhanced proliferation of dECM expanded hSDSCs. (A) Scanning electron microscopy was used to characterize surface topography of both Plastic and dECM (scale bar: 20 μm) and morphology of expanded hSDSCs (scale bar: 200 μm). (B) Human SDSCs were expanded on either dECM or Plastic for one passage (8 days) with or without preconditioning of sb203580. Flow cytometry was used to measure proliferation index of expanded cells. (C) A hemocytometer was used to measure cell numbers in T175 flasks (n=6) from each group. (D) Flow cytometry was used to measure both percentage and median fluorescence intensity of MSC surface markers (CD29, CD90, and CD105) and SSEA4 of expanded cells.
3.2. sb203580 preconditioning enhanced dECM expanded hSDSC chondrogenic potential while use of sb203580 in the induction phase arrested hSDSC chondrogenic differentiation
To determine whether p38 MAPK inhibitor preconditioning played a role in hSDSC expansion and chondrogenic potential, 10 μM of sb203580 was added to culture medium for hSDSC expansion on either Plastic or dECM. Both dECM expansion and sb203580 preconditioning alike led to chondrogenically differentiated 35-day pellets of a larger size with more intense staining of sulfated GAGs and type II collagen (Fig. 2A) and higher cell viability (DNA ratio adjusted by day 0) and GAG amount (chondrogenic marker) (Fig. 2B) compared to the corresponding Plastic group. Co-preconditioning with dECM and sb203580 yielded the largest hSDSC pellets (Fig. 2A) with the highest cell viability and GAG amount followed by the dECM group with the Plastic group having the lowest viability (Fig. 2B).
Fig. 2.
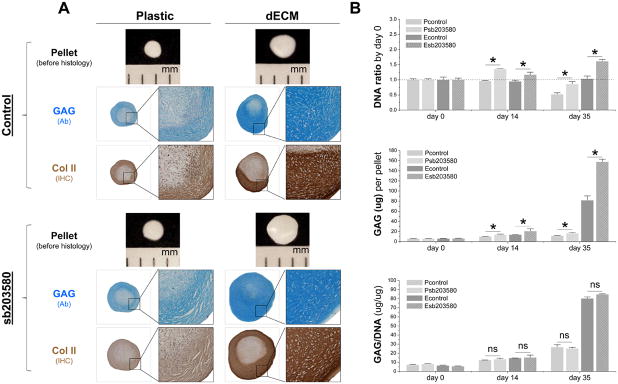
Preconditioning using sb203580 enhanced chondrogenesis of dECM expanded hSDSCs. Human SDSCs expanded on either dECM or Plastic for one passage (8 days) with or without preconditioning of sb203580 were followed by a 35-day chondrogenic induction in a pellet system. (A) Pellets were photographed before histology; Alcian blue (Ab) was used to stain sulfated GAGs and immunohistochemical staining (IHC) was for Col II. (B) Biochemical analysis was used for DNA and GAG amounts in chondrogenic pellets. Cell proliferation and viability were evaluated using DNA ratio (DNA amount at days 35 and 14 adjusted by that at day 0). Chondrogenic index was evaluated using ratio of GAG to DNA. Data are shown as average ± SD for n = 4. * p < 0.05 suggests a statistically significant difference. NS indicates no significant difference.
To determine whether p38 MAPK inhibitor played a role in hSDSC chondrogenic differentiation, 10 μM of sb203580 was supplemented in a serum-free chondrogenic medium. Due to a dramatic decrease in pellet size from both the Plastic and dECM groups, chondrogenic induction was ended at day 28. Despite enhanced chondrogenic staining in the pellets from the dECM group, the addition of sb203580 in the induction medium significantly decreased both staining and size of pellets in both groups, even as early as day 14 (Fig. 3A). The above findings were corroborated by biochemical analysis data, in which sb203580 arrested cell viability and reduced both GAG amount per pellet and ratio of GAG to DNA in both the Plastic and dECM groups (Fig. 3B).
Fig. 3.
