

Abstract
In light of greatly improved long-term patient and graft survival after renal transplantation, improving other clinical outcomes such as risk of fracture and cardiovascular disease is of paramount importance. After renal transplantation, a large percentage of patients lose bone. This loss of bone results from a combination of factors that include pre-existing renal osteodystrophy, immunosuppressive therapy, and the effects of chronically reduced renal function after transplantation. In addition to low bone volume, histological abnormalities include decreased bone turnover and defective mineralization. Low bone volume and low bone turnover were recently shown to be associated with cardiovascular calcifications, highlighting specific challenges for medical therapy and the need to prevent low bone turnover in the pretransplant patient. This Review discusses changes in bone histology and mineral metabolism that are associated with renal transplantation and the effects of these changes on clinical outcomes such as fractures and cardiovascular calcifications. Therapeutic modalities are evaluated based on our understanding of bone histology.
Introduction
The development of novel immunosuppressive therapies has led to a >90% increase in the 1-year survival rates of renal allografts after transplantation.1 In addition, long-term follow-up studies report renal allograft survival rates of ≥70% at 5 years and ≥50% at 10 years after transplantation.2 Accordingly, improving the long-term survival and quality of life for renal transplant recipients has become a major focus of post-transplantation patient care and includes prevention of cardiovascular complications, bone disease and fractures associated with bone disease.
Post-Transplant Changes in Bone
Disturbances of bone metabolism are common complications that affect patients after successful renal transplantation and represent important causes of morbidity and mortality. Post-transplantation bone disease is distinctly different from common forms of osteoporosis. Three major components contribute to bone metabolic disturbances in patients after renal transplantation: pre-existing renal osteodystrophy at time of renal transplantation, effects and consequences of transplantation-specific therapies on bone, and the effects of reduced renal function after renal transplantation.
Pre-existing renal osteodystrophy
Renal osteodystrophy was traditionally classified into four major groups: hyperparathyroid bone disease, adynamic bone disease, mixed renal osteodystrophy, and osteomalacia.3-5 Hyperparathyroid bone disease is characterized by a marked increase in bone turnover. Irregularly-shaped trabecules display numerous abnormal remodelling sites, and an unusually high number of bone cells with irregular arrangement and shape. Representing the other end of the spectrum, low turnover renal osteodystrophy is characterized by a profound decrease in active remodelling sites. Two separate entities of low turnover renal osteodystrophy exist: low turnover osteomalacia and adynamic bone disease. Low turnover osteomalacia is characterized by an accumulation of unmineralized matrix in which a decrease in mineralization precedes or is more pronounced than the inhibition of collagen deposition. Although the bone volume fraction of total tissue volume (bone volume/tissue volume6) can vary, mineralized bone volume is always low in these patients. Similarly, bone volume is frequently low in adynamic bone disease. In adynamic bone disease, reduced mineralization is paralleled by a decrease in bone formation. Adynamic bone disease is characterized by the presence of few osteoid seams and few osteoblasts. Osteoclast number may be low, normal or high. Mixed uremic osteodystrophy is primarily caused by defective mineralization with or without increased bone formation and by increased parathyroid hormone activity in bone. These features coexist to varying degrees in different patients. Bone volume is extremely variable and depends on a dominant pathogenic cause. Other features of mixed uremic osteodystrophy include increased numbers of heterogeneous remodelling sites and, typically, an increase in osteoclast number.
Since 2000, this complex classification system has been streamlined to focus on the underlying histological abnormalities in bone. These abnormalities include changes in bone turnover, mineralization and bone balance, which result in changes in bone volume.7,8 Bone balance describes the equilibrium between bone formation and bone resorption. Loss of bone volume occurs when bone resorption exceeds bone formation, whereas an increase in bone volume is observed when the rate of bone resorption falls below the rate at which bone is formed. Entities that are associated with changes in bone turnover include mixed renal osteodystrophy, hyperparathyroid bone disease and adynamic bone disease. Mineralization abnormalities describe the histological changes seen in patients with osteomalacia and mixed renal osteodystrophy. Bone volume changes occur mainly in hyperparathyroid and adynamic bone disease.
In a 2008 survey of 544 bone biopsy samples from patients with chronic kidney disease (CKD) stage 5 on dialysis, bone turnover was low in 52%, normal in 21% and high in 27% of biopsies. Defective mineralization was found in only 3%. Cancellous bone volume was low in 32%, normal in 30%, and high in 38%.8 In the assessment of bone volume abnormalities, it is important to differentiate between cortical and cancellous bone. Loss of cortical bone occurs mainly in patients with high turnover bone disease, while loss of cancellous bone is often seen in patients with low bone turnover.9 These abnormalities cause disturbances in the two critical functions of bone—mechanical strength and mineral metabolic activity. Disturbances in the latter function in patients with abnormal bone turnover are evidenced by abnormal calcium kinetics in low and high bone turnover and frequent hyperphosphatemia in high bone turnover.9,10 The mechanical function of bone is served mainly by cortical bone, whereas the metabolic function is primarily served by cancellous bone.11,12 The clinical outcome of decreased bone strength is fracture, while abnormal metabolic activity results in the inability to maintain mineral homeostasis, which is associated with vascular and soft tissue calcifications.13, 14
Precise diagnosis of bone turnover abnormalities in patients with renal disease is of paramount importance since the therapeutic approaches to the different abnormalities are distinctly different and misdiagnosis can lead to serious adverse clinical outcomes. In patients with advanced CKD, high bone turnover is managed therapeutically by the suppression of parathyroid hormone (PTH) by use of vitamin D analogues and/or calcimimetics.15 Management of low bone turnover consists mainly of preventative measures such as avoidance of excessive doses of calcium and/or vitamin D. No prospective studies have evaluated the effects of anabolic agents for the treatment of low bone turnover. Low bone mineralization requires vitamin D replacement therapy including administration of both dihydroxylated and hydroxylated vitamin D since both metabolites are needed for normal bone mineralization.16,17 Bone volume abnormalities represent a therapeutic challenge in transplant recipients as the most commonly used antiosteoporotic treatment modality, bisphosphonates, have a rather strong and lasting suppressive effect on bone turnover.