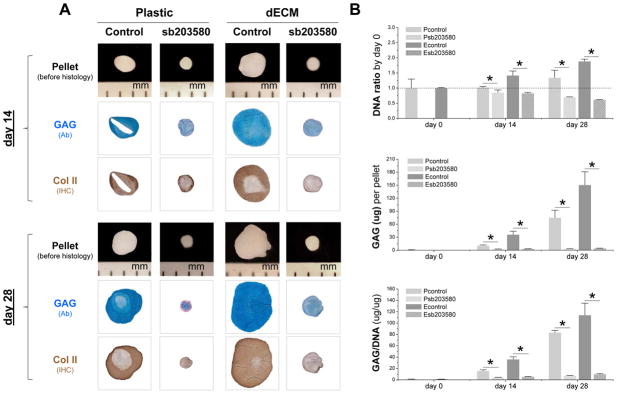
Human SDSCs expanded on either dECM or Plastic for one passage were followed by a 28-day chondrogenic induction in a pellet system with or without preconditioning of sb203580. (A) Pellets were photographed before histology; Alcian blue (Ab) was used to stain sulfated GAGs and immunohistochemical staining (IHC) was for Col II. (B) Biochemical analysis was used for DNA and GAG amounts in chondrogenic pellets. Cell proliferation and viability was evaluated using DNA ratio (DNA amount at days 28 and 14 adjusted by that at day 0). Chondrogenic index was evaluated using ratio of GAG to DNA. Data are shown as average ± SD for n = 4. * p < 0.05 suggests a statistically significant difference.
3.3. dECM expansion protects chondrogenically induced hSDSCs from IL-1β induced inflammatory stress
To determine whether dECM expanded hSDSCs had the ability to resist inflammatory stress, two doses of IL-1β, 0.1 and 1 ng/mL, were used to treat chondrogenically induced hSDSCs after expansion on either dECM or Plastic. Continuous treatment with IL-1β from day 0 to day 15 of chondrogenic induction yielded small pellets with less intensive staining of sulfated GAGs and type II collagen compared to the untreated control in both the dECM and Plastic groups, particularly for treatment with 1 ng/mL of IL-1β (Fig. 4A). Without IL-1β treatment, dECM expanded hSDSCs yielded pellets with higher GAG amount per pellet and ratio of GAG to DNA than those from Plastic expansion. With IL-1β treatment, both GAG amount per pellet and ratio of GAG to DNA decreased in a dose-dependent fashion; however, dECM expanded hSDSCs yielded pellets with a significantly higher ratio of GAG to DNA than those from Plastic expansion (Fig. 5A).
Fig. 4.

Chondrogenic induction of dECM-expanded hSDSCs in the presence of IL-1β. Human SDSCs were expanded on either Plastic or dECM for one passage followed by a pellet culture in a serum-free chondrogenic medium supplemented with either 0.1 or 1 ng/mL of IL-1β treatment for 15 days (A), 24 days (B), or the later stage between the 15th and 24th days (C). Chondrogenic induction without IL-1β treatment served as a control. Alcian blue (Ab) was used to stain sulfated GAGs. Immunohistochemistry staining (IHC) was used to detect collagen II (Col II).
Fig. 5.

Chondrogenic induction of dECM-expanded hSDSCs in the presence of IL-1β. Human SDSCs were expanded on either Plastic or dECM for one passage followed by a pellet culture in a serum-free chondrogenic medium supplemented with either 0.1 or 1 ng/mL of IL-1β treatment for 15 days (A), 24 days, or the later stage between the 15th and 24th days (B). Biochemical analysis was used for DNA and GAG amounts in the chondrogenically-induced pellets. Cell proliferation and viability was evaluated using DNA ratio (DNA amount adjusted by that at day 0). Chondrogenic index was evaluated using a ratio of GAG to DNA. (C) Real-time PCR was used to evaluate chondrogenic marker gene expression (SOX9, ACAN, and COL2A1) in day 24 pellets. Data are shown as average ± SD for n = 4. * p < 0.05 compared with the corresponding Plastic group. # p < 0.05 compared with the corresponding group with the same dose treatment of IL-1β.
Continuous treatment with IL-1β from day 0 to day 24 of chondrogenic induction yielded pellets with a dramatic decrease in both size and chondrogenic staining when 1 ng/mL of IL-1β was applied; there was no significant difference when 0.1 ng/mL of IL-1β was used, particularly for dECM expanded cells (Fig. 4B). Interestingly, treatment with IL-1β from day 15 to day 24 of chondrogenic induction yielded pellets with a significant improvement in both size and chondrogenic staining for both dECM and Plastic expansion even in the presence of 1 ng/mL of IL-1β (Fig. 4C). The above histology result was supported by quantification data. Biochemical analysis data showed that continuous treatment with 1 ng/mL of IL-1β yielded pellets with a significant decrease in both DNA ratio and ratio of GAG to DNA (Fig. 5B). However, treatment with 1 ng/mL of IL-1β from day 15 to day 24 of chondrogenic induction yielded pellets with a significant increase in both DNA ratio and GAG amount compared to the corresponding group with continuous IL-1β treatment (Fig. 5B). The above protein level data were also consistent with the mRNA data using real-time PCR for chondrogenic marker genes including SOX9, ACAN, and COL2A1 (Fig. 5C).
3.4. sb203580 preconditioning promotes dECM rejuvenated hSDSCs’ ability against inflammation during chondrogenic induction
The above results indicated that continuous treatment with 1 ng/mL of IL-1β from day 0 to day 24 of chondrogenic induction yielded pellets with diminished chondrogenic differentiation in hSDSCs from dECM expansion compared with the corresponding control without IL-1β treatment. In this study, we assumed that co-preconditioning using dECM and sb203580 would retard the diminished chondrogenic differentiation of dECM expanded hSDSCs in the presence of a high concentration of IL-1β. Continuous treatment for 35 days with 1 ng/mL of IL-1β reduced the difference in chondrogenic capacity in hSDSCs from dECM and Plastic expansion. Compared to the slight increase in pellet size observed in Plastic expanded cells, preconditioning using sb203580 dramatically increased both size and chondrogenic staining in the 35-day IL-1β treated pellets from dECM expanded hSDSCs (Fig. 6A). This histology result was also supported by both biochemical analysis and real-time PCR data. dECM expanded cells yielded pellets with a higher amount of GAG and a higher ratio of GAG to DNA compared to those from Plastic expanded cells in the presence of 1 ng/mL of IL-1β (Fig. 6B) despite no difference in ACAN and COL2A1 (Fig. 6C). Preconditioning using sb203580 dramatically increased both GAG amount and ratio of GAG to DNA (Fig. 6B) as well as ACAN and COL2A1 (Fig. 6C) in hSDSC pellets from both dECM and Plastic expansion with the co-preconditioning group being the highest.
Fig. 6.

35-day-chondrogenic induction of dECM-expanded hSDSCs preconditioned with sb203580 in the presence of 1 ng/mL IL-1β. Human SDSCs were expanded on either Plastic or dECM for one passage with or without preconditioning using sb203580 followed by a 35-day pellet culture in a serum-free chondrogenic medium supplemented with continuous treatment of IL-1β at 1 ng/mL. (A) Alcian blue (Ab) was used to stain sulfated GAGs. Immunohistochemistry staining (IHC) was used to detect collagen II (Col II). (B) Biochemical analysis was used for DNA and GAG amounts in the 35-day chondrogenically-induced pellets. Chondrogenic index was evaluated using a ratio of GAG to DNA. (C) Real-time PCR was used to evaluate chondrogenic marker gene expression (ACAN and COL2A1) in day 35 pellets. (D) Real-time PCR was used to evaluate inflammatory marker gene expression (PTGS2, MMP13, and VEGFA) in day 35 pellets. Data are shown as average ± SD for n = 4. * p < 0.05 compared with the corresponding Plastic group. # p < 0.05 compared with the corresponding group without sb203580 treatment.
To determine whether sb203580 preconditioning had any inhibitory effect on inflammation during subsequent chondrogenic induction, real-time PCR data (Fig. 6D) showed that, without sb203580 preconditioning, dECM expanded hSDSCs yielded 35-day pellets with a significantly lower VEGFA level compared to those from Plastic expanded cells. With sb203580 preconditioning, dECM expanded hSDSCs yielded pellets with significantly lower levels of PTGS2, MMP13, and VEGFA; a notable increase in these gene expressions occurred in pellets from Plastic expanded hSDSCs.