Effects of transplant-specific therapies
Glucocorticoids
The role of glucocorticoids in the development of secondary osteoporosis in renal transplant recipients is well established.18-22 During the first 6 months after transplantation, rapid bone loss secondary to a glucocorticoid-induced acceleration in bone remodeling occurs in cancellous bone.23 A study that involved serial bone biopsies at 22 days and 160 after transplantation reported impaired osteoblastogenesis and early osteoblast apoptosis.24 Glucocorticoids inhibit bone formation by reducing osteoblast proliferation, function (by reducing levels of type 1 collagen, IGF-I and osteocalcin),25 and life span (by induction of osteoblast apoptosis).26 Glucocorticoids also promote osteoclastogenesis by increasing levels of RANKL and decreasing levels of the RANKL decoy receptor osteoprotegerin.25 The negative effects of glucocorticoids on bone turnover and bone volume are related to the cumulative dose of glucocorticoids in renal transplant recipients (Figure 1).22
Figure 1.


Glucocorticoids reduce bone turnover. a) Relationship between the cumulative dose of prednisone and activation frequency in 53 patients after renal transplantation. Coefficient of correlation = 0.43, P < 0.05. b) Relationship between cumulative dose of prednisone and BV/TV in 53 patients after renal transplantation. Coefficient of correlation = 0.54, P < 0.05. Abbreviation: BV/TV, bone volume fraction of total tissue volume. Permission obtained from American Society of Nephrology © Monier-Faugere, M. C. et al. High prevalence of low bone turnover and occurrence of osteomalacia after kidney transplantation. J. A. Soc. Nephrol. 11, 1093-1099 (2000).
The additional effects of glucocorticoids to reduce intestinal calcium absorption and increase urinary calcium excretion (resulting in a negative calcium balance) can foster the development of persistent hyperparathyroidism,27,28 which in combination with hypogonadotropic hypogonadism promotes further bone loss.29,30 Although an increased loss of lumbar vertebrae mass was reported to correlate with high daily and cumulative glucocorticoid doses as well as with more frequent rejection episodes (requiring glucocorticoid pulse therapy),20 studies that have focused on fracture risk in renal transplant recipients have not found an association between corticosteroid use and fracture risk even in very long term (>6 years) studies.31,32 Despite current data not supporting the concept of a mean daily or cumulative glucocorticoid ‘threshold’ that might increase the risk of fracture in renal transplant recipients, the multiple detrimental effects of glucocorticoids on bone and mineral metabolism warrant the use of the lowest possible glucocorticoid doses or the avoidance of glucocorticoids.
Calcineurin inhibitors
Both, ciclosporine and tacrolimus use have been linked to osteoporosis in clinical studies,33,34 and progression of bone loss with calcineurin inhibitor monotherapies has been demonstrated in rodent models.35,36 Despite these findings, population-based studies that have focused on fracture risk could not establish an association between use of calcineurin inhibitors and fracture risk.32,37
In murine models, ciclosporin stimulates osteoclastic activity more than that of osteoblasts, which results in bone loss.38,39 This effect is, however, attenuated by parathyroidectomy.40 Other data that compared the effects of ciclosporin treatment showed no differences in bone volume between normal and moderately uremic rats. When compared to vehicle-treated controls, ciclosporin treatment resulted in higher mineralized bone volume fraction of total tissue volume and in shorter mineralization lag time.41 In 139 renal transplant recipients who received ciclosporin, a transient rise in serum alkaline phosphatase levels that peaked after 8±6 months and that exceeded the normal range for 16±10 months post-transplantation was demonstrated.42 In another clinical study that combined ciclosporin treatment with no or very low dosing of glucocorticoids, bone loss after renal transplantation was prevented by glucocorticoid sparing.19 In these patients, serum PTH remained elevated 1–2 years after transplantation despite normalization of serum 1,25(OH)2 vitamin D levels. Bone histological abnormalities were consistent with hyperparathyroid bone changes suggesting a resistance of bone to normal levels of 1,25(OH)2 vitamin D.19
Tacrolimus treatment was reported to result in severe cancellous bone loss associated with high bone turnover in a rat model.36 This agent induces osteoblastic differentiation concurrently with the activation of alkaline phosphatase and osteocalcin transcription35 and has been shown to induce osteoclast differentiation in the presence of elevated PTH levels.36 A 2008 study that used serum markers to measure bone turnover did not find a difference between tacrolimus and ciclosporin-treated renal transplant recipients, suggesting that bone turnover abnormalities might be similar in these patient groups.43
The isolated effects of calcineurin inhibitors are difficult to evaluate in human studies as these immunosuppressants are usually used concomitantly with glucocorticoids. The development of steroid-free protocols might, however, offer new opportunities to evaluate some of the clinical questions related to bone metabolism in renal transplant recipients.
Other immunosuppressive agents
Although clinical data are scarce, the use of mycophenolate mofetil, sirolimus, azathioprine, and induction therapies might exert protective effects on the skeleton in addition to reducing glucocorticoid use. Although mycophenolate mofetil, sirolimus, and azathioprine did not affect bone volume in rodents,44-46 a recent in vitro study suggests sirolimus might interfere with the proliferation and differentiation of osteoblasts.47 Everolimus, an orally available derivative of sirolimus, inhibited osteoclast formation and activity and osteoblastic differentiation in mouse and human cell lineages in an in vitro study. In that study, everolimus reduced cancellous bone loss in ovarectomized rats by decreasing osteoclast-mediated bone resorption.48
Effects of reduced renal function
Despite a paucity of data on the isolated effects of reduced renal function on bone disease after renal transplantation, it has been postulated that patients with a post-transplantation glomerular filtration rate (GFR) <70 ml/min/1.73m2 are at increased risk for the persistent or de novo development of hyperparathyroidism.49,50 A study that evaluated the associations between serial bone mineral density measurements, serum PTH levels and GFR in long-term (>5 years) renal transplant recipients, reported higher PTH values in patients with a GFR 40–50 ml/min/1.73m2 than in those with a GFR >50 ml/min/1.73m2. High PTH values correlated with significant bone loss at the hip.51 Chronic progressive loss of renal function after renal transplantation leads to bone histological abnormalities similar to those observed before transplantation, described in the previous section. By use of bone histomorphometry in a cross-sectional study of 57 renal transplant recipients (post transplantation time range: 6 months to 27 years), we found low cancellous bone volume in 56% of patients, low bone turnover (Figure 2) in 60% of patients, and abnormal mineralization (Figure 3) in 37% of patients (focal osteomalacia in 21% and generalized osteomalacia in 16%).22
Figure 2.
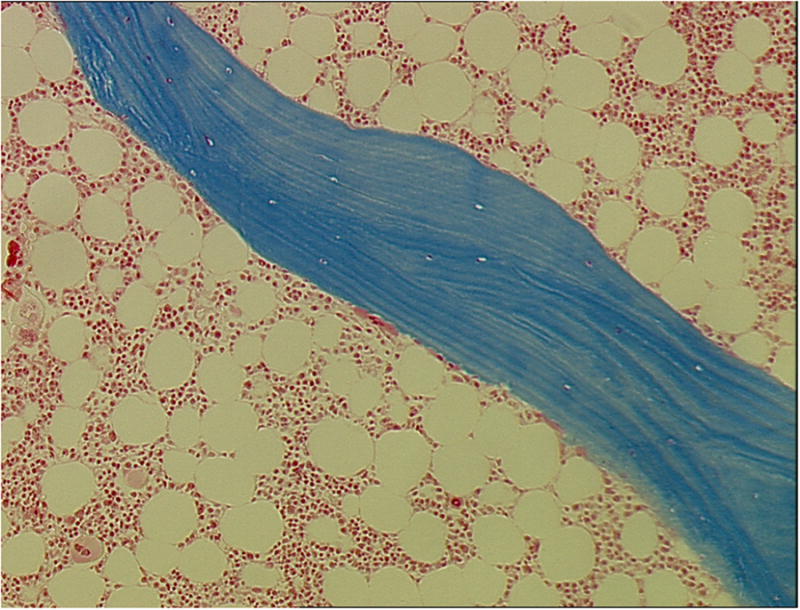
Low turnover bone disease (adynamic bone disease) in a 35-year-old white male renal transplant recipient. Bone biopsy was performed 24 months after kidney transplantation. No evidence of matrix apposition (osteoid) can be visualized. Only one osteoclast engaging in shallow resorption at the trabecular surface is evident (arrow). Mineralized, 4 μm thick bone section, stained with modified Masson-Goldner trichrome, original magnification ×100.
Figure 3.
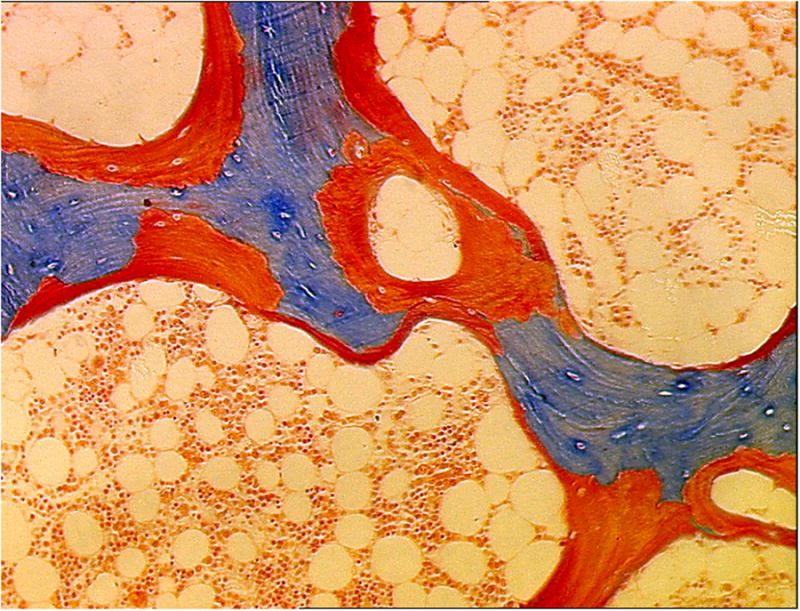
Low turnover osteomalacia in a 49-year-old white female renal transplant recipient. This bone biopsy was performed 14 months after kidney transplantation. High osteoid volume (arrows) secondary to increased osteoid surface and thick osteoid seams is evident. No osteoblasts or osteoclasts are present. Mineralized, 4 μm thick bone section, stained with modified Masson-Goldner trichrome, original magnification ×100.
Clinical Implications
Bone metabolic consequences
Parathyroid hormone
PTH levels usually decline rapidly (>50%) during the first 3–6 months after renal transplantation because of a reduction in functional parathyroid gland mass.52 This effect is thought to be the result of improved calcium, phosphorus and 1,25(OH)2 vitamin D levels. The rapid initial decline in PTH levels is followed by a more gradual decline that is probably attributable to the very slow and incomplete involution of the parathyroid gland.53,54 Persistently elevated levels of serum PTH despite normalization of renal function have been reported in >25% of renal transplant recipients 1 year after transplantation;54.55 these elevated levels may be secondary to monoclonal glandular hyperplasia.56,57 Prolonged renal failure before transplantation combined with high levels of PTH, phosphorus, and/or alkaline phosphatase has been suggested to be associated with persistent post-transplantation hyperparathyroidism despite parathyroid gland involution.54,58,59 Low levels of 1,25(OH)2 vitamin D and 25(OH) vitamin D and reduced expression of vitamin D and calcium sensing receptors have also been implicated in the development of post-transplantation hyperparathyroidism.55,60,61 Of note, two studies that assessed bone biopsy samples in renal transplant recipients did not find a correlation between serum PTH levels and bone turnover.22,62
Calcium metabolism
Persistently elevated serum PTH levels after renal transplantation may result in marked hypercalcemia and hypophosphatemia. In a 2009 study of 201 renal transplant recipients, a biphasic pattern of serum calcium with hypocalcemia immediately after transplantation (<1 week) and subsequent hypercalcemia was described.63 Successful renal engraftment can be expected to be associated with resolution of uremia, improvement in hyperphosphatemia, improved 1,25(OH)2 vitamin D production, and possibly excretion of previously retained antagonistic PTH fragments. All these entities may contribute to hypercalcemia by increasing calcium release from bone. Although severe hypercalcemia (>3 mmol/l) is seldom observed, hypercalcemic episodes (defined as total serum calcium >2.62 mmol/l) were reported in 30% and 12% of renal transplant recipients, 1 and 5 years after transplantation, respectively.54 A 2007 study of renal transplant recipients with hypercalcemia and persistently elevated serum PTH levels did not find an association between hypercalcemia and a specific bone turnover abnormality, which suggests defective tubular calcium handling as an additional pathogenetic mechanism for the development of hypercalcemia.62 Persistently high serum calcium and PTH levels were also shown to correlate with interstitial microcalcifications in the renal allograft and poor long-term graft outcomes.64
Phosphate metabolism
Hypophosphatemia related to hyperphosphaturia is present in up to 90% of patients after renal transplantation65 and can develop through multiple pathways: PTH excess secondary to pre-existing chronic or end-stage renal failure, as discussed above, dysfunctional proximal tubule phosphate transport, and/or tubular PTH hypersensitivity after normalization of kidney function. PTH-independent alterations in the expression of the sodium–phosphate co-transporter in the proximal tubule (NaPi II) are postulated to be a crucial factor in the dysfunction of proximal tubular phosphate handling in the renal allograft.66 Studies from the past few years have identified serum levels of the phosphatonin FGF23 to be the best predictor of serum phosphate nadir after renal transplantation and have demonstrated a correlation between resolution of ‘hyperphosphatoninism’ and renal phosphorus wasting 1 year after renal transplantation.65,67,68 The multifactorial etiology of phosphate wasting in renal allografts is highlighted by data that show a marked decrease in urinary phosphate excretion after administration of the calcimimetic agent cinacalcet that correlated with decreased serum PTH but not with FGF23 levels.69
Hyperphosphaturia has also been shown to occur as a result of renal denervation via functional damage or surgical obliteration of the branches of the renal vascular pedicle nerves. This effect is independent of serum PTH levels.70-73 Other studies have suggested that neural pathways can regenerate in transplanted kidneys but the selective contribution of neural regeneration to tubular phosphate handling is unknown.74,45 In a rodent model of renal denervation, fecal excretion of phosphate has been shown to decrease, possibly as a regulatory mechanism to prevent hypophosphatemia.73 The degree to which this mechanism is present in renal transplant recipients and the degree to which it may counteract urinary phosphate losses in these patients is unknown.