3.5. Allogeneic dECM expanded hSDSCs exhibited a non-detectable HLA-DR expression
Flow cytometry was also used to determine whether expansion of hSDSCs on Plastic, HECM (allogeneic substrate), or PECM (xenogeneic substrate) increased HLA-DR expression. After subtracting the respective unstained negative controls (Fig. 7A/B) from the percent HLA-DR-positive values for both Plastic (Fig. 7C) and dECM substrates (Fig. 7D/E), the percent HLA-DR-positive cells in each population were 1.08% for Plastic expanded hSDSCs (Fig. 7C), 0.81% for HECM expanded hSDSCs (Fig. 7D), and 6.48% for PECM expanded hSDSCs (Fig. 7E). These cells also exhibited a similar trend when comparing the mean fluorescence intensity (MFI) values: the Plastic group was 474.19, the HECM group was 357.23, and the PECM group was 583.29. Additionally, all hSDSC samples were CD45 negative, ensuring there was no persisting contamination in the hSDSC populations with HLA-DR+ immune cells following their isolation from synovial tissue.
Fig. 7.

Flow cytometry analysis of CD45 and HLA-DR surface marker expression following cell expansion. Unstained Plastic-expanded (A) and dECM-expanded (B) hSDSCs were used as negative controls. CD45 and HLA-DR surface marker expression was analyzed for each sample of hSDSCs expanded on Plastic (C), HECM (D), and PECM substrates (E). Additionally, a separate macrophage/monocyte population was analyzed for CD45 expression (F) as a positive control.
4. Discussion
Cartilage tissue engineering has emerged as a potential therapeutic option for cartilage repair; however, once transplanted into the joints, cells or premature cartilage often face an inflammatory environment, making cartilage resurfacing challenging [13]. Among the proinflammatory cytokines, IL-1β acts as an important mediator of increased ECM degradation by stimulating a number of events, such as an increase of MMPs and nitric oxide production [23] and by suppressing type II collagen and proteoglycan synthesis [24]. In osteoarthritic joints, the IL-1β concentration was reported to range from 1-10 pg/mL [25]. Xie et al. reported that the mean concentration of IL-1β in synovial fluid from early osteoarthritis in dysplastic hips was 55 (SD 12), ranging from 24 to 74 pg/mL [26]. Attur et al. reported that 5 ng/mL of IL-1β could significantly suppress proteoglycan synthesis from both human and bovine chondrocytes [27]. Yudoh et al. reported a significant reduction in production of type II collagen by rabbit chondrocytes incubated in the presence of 10 ng/mL of IL-1β [28]. Thus, in this study, IL-1β was added into chondrogenic medium to simulate the inflammatory environment often encountered in joint capsule; 0.1 ng/mL of IL-1β was chosen to represent a relatively low concentration despite the fact that this dose is still higher than the reported pathological concentration of IL-1β in osteoarthritis [25]. One ng/mL of IL-1β was chosen to represent a relatively high concentration.
In this report, we found that dECM expansion could maintain an advantage over Plastic expansion in chondrogenic capacity of hSDSCs in the presence of a low concentration (0.1 ng/mL) of IL-1β. However, this advantage was diminished when a high concentration (1 ng/mL) of IL-1β was applied during chondrogenic induction. Interestingly, premature tissue pellets from dECM expanded hSDSCs could, to a certain degree, resist the decline of chondrogenic differentiation resulting from a high concentration of IL-1β. This finding is consistent with a previous report in which more mature cartilaginous tissue was more resistant to 1 ng/mL of IL-1β exposure [29]. Previous investigations showed that inflammatory-related genes such as MMP13, PTGS2, and VEGF were upregulated in the presence of IL-1β but were blocked by application of sb203580 [30–32]. In this study, despite a higher expression in the preconditioning group by sb203580, these inflammatory genes were significantly downregulated in chondrogenically induced hSDSCs when a co-preconditioning strategy with dECM expansion was applied. This finding might be attributable to the anti-inflammatory function of sb203580 [16,33] and antioxidant characteristics of dECM during cell expansion [10].