Vitamin D levels
Improvement of renal function after renal transplantation often reveals suboptimal reserves of 25(OH) vitamin D. Normalization of serum 25(OH) vitamin D levels by supplementation can lead to an increase in serum levels of 1,25(OH)2 vitamin D.55,76 However, when circulating 1,25(OH)2 vitamin D levels are compared with circulating PTH levels in renal transplant recipients, it becomes apparent that 1,25(OH)2 vitamin D levels are lower than expected despite the presence of physiological signals that should stimulate its production, such as elevated serum PTH levels.77
Low levels of 1,25(OH)2 vitamin D can also be related to immunosuppressive therapy, a deficit in 1α-hydroxylase activity, and/or gonadal steroid deficiency. A 2007 study identified good renal graft function to be the most important predictor of improved 1,25 (OH)2 vitamin D serum levels.65 This study also found elevated pretransplantation PTH levels and low post-transplantation levels of FGF23 to be additional predictors of improved post-transplantation 1,25(OH)2 vitamin D levels, although weaker than renal graft function. Despite an improvement in circulating levels of 1,25(OH)2 vitamin D, signs of defective bone mineralization as evidenced by focal or even generalized osteomalacia were reported to occur after renal transplantation.22 These bone histological findings might indicate target organ resistance to vitamin D or could be related to low 25(OH) vitamin D levels.
Bone mineral density and fractures
The magnitude of bone loss after transplantation depends largely on the time interval after renal transplantation; a rapid decrease in mean bone mineral density (BMD) measured by dual-energy X-ray absorptiometry (DXA) is noted during the first 6 months after transplantation and seems to slow thereafter, possibly reflecting a decreasing use of corticosteroids. BMD has been reported to decrease a mean of 5.5–19.5% during the first 6 months,23,78,79 but only 2.6–8.2% between months 6 and 12 after transplantation.80,81 Long-term follow-up studies (≥6 years) noted an annual lumbar BMD decrease of 0.4–4.5%.82,83 Prospective studies from the past 5 years that have focused on renal transplant recipients who receive low-dose prednisone therapy but no specific antiosteoporotic therapy have reported bone loss of 2.0–3.2% at the lumbar spine and 1.8–2.0% at the distal radius 1 year after renal engraftment.84-86
BMD measured by DXA correlates with fracture rates in renal transplant recipients, although this association is not as robust as in patients without kidney disease.87 According to estimates, approximately 7–10% of all renal transplant recipients will suffer one or more fractures over their lifetime. The overall fracture risk after renal transplantation is 360–380% higher than in healthy individuals,32,88 and is 30% higher during the first 3 years after transplantation than in patients on dialysis.88 The fracture risk seems to remain unchanged during the first 10 years after renal transplantation, but more than 10 years post-transplantation, decreases to about twice that of healthy individuals.32 Renal transplant recipients are at particular risk of vertebral fracture; this risk is greater than their risk of lower extremity fractures.32 This increased risk might be because of the higher amount of cancellous bone present in the vertebrae and the effect of immunosuppression on cancellous bone. Although in one study the incidence of fractures requiring hospitalization in renal transplant recipients was reported to be 8.9 per 1,000 person-years,88 a population-based study that also considered fractures treated on an outpatient basis reported a much higher incidence rate (128 fractures per 1,000 patient years).32 This population-based study identified the presence of peripheral neuropathy, peripheral vascular disease and impaired vision but not cumulative corticosteroid use as factors that were associated with fracture risk after renal transplantation.32 Additional risk factors for fracture that have been identified are older age, female gender (especially when combined with post-menopausal status), diabetes mellitus, and combined kidney–pancreas transplantation.20,90-92
Cardiovascular calcifications
Cardiovascular disease is recognized as a major cause of mortality not only in patients with CKD before renal transplantation,93 but also in renal transplant recipients.94 We have known for over a decade that patients with stage 5 CKD on dialysis present with higher coronary calcification scores than nondialysis patients,95 and that these vascular calcifications are associated with cardiovascular morbidity and mortality.96 A 2007 study of 281 renal transplant recipients who were undergoing multislice CT (MSCT) reported coronary calcifications to be prevalent in 81% of patients after >1 year after renal engraftment.97 Interestingly, coronary calcifications were predicted by history of parathyroidectomy but not by glucocorticoid use. Although proatherogenic effects of immunosuppressive agents are known,98 a longitudinal study of 23 renal transplant recipients who underwent MSCT at time of transplantation and again at 15–20 months after transplantation, reported no net change in coronary calcifications.99 Whether progression of cardiovascular calcifications will be revealed by improved sensitivity of imaging modalities awaits further study.
Evidence from the past decade shows that vascular calcifications in CKD occur early and that deposition of calcium in the vascular wall is a complex and tightly regulated process that is akin to bone mineralization.100,101 Although associations between low bone turnover and/or low bone volume and increased cardiovascular calcifications were reported in patients with stage 5 CKD on dialysis,13,14,102,103 the relationship between histomorphometric parameters of bone turnover and volume with quantitative determinations of cardiovascular calcifications in renal transplant recipients has not been investigated.
Therapy
A detailed discussion of therapeutic interventions for renal transplant recipients is beyond the scope of this Review and is provided elsewhere.104-107 The 2009 Kidney Disease: Improving Global Outcomes practice guidelines provide detailed recommendations for the evaluation and treatment of bone disease associated with renal transplantation.15 Studies on currently available therapies focus on BMD changes measured by DXA as clinical endpoints. Since extraosseous calcifications are frequent complications in patients with renal insufficiency and after renal transplantation, the use of improved BMD as an endpoint is potentially problematic since DXA can not differentiate between calcium accumulation in the bone and that in surrounding soft tissue. Studies that have used estrogens, calcitonin, bisphosphonates and vitamin D replacement therapy report mostly conflicting; however, the majority of studies have not reported positive findings. Although a favorable effect of cinacalcet on BMD in renal transplant patients was reported by two small studies that included just nine and 11 patients, respectively, these findings will need to be confirmed in larger, prospective trials.108,109
Given the frequent finding of low bone turnover in bone disease associated with renal transplantation, it is not surprising that antiresorptive agents will probably be of limited value for the improvement of BMD. Indeed, a prospective study that used bone biopsies to evaluate the effects of pamidronate therapy showed that 52% of patients had low bone turnover at the time of renal transplantation.110 Although pamidronate treatment combined with low dose calcium and calcitriol preserved bone mass at 6 months and 12 months as measured by BMD at the lumbar spine and by histomorphometry, all patients who received this treatment developed low bone turnover after 6 months (Figure 4). In the group of patients who received only low-dose calcium and calcitriol treatment, just 25% continued to have low bone turnover, 50% developed low bone turnover, and 25% showed a mild increase in turnover after 6 months.
Figure 4.

Administration of pamidronate reduces bone turnover after renal transplantation.113 Activation frequency was assessed by histomorphometry in patients 6 months after renal transplantation. Patients in both the control group and the pamidronate group received low-dose calcitriol and calcium carbonate. Patients in the pamidronate group received 60 mg intravenous pamidronate immediately after renal transplantation followed by 30 mg at months 1, 2 and 6.
In light of the known association between low bone turnover and cardiovascular calcifications in patients with CKD, therapeutic agents without negative effects on bone turnover are of great clinical importance. Teriparatide, given as daily subcutaneous injections, is the only agent that fits this profile and is currently approved by the FDA for use in osteoporotic patients but not in patients with organ transplantation. Although continuous administration of PTH may result in bone loss or no change in bone volume,111 pulsatile administration of PTH or an N-terminal PTH fragment such as teriparatide leads to gains in bone volume.112,113 Despite stabilization of BMD in the femoral neck and increased cortical width as measured by histology, a 2008 study of 26 renal transplant recipients who received daily teriparatide injections could not demonstrate improvement in bone turnover or bone mineralization as measured histologically.114 In addition, as the clinical safety of teriparatide in renal transplant recipients has not been established, recommendations for the use of teriparatide in this patient population cannot be given.
At our institution, the current therapeutic approach for renal transplant recipients with fractures, unexplained hypercalcemia or bone pain, is the performance of a bone biopsy because of the high risk of low bone turnover after renal transplantation. In patients with low bone volume and in those with an increased bone resorption to formation ratio, we use antiresorptive agents adjusted for GFR. Patients with mineralization defects as determined by histology are treated with both 25(OH) vitamin D and active vitamin D analogues. Calcium supplementation is also given but should not exceed 800–1,200 mg elemental calcium daily. In patients with low bone turnover and severely suppressed rates of bone resorption and formation, we consider antiresorptive agents to be contraindicated. We also do not use teriparatide in these patients because of the lack of safety data in renal transplant patients.
Conclusions
The clinical consequences associated with fractures and cardiovascular calcification in recipients of renal allografts calls for the early recognition of bone disease associated with renal transplantation. This bone disease is characterized by loss of bone volume, the presence of mineralization defects that can be extensive and low bone turnover. At this time, no well-established therapeutic approaches that provide bone anabolic effects in renal transplant recipients are available. In our opinion, early evaluation and primary prevention of bone loss, low bone turnover, and defective mineralization in prospective renal transplant patients is warranted. Therapy should be individualized, and based on biochemical and imaging results. If these results are inconclusive, a bone biopsy should be considered.
Key Points.
Metabolic abnormalities are common in patients after renal transplantation
Pathogenic mechanisms associated with bone loss in renal transplant recipients are different to those involved in postmenopausal bone loss
Low bone turnover is common in patients with bone disease associated with renal transplantation
Focal or generalized osteomalacia may coexist with bone loss after renal transplantation
Cardiovascular calcifications may be associated with low bone volume and low bone turnover in recipients of renal transplants
Use of bisphosphonates for treatment of bone loss associated with renal transplantation may have deleterious effects because of reduced glomerular filtration rate and the presence of low bone turnover
Acknowledgments
This project was supported by grants NIH R01 DK51530 (H.H.Malluche), by the Kentucky Nephrology Research Trust (M.-C.Monier-Faugere), and by the Dean's Clinical Research Scholar Program, University of Kentucky, no 1012112710 (J.Herberth). The authors want to thank Dr. Jessica McAbee, MD, Department of Internal Medicine, University of Kentucky, for her assistance with literature review.
Footnotes
Competing interest: The authors declare no competing interests.
Review Criteria: The PubMed database was searched for articles published up to July 31 2009 using the search terms “kidney”, “transplant”, “bone disease”, “fracture”, and “calcifications”. All full-text, English-language publications were evaluated for their relevance for inclusion in the Review. The reference lists of publications were also reviewed to identify additional relevant articles.