Consistent with previous reports [34,35], we found that the use of sb203580 in the induction medium significantly diminished TGFβ mediated hSDSC chondrogenic differentiation. It is well known that TGFβ induces chondrogenic differentiation classically through the SMAD pathway and also via p38 MAPK signaling [36]. For instance, TGFβ induced rapid phosphorylation of SMAD2, extracellular signal-activated kinase 1/2 (ERK1/2), and p38 MAPK in ATDC5 cells; however, incubation with sb203580 repressed TGFβ-induced ACAN, suggesting an important role for transcriptional cross-talk between the TGFβ and p38 MAPK pathways in expression of early chondrocyte differentiation [37]. A recent study suggested that, following activation by TGFβ, MAPKs (in particular the p38 MAPK pathway) were involved in the control of post-translational modification of SMADs by a direct phosphorylation or through their downstream effector molecules [38]. Another report indicated that SOX9 is likely a downstream target of the p38 MAPK pathway because the increased activity of the SOX9-dependent COL2A1 enhancer in primary chondrocytes was abolished by sb203580 [39].
Our recent study demonstrated that dECM deposited by young and healthy hSDSCs could rejuvenate older hSDSCs in chondrogenic potential [22], indicating that allogeneic dECM can be a promising approach to expand patient hSDSCs for autologous transplantation. To solve the immune concerns in hSDSCs after expansion on allogeneic dECMs, expression of HLA-DR was used as an indicator since HLA-DR is known to be involved in graft rejection and other adverse immune events [40,41]. The flow cytometry data showed no increase in HLA-DR expression in the HECM (allogeneic control) expanded cells compared to the Plastic group (negative control); however, there was a greater than 6-fold increase in the percent HLA-DR-positive expression when hSDSCs were expanded on PECM (xenogeneic control) versus Plastic or HECM substrates. Not only does the PECM-expansion of hSDSCs seem to produce more HLA-DR+ cells than the Plastic or HECM-expansion, but the MFI values also suggest that these PECM-expanded cells seem to express more HLA-DR per cell. These increases in HLA-DR+ expression could be troubling for future clinical use of PECM substrates for hSDSC expansion strategies, as studies have demonstrated the ability of MHC class II-positive antigen-presenting cells, and more recently synovial fibroblasts, to cause T-cell activation [42,43]. In order to verify that all hSDSC populations were void of immune cells whose presence could create false positives for HLA-DR expression, CD45 expression was also examined. All hSDSCs, regardless of expansion condition, were comparatively negative for CD45 expression, leading us to conclude that the hSDSCs were the cells expressing HLA-DR. Compared to potential immunogenicity from PECM-expanded hSDSCs, allogeneic HECM-expansion seems a potentially more attractive strategy for future clinical use; however, further in vivo studies will be necessary to investigate this issue.
5. Conclusion
This study is the first to investigate p38 MAPK inhibitor for its preconditioning in hSDSC proliferation and chondrogenic potential as well as its inhibitory effect on hSDSC chondrogenic differentiation. The joint preconditioning effect of p38 MAPK inhibitor and dECM expansion on hSDSC chondrogenesis in the presence of IL-1β was also explored. We found that preconditioning with sb203580 favored hSDSC proliferation and chondrogenic capacity, which was enhanced in combination with dECM expansion; however, the application of sb203580 in the induction medium suppressed the growth of chondrogenic pellets in both the Plastic and dECM groups. The inhibitory effect of IL-1β on chondrogenic differentiation of expanded hSDSCs was in a dose- and phase-dependent manner. Despite the fact that pre-differentiated hSDSCs acquired an enhanced capacity against inflammatory insult, expanded hSDSCs could only survive chondrogenic induction in the presence of a low dose of IL-1β; interestingly, we found that sb203580 preconditioning could enhance matrix expanded hSDSC chondrogenesis even under stimulation by a high dose of IL-1β. This preconditioning strategy could enhance the successful rate of implantation of undifferentiated stem cells or premature tissue constructs in treating cartilage defects in which a harsh inflammatory environment exists. Non-detectable expression of HLA-DR in hSDSCs grown on allogeneic dECM indicates the feasibility of commercial preparation of these dECMs from healthy, young donors for patients who need autologous transplantation.
Acknowledgments
We thank Suzanne Danley for editing the manuscript. This project was partially supported by Research Grants from the AO Foundation (S-12-19P) and the National Institutes of Health (1 R03 AR062763-01A1). We also appreciate support from the WVU Flow Cytometry Core (MBRCC CoBRE grant: GM 103488/RR032138; ARIA S10 grant: RR020866; Fortess S10 grant: OD016165; WV InBRE grant: GM103434).
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pizzute T, Lynch K, Pei M. Impact of tissue-specific stem cells on lineage-specific differentiation: a focus on the musculoskeletal system. Stem Cell Rev. 2015;11:119–32. doi: 10.1007/s12015-014-9546-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones B, Pei M. Synovium-derived stem cells: a tissue-specific stem cell for cartilage tissue engineering and regeneration. Tissue Eng Part B Rev. 2012;18:301–11. doi: 10.1089/ten.TEB.2012.0002. [DOI] [PubMed] [Google Scholar]
- 3.Lee JC, Min HJ, Park HJ, Lee S, Seong SC, Lee MC. Synovial membrane-derived mesenchymal stem cells supported by platelet-rich plasma can repair osteochondral defects in a rabbit model. Arthroscopy. 2013;29:1034–46. doi: 10.1016/j.arthro.2013.02.026. [DOI] [PubMed] [Google Scholar]
- 4.Pei M, He F, Boyce BM, Kish VL. Repair of full-thickness femoral condyle cartilage defects using allogeneic synovial cell-engineered tissue constructs. Osteoarthritis Cartilage. 2009;17:714–22. doi: 10.1016/j.joca.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 5.Pei M, He F, Li J, Tidwell JE, Jones AC, McDonough EB. Repair of large animal partial-thickness cartilage defects through intraarticular injection of matrix-rejuvenated synovium-derived stem cells. Tissue Eng Part A. 2013;19:1144–54. doi: 10.1089/ten.TEA.2012.0351. [DOI] [PubMed] [Google Scholar]
- 6.Lynch K, Pei M. Age associated communication between cells and matrix: a potential impact on stem cell-based tissue regeneration strategies. Organogenesis. 2014;10:289–98. doi: 10.4161/15476278.2014.970089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li JT, Pei M. Cell senescence: a challenge in cartilage engineering and regeneration. Tissue Eng Part B. 2012;18:270–87. doi: 10.1089/ten.TEB.2011.0583. [DOI] [PubMed] [Google Scholar]
- 8.He F, Chen XD, Pei M. Reconstruction of an in vitro tissue-specific microenvironment to rejuvenate synovium-derived stem cells for cartilage tissue engineering. Tissue Eng Part A. 2009;15:3809–21. doi: 10.1089/ten.TEA.2009.0188. [DOI] [PubMed] [Google Scholar]
- 9.Pei M, Li JT, Shoukry M, Zhang Y. A Review of Decellularized Stem Cell Matrix: a Novel Cell Expansion System for Cartilage Tissue Engineering. Eur Cell Mater. 2011;22:333–43. doi: 10.22203/ecm.v022a25. [DOI] [PubMed] [Google Scholar]
- 10.Pei M, Zhang Y, Li JT, Chen DQ. Antioxidation of decellularized stem cell matrix promotes human synovium-derived stem cell-based chondrogenesis. Stem Cells Dev. 2013;22:889–900. doi: 10.1089/scd.2012.0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gruber J, Vincent TL, Hermansson M, Bolton M, Wait R, Saklatvala J. Induction of interleukin-1 in articular cartilage by explantation and cutting. Arthritis Rheum. 2004;50:2539–46. doi: 10.1002/art.20369. [DOI] [PubMed] [Google Scholar]
- 12.Hembry RM, Dyce J, Driesang I, Hunziker EB, Fosang AJ, Tyler JA, et al. Immunolocalization of matrix metalloproteinases in partial-thickness defects in pig articular cartilage. A preliminary report. J Bone Joint Surg Am. 2001;83A:826–38. doi: 10.2106/00004623-200106000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Pizzute T, Pei M. Anti-inflammatory strategies in cartilage repair. Tissue Eng Part B Rev. 2014;20:655–68. doi: 10.1089/ten.teb.2014.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 15.Schindler JF, Monahan JB, Smith WG. p38 pathway kinases as anti-inflammatory drug targets. J Dent Res. 2007;86:800–11. doi: 10.1177/154405910708600902. [DOI] [PubMed] [Google Scholar]