References
- 1.Nishikawa K, Terasaki PI. Annual trends and triple therapy--1991-2000. Clin Transpl. 2001:247–69. [PubMed] [Google Scholar]
- 2.Marcen R, Teruel JL. Patient outcomes after kidney allograft loss. Transplant Rev (Orlando) 2008;22:62–72. doi: 10.1016/j.trre.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Malluche HH, Faugere MC. Atlas of Mineralized Bone Histology. Karger; New York: 1986. [Google Scholar]
- 4.Hruska KA, Teitelbaum SL. Renal osteodystrophy. N Engl J Med. 1995;333:166–74. doi: 10.1056/NEJM199507203330307. [DOI] [PubMed] [Google Scholar]
- 5.Wang M, et al. Relationship between intact 1-84 parathyroid hormone and bone histomorphometric parameters in dialysis patients without aluminum toxicity. Am J Kidney Dis. 1995;26:836–44. doi: 10.1016/0272-6386(95)90453-0. [DOI] [PubMed] [Google Scholar]
- 6.Parfitt AM. Bone histomorphometry: proposed system for standardization of nomenclature, symbols, and units. Calcif Tissue Int. 1988;42:284–286. doi: 10.1007/BF02556360. [DOI] [PubMed] [Google Scholar]
- 7.Malluche HH, Monier-Faugere MC. Renal osteodystrophy: what's in a name? Presentation of a clinically useful new model to interpret bone histologic findings. Clin Nephrol. 2006;65:235–42. doi: 10.5414/cnp65235. [DOI] [PubMed] [Google Scholar]
- 8.Moe S, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO) Kidney Int. 2006;69:1945–53. doi: 10.1038/sj.ki.5000414. [DOI] [PubMed] [Google Scholar]
- 9.Malluche H, Lee J, Wang G, Herberth J, Faugere MC. Usefulness of the new TMV classification of renal osteodystrophy. J Am Soc Nephrol. 2008;19:38A. [Google Scholar]
- 10.Kurz P, et al. Evidence for abnormal calcium homeostasis in patients with adynamic bone disease. Kidney Int. 1994;46:855–61. doi: 10.1038/ki.1994.342. [DOI] [PubMed] [Google Scholar]
- 11.Bell KL, et al. A novel mechanism for induction of increased cortical porosity in cases of intracapsular hip fracture. Bone. 2000;27:297–304. doi: 10.1016/s8756-3282(00)00318-5. [DOI] [PubMed] [Google Scholar]
- 12.Greenspan SL, Beck TJ, Resnick NM, Bhattacharya R, Parker RA. Effect of hormone replacement, alendronate, or combination therapy on hip structural geometry: a 3-year, double-blind, placebo-controlled clinical trial. J Bone Miner Res. 2005;20:1525–32. doi: 10.1359/JBMR.050508. [DOI] [PubMed] [Google Scholar]
- 13.London GM, et al. Arterial calcifications and bone histomorphometry in end-stage renal disease. J Am Soc Nephrol. 2004;15:1943–51. doi: 10.1097/01.asn.0000129337.50739.48. [DOI] [PubMed] [Google Scholar]
- 14.Adragao T, et al. Low bone volume--a risk factor for coronary calcifications in hemodialysis patients. Clin J Am Soc Nephrol. 2009;4:450–5. doi: 10.2215/CJN.01870408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Kidney Int Suppl. 2009:S1–130. doi: 10.1038/ki.2009.188. [DOI] [PubMed] [Google Scholar]
- 16.Bordier P, et al. Vitamin D metabolites and bone mineralization in man. J Clin Endocrinol Metab. 1978;46:284–94. doi: 10.1210/jcem-46-2-284. [DOI] [PubMed] [Google Scholar]
- 17.Malluche HH, Goldstein DA, Massry SG. Osteomalacia and hyperparathyroid bone disease in patients with nephrotic syndrome. J Clin Invest. 1979;63:494–500. doi: 10.1172/JCI109327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolpaw T, et al. Factors influencing vertebral bone density after renal transplantation. Transplantation. 1994;58:1186–9. [PubMed] [Google Scholar]
- 19.Briner VA, et al. Prevention of cancellous bone loss but persistence of renal bone disease despite normal 1,25 vitamin D levels two years after kidney transplantation. Transplantation. 1995;59:1393–400. doi: 10.1097/00007890-199505270-00006. [DOI] [PubMed] [Google Scholar]
- 20.Grotz WH, et al. Bone loss after kidney transplantation: a longitudinal study in 115 graft recipients. Nephrol Dial Transplant. 1995;10:2096–100. [PubMed] [Google Scholar]
- 21.Grotz WH, et al. Bone mineral density after kidney transplantation. A cross-sectional study in 190 graft recipients up to 20 years after transplantation. Transplantation. 1995;59:982–6. [PubMed] [Google Scholar]
- 22.Monier-Faugere MC, Mawad H, Qi Q, Friedler RM, Malluche HH. High prevalence of low bone turnover and occurrence of osteomalacia after kidney transplantation. J Am Soc Nephrol. 2000;11:1093–9. doi: 10.1681/ASN.V1161093. [DOI] [PubMed] [Google Scholar]
- 23.Julian BA, et al. Rapid loss of vertebral mineral density after renal transplantation. N Engl J Med. 1991;325:544–50. doi: 10.1056/NEJM199108223250804. [DOI] [PubMed] [Google Scholar]
- 24.Rojas E, et al. The pathogenesis of osteodystrophy after renal transplantation as detected by early alterations in bone remodeling. Kidney Int. 2003;63:1915–23. doi: 10.1046/j.1523-1755.2003.00938.x. [DOI] [PubMed] [Google Scholar]
- 25.Canalis E, Delany AM. Mechanisms of glucocorticoid action in bone. Ann N Y Acad Sci. 2002;966:73–81. doi: 10.1111/j.1749-6632.2002.tb04204.x. [DOI] [PubMed] [Google Scholar]
- 26.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274–82. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki Y, Ichikawa Y, Saito E, Homma M. Importance of increased urinary calcium excretion in the development of secondary hyperparathyroidism of patients under glucocorticoid therapy. Metabolism. 1983;32:151–6. doi: 10.1016/0026-0495(83)90221-4. [DOI] [PubMed] [Google Scholar]