- 16.Brown KK, Heitmeyer SA, Hookfin EB, Hsieh L, Buchalova M, Taiwo YO, et al. P38 MAP kinase inhibitors as potential therapeutics for the treatment of joint degeneration and pain associated with osteoarthritis. J Inflamm (Lond) 2008;5:22. doi: 10.1186/1476-9255-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braem K, Luyten FP, Lories RJ. Blocking p38 signalling inhibits chondrogenesis in vitro but not ankylosis in a model of ankylosing spondylitis in vivo. Ann Rheum Dis. 2012;71:722–8. doi: 10.1136/annrheumdis-2011-200377. [DOI] [PubMed] [Google Scholar]
- 18.Prasadam I, Mao X, Wang Y, Shi W, Crawford R, Xiao Y. Inhibition of p38 pathway leads to OA-like changes in a rat animal model. Rheumatology. 2012;51:813–23. doi: 10.1093/rheumatology/ker360. [DOI] [PubMed] [Google Scholar]
- 19.Yong HY, Koh MS, Moon A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin Investig Drugs. 2009;18:1893–905. doi: 10.1517/13543780903321490. [DOI] [PubMed] [Google Scholar]
- 20.Li JT, Jones B, Zhang Y, Vinardell T, Pei M. Low density expansion rescues human synovium-derived stem cells from replicative senescence. Drug Deliv Transl Res. 2012;2:363–74. doi: 10.1007/s13346-012-0094-y. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Li J, Davis ME, Pei M. Delineation of an in vitro chondrogenesis of human synovial stem cells following preconditioning using decellularized matrix. Acta Biomater. 2015;20:39–50. doi: 10.1016/j.actbio.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li JT, Hansen K, Zhang Y, Dong CB, Dinu C, Dzieciatkowska M, et al. Rejuvenation of chondrogenic potential by young stem cell microenvironment. Biomaterials. 2014;35:642–53. doi: 10.1016/j.biomaterials.2013.09.099. [DOI] [PubMed] [Google Scholar]
- 23.Van der Kraan PM, van den Berg WB. Anabolic and destructive mediators in osteoarthritis. Curr Opin Clin Nutr Metab Care. 2000;3:205–11. doi: 10.1097/00075197-200005000-00007. [DOI] [PubMed] [Google Scholar]
- 24.Chevalier X. Upregulation of enzymatic activity by interleukin-1 in osteoarthritis. Biomed Pharmacother. 1997;51:58–62. doi: 10.1016/s0753-3322(97)87727-x. [DOI] [PubMed] [Google Scholar]
- 25.Barakat AF, Elson CJ, Westacott CI. Susceptibility to physiological concentrations of IL-1beta varies in cartilage at different anatomical locations on human osteoarthritic knee joints. Osteoarthritis Cartilage. 2002;10:264–9. doi: 10.1053/joca.2002.0515. [DOI] [PubMed] [Google Scholar]
- 26.Xie J, Naito M, Maeyama A. Intracapsular pressure and interleukin-1beta cytokine in hips with acetabular dysplasia. Acta Orthop. 2010;81:189–92. doi: 10.3109/17453671003717807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attur MG, Dave MN, Clancy RM, Patel IR, Abramson SB, Amin AR. Functional genomic analysis in arthritis-affected cartilage: yin-yang regulation of inflammatory mediators by alpha 5 beta 1 and alpha V beta 3 integrins. J Immunol. 2000;164:2684–91. doi: 10.4049/jimmunol.164.5.2684. [DOI] [PubMed] [Google Scholar]
- 28.Yudoh K, Shishido K, Murayama H, Yano M, Matsubayashi K, Takada H, et al. Water-soluble C60 fullerene prevents degeneration of articular cartilage in osteoarthritis via down-regulation of chondrocyte catabolic activity and inhibition of cartilage degeneration during disease development. Arthritis Rheum. 2007;56:3307–18. doi: 10.1002/art.22917. [DOI] [PubMed] [Google Scholar]