- 28.Canalis E, Mazziotti G, Giustina A, Bilezikian JP. Glucocorticoid-induced osteoporosis: pathophysiology and therapy. Osteoporos Int. 2007;18:1319–28. doi: 10.1007/s00198-007-0394-0. [DOI] [PubMed] [Google Scholar]
- 29.Sakakura M, Takebe K, Nakagawa S. Inhibition of luteinizing hormone secretion induced by synthetic LRH by long-term treatment with glucocorticoids in human subjects. J Clin Endocrinol Metab. 1975;40:774–9. doi: 10.1210/jcem-40-5-774. [DOI] [PubMed] [Google Scholar]
- 30.Brandenburg VM, et al. Lumbar bone mineral density in very long-term renal transplant recipients: impact of circulating sex hormones. Osteoporos Int. 2005;16:1611–20. doi: 10.1007/s00198-005-1884-6. [DOI] [PubMed] [Google Scholar]
- 31.O'Shaughnessy EA, Dahl DC, Smith CL, Kasiske BL. Risk factors for fractures in kidney transplantation. Transplantation. 2002;74:362–6. doi: 10.1097/00007890-200208150-00012. [DOI] [PubMed] [Google Scholar]
- 32.Vautour LM, et al. Long-term fracture risk following renal transplantation: a population-based study. Osteoporos Int. 2004;15:160–7. doi: 10.1007/s00198-003-1532-y. [DOI] [PubMed] [Google Scholar]
- 33.Ugur A, et al. Osteoporosis after renal transplantation: single center experience. Transplantation. 2001;71:645–9. doi: 10.1097/00007890-200103150-00011. [DOI] [PubMed] [Google Scholar]
- 34.Marcen R, et al. Lumbar bone mineral density in renal transplant patients on neoral and tacrolimus: a four-year prospective study. Transplantation. 2006;81:826–31. doi: 10.1097/01.tp.0000203557.36884.e3. [DOI] [PubMed] [Google Scholar]
- 35.Tang L, et al. FK506 enhanced osteoblastic differentiation in mesenchymal cells. Cell Biol Int. 2002;26:75–84. doi: 10.1006/cbir.2001.0812. [DOI] [PubMed] [Google Scholar]
- 36.Kirino S, et al. Regulation of bone metabolism in immunosuppressant (FK506)-treated rats. J Bone Miner Metab. 2004;22:554–60. doi: 10.1007/s00774-004-0523-1. [DOI] [PubMed] [Google Scholar]
- 37.Patel S, et al. Prevalence and causes of low bone density and fractures in kidney transplant patients. J Bone Miner Res. 2001;16:1863–70. doi: 10.1359/jbmr.2001.16.10.1863. [DOI] [PubMed] [Google Scholar]
- 38.Movsowitz C, Epstein S, Fallon M, Ismail F, Thomas S. Cyclosporin-A in vivo produces severe osteopenia in the rat: effect of dose and duration of administration. Endocrinology. 1988;123:2571–7. doi: 10.1210/endo-123-5-2571. [DOI] [PubMed] [Google Scholar]
- 39.Schlosberg M, et al. The effect of cyclosporin A administration and its withdrawal on bone mineral metabolism in the rat. Endocrinology. 1989;124:2179–84. doi: 10.1210/endo-124-5-2179. [DOI] [PubMed] [Google Scholar]
- 40.Epstein S, et al. Effect of the interaction of parathyroid hormone and cyclosporine a on bone mineral metabolism in the rat. Calcif Tissue Int. 2001;68:240–7. doi: 10.1007/s002230001167. [DOI] [PubMed] [Google Scholar]
- 41.Rastogi R, Faugere MC, Geng Z, Bognar B, Malluche HH. Cyclosporin does not induce bone loss but enhances mineralization in normal and nephrectomized rats. J Bone Miner Res. 1997;12(Suppl 1):402. [Google Scholar]
- 42.Briner VA, Landmann J, Brunner FP, Thiel G. Cyclosporin A-induced transient rise in plasma alkaline phosphatase in kidney transplant patients. Transpl Int. 1993;6:99–107. doi: 10.1007/BF00336653. [DOI] [PubMed] [Google Scholar]
- 43.Bozkaya G, et al. Impact of calcineurin inhibitors on bone metabolism in primary kidney transplant patients. Transplant Proc. 2008;40:151–5. doi: 10.1016/j.transproceed.2007.11.040. [DOI] [PubMed] [Google Scholar]
- 44.Joffe I, et al. Lack of change of cancellous bone volume with short-term use of the new immunosuppressant rapamycin in rats. Calcif Tissue Int. 1993;53:45–52. doi: 10.1007/BF01352014. [DOI] [PubMed] [Google Scholar]
- 45.Bryer HP, et al. Azathioprine alone is bone sparing and does not alter cyclosporin A-induced osteopenia in the rat. J Bone Miner Res. 1995;10:132–8. doi: 10.1002/jbmr.5650100119. [DOI] [PubMed] [Google Scholar]
- 46.Dissanayake IR, et al. Mycophenolate mofetil: a promising new immunosuppressant that does not cause bone loss in the rat. Transplantation. 1998;65:275–8. doi: 10.1097/00007890-199801270-00025. [DOI] [PubMed] [Google Scholar]
- 47.Singha UK, et al. Rapamycin inhibits osteoblast proliferation and differentiation in MC3T3-E1 cells and primary mouse bone marrow stromal cells. J Cell Biochem. 2008;103:434–46. doi: 10.1002/jcb.21411. [DOI] [PubMed] [Google Scholar]
- 48.Kneissel M, et al. Everolimus suppresses cancellous bone loss, bone resorption, and cathepsin K expression by osteoclasts. Bone. 2004;35:1144–56. doi: 10.1016/j.bone.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 49.Malluche H, et al. Bone histology in incipient and advanced renal failure. Kidney Int. 1976;9:355–362. doi: 10.1038/ki.1976.42. [DOI] [PubMed] [Google Scholar]
- 50.Bellorin-Font E, Rojas E, Carlini RG, Suniaga O, Weisinger JR. Bone remodeling after renal transplantation. Kidney Int Suppl. 2003:S125–8. doi: 10.1046/j.1523-1755.63.s85.30.x. [DOI] [PubMed] [Google Scholar]
- 51.Akaberi S, Lindergard B, Simonsen O, Nyberg G. Impact of parathyroid hormone on bone density in long-term renal transplant patients with good graft function. Transplantation. 2006;82:749–52. doi: 10.1097/01.tp.0000230130.50451.78. [DOI] [PubMed] [Google Scholar]