- 29.Francioli S, Cavallo C, Grigolo B, Martin I, Barbero A. Engineered cartilage maturation regulates cytokine production and interleukin-1β response. Clin Orthop Relat Res. 2011;469:2773–84. doi: 10.1007/s11999-011-1826-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joos H, Albrecht W, Laufer S, Brenner RE. Differential effects of p38MAP kinase inhibitors on the expression of inflammation-associated genes in primary, interleukin-1beta-stimulated human chondrocytes. Br J Pharmacol. 2010;160:1252–62. doi: 10.1111/j.1476-5381.2010.00760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joos H, Albrecht W, Laufer S, Brenner RE. Influence of p38MAPK inhibition on IL-1beta-stimulated human chondrocytes: a microarray approach. Int J Mol Med. 2009;23:685–93. doi: 10.3892/ijmm_00000181. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka T, Kanai H, Sekiguchi K, Aihara Y, Yokoyama T, Arai M, et al. Induction of VEGF gene transcription by IL-1 beta is mediated through stress-activated MAP kinases and Sp1 sites in cardiac myocytes. J Mol Cell Cardiol. 2000;32:1955–67. doi: 10.1006/jmcc.2000.1228. [DOI] [PubMed] [Google Scholar]
- 33.Han SK, Jeon SJ, Miyazawa K, Yi SY, Yoo YS. Enhancement of anti-inflammatory tendency by SB203580, p38alpha specific inhibitor, in human fibroblast-like synoviocyte cell line, MH7A. Rheumatol Int. 2006;26:972–8. doi: 10.1007/s00296-006-0109-4. [DOI] [PubMed] [Google Scholar]
- 34.Oh CD, Chang SH, Yoon YM, Lee SJ, Lee YS, Kang SS, et al. Opposing role of mitogen-activated protein kinase subtypes, erk-1/2 and p38, in the regulation of chondrogenesis of mesenchymes. J Biol Chem. 2000;275:5613–9. doi: 10.1074/jbc.275.8.5613. [DOI] [PubMed] [Google Scholar]
- 35.Prasadam I, Mao X, Wang Y, Shi W, Crawford R, Xiao Y. Inhibition of p38 pathway leads to OA-like changes in a rat animal model. Rheumatology (Oxford) 2012;51:813–23. doi: 10.1093/rheumatology/ker360. [DOI] [PubMed] [Google Scholar]
- 36.Horbelt D, Denkis A, Knaus P. A portrait of Transforming Growth Factor β superfamily signalling: Background matters. Int J Biochem Cell Biol. 2012;44:469–74. doi: 10.1016/j.biocel.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe H, de Caestecker MP, Yamada Y. Transcriptional cross-talk between Smad, ERK1/2, and p38 mitogen-activated protein kinase pathways regulates transforming growth factor-beta-induced aggrecan gene expression in chondrogenic ATDC5 cells. J Biol Chem. 2001;276:14466–73. doi: 10.1074/jbc.M005724200. [DOI] [PubMed] [Google Scholar]
- 38.Javelaud D, Mauviel A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene. 2005;24:5742–50. doi: 10.1038/sj.onc.1208928. [DOI] [PubMed] [Google Scholar]
- 39.Zhang R, Murakami S, Coustry F, Wang Y, de Crombrugghe B. Constitutive activation of MKK6 in chondrocytes of transgenic mice inhibits proliferation and delays endochondral bone formation. Proc Natl Acad Sci U S A. 2006;103:365–70. doi: 10.1073/pnas.0507979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Claesson K. Mechanisms in organ allograft rejection. An experimental and clinical study. Scand J Urol Nephrol Suppl. 1987;103:1–42. [PubMed] [Google Scholar]
- 41.Döring M, Rohrer KM, Erbacher A, Gieseke F, Schwarze CP, Bader P, et al. Human leukocyte antigen DR surface expression on CD14+ monocytes during adverse events after hematopoietic stem cell transplantation. Ann Hematol. 2015;94:265–73. doi: 10.1007/s00277-014-2185-y. [DOI] [PubMed] [Google Scholar]
- 42.Conte E, Gili E, Fruciano M, Fagone E, Vancheri C. Human lung fibroblasts increase CD4(+)CD25(+)Foxp3(+) T cells in co-cultured CD4(+) lymphocytes. Cell Immunol. 2013;285:55–61. doi: 10.1016/j.cellimm.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 43.Kraft M, Filsinger S, Krämer KL, Kabelitz D, Hänsch GM, Schoels M. Synovial fibroblasts as accessory cells for staphylococcal enterotoxin-mediated T-cell activation. Immunology. 1995;85:461–6. [PMC free article] [PubMed] [Google Scholar]