- 52.Bonarek H, et al. Reduced parathyroid functional mass after successful kidney transplantation. Kidney Int. 1999;56:642–9. doi: 10.1046/j.1523-1755.1999.00589.x. [DOI] [PubMed] [Google Scholar]
- 53.Parfitt AM. Hypercalcemic hyperparathyroidism following renal transplantation: differential diagnosis, management, and implications for cell population control in the parathyroid gland. Miner Electrolyte Metab. 1982;8:92–112. [PubMed] [Google Scholar]
- 54.Evenepoel P, et al. Natural history of parathyroid function and calcium metabolism after kidney transplantation: a single-centre study. Nephrol Dial Transplant. 2004;19:1281–7. doi: 10.1093/ndt/gfh128. [DOI] [PubMed] [Google Scholar]
- 55.Reinhardt W, et al. Sequential changes of biochemical bone parameters after kidney transplantation. Nephrol Dial Transplant. 1998;13:436–42. doi: 10.1093/oxfordjournals.ndt.a027843. [DOI] [PubMed] [Google Scholar]
- 56.Messa P, et al. Persistent secondary hyperparathyroidism after renal transplantation. Kidney Int. 1998;54:1704–13. doi: 10.1046/j.1523-1755.1998.00142.x. [DOI] [PubMed] [Google Scholar]
- 57.Kruse AE, Eisenberger U, Frey FJ, Mohaupt MG. The calcimimetic cinacalcet normalizes serum calcium in renal transplant patients with persistent hyperparathyroidism. Nephrol Dial Transplant. 2005;20:1311–4. doi: 10.1093/ndt/gfh924. [DOI] [PubMed] [Google Scholar]
- 58.Torres A, Lorenzo V, Salido E. Calcium metabolism and skeletal problems after transplantation. J Am Soc Nephrol. 2002;13:551–8. doi: 10.1681/ASN.V132551. [DOI] [PubMed] [Google Scholar]
- 59.Lewin E. Involution of the parathyroid glands after renal transplantation. Curr Opin Nephrol Hypertens. 2003;12:363–71. doi: 10.1097/00041552-200307000-00004. [DOI] [PubMed] [Google Scholar]
- 60.Caravaca F, et al. Are plasma 1,25-dihydroxyvitamin D3 concentrations appropriate after successful kidney transplantation? Nephrol Dial Transplant. 1998;13(Suppl 3):91–3. doi: 10.1093/ndt/13.suppl_3.91. [DOI] [PubMed] [Google Scholar]
- 61.Drueke TB. Primary and secondary uraemic hyperparathyroidism: from initial clinical observations to recent findings. Nephrol Dial Transplant. 1998;13:1384–7. doi: 10.1093/ndt/13.6.1384. [DOI] [PubMed] [Google Scholar]
- 62.Borchhardt K, et al. Low-turnover bone disease in hypercalcemic hyperparathyroidism after kidney transplantation. Am J Transplant. 2007;7:2515–21. doi: 10.1111/j.1600-6143.2007.01950.x. [DOI] [PubMed] [Google Scholar]
- 63.Evenepoel P, et al. Calcium metabolism in the early posttransplantation period. Clin J Am Soc Nephrol. 2009;4:665–72. doi: 10.2215/CJN.03920808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gwinner W, et al. Early calcification of renal allografts detected by protocol biopsies: causes and clinical implications. Am J Transplant. 2005;5:1934–41. doi: 10.1111/j.1600-6143.2005.00938.x. [DOI] [PubMed] [Google Scholar]
- 65.Evenepoel P, Naesens M, Claes K, Kuypers D, Vanrenterghem Y. Tertiary ‘hyperphosphatoninism’ accentuates hypophosphatemia and suppresses calcitriol levels in renal transplant recipients. Am J Transplant. 2007;7:1193–200. doi: 10.1111/j.1600-6143.2007.01753.x. [DOI] [PubMed] [Google Scholar]
- 66.Green J, et al. Evidence for a PTH-independent humoral mechanism in post-transplant hypophosphatemia and phosphaturia. Kidney Int. 2001;60:1182–96. doi: 10.1046/j.1523-1755.2001.0600031182.x. [DOI] [PubMed] [Google Scholar]
- 67.Bhan I, et al. Post-transplant hypophosphatemia: Tertiary ‘Hyper-Phosphatoninism’? Kidney Int. 2006;70:1486–94. doi: 10.1038/sj.ki.5001788. [DOI] [PubMed] [Google Scholar]
- 68.Evenepoel P, et al. Recovery of hyperphosphatoninism and renal phosphorus wasting one year after successful renal transplantation. Clin J Am Soc Nephrol. 2008;3:1829–36. doi: 10.2215/CJN.01310308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Serra AL, Wuhrmann C, Wuthrich RP. Phosphatemic effect of cinacalcet in kidney transplant recipients with persistent hyperparathyroidism. Am J Kidney Dis. 2008;52:1151–7. doi: 10.1053/j.ajkd.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 70.Mann KJ, et al. Renal denervation enhances the phosphaturic effect of parathyroid hormone. Miner Electrolyte Metab. 1991;17:16–20. [PubMed] [Google Scholar]
- 71.Mann KJ, Dousa DM, Kerrigan RJ, Berndt TJ, Knox FG. Acute renal denervation decreases tubular phosphate reabsorption. Miner Electrolyte Metab. 1992;18:354–8. [PubMed] [Google Scholar]
- 72.Berndt TJ, Khraibi AA, Knox FG. Interaction of the renal nerves and prostaglandins on the phosphaturic response to PTH in phosphate-deprived rats. Am J Physiol. 1995;268:R731–5. doi: 10.1152/ajpregu.1995.268.3.R731. [DOI] [PubMed] [Google Scholar]
- 73.Straub B, et al. Hyperphosphaturia after kidney transplantation in syngeneic rats: effects on nephrocalcinosis and bone metabolism? Transplant Proc. 2003;35:1575–80. doi: 10.1016/s0041-1345(03)00526-8. [DOI] [PubMed] [Google Scholar]
- 74.Gazdar AF, Dammin GJ. Neural degeneration and regeneration in human renal transplants. N Engl J Med. 1970;283:222–4. doi: 10.1056/NEJM197007302830502. [DOI] [PubMed] [Google Scholar]
- 75.Sankari B, et al. Studies on the afferent and efferent renal nerves following autotransplantation of the canine kidney. J Urol. 1992;148:206–10. doi: 10.1016/s0022-5347(17)36554-0. [DOI] [PubMed] [Google Scholar]
- 76.Amin H, Wall BM, Cooke CR. Osteomalacia and secondary hyperparathyroidism after kidney transplantation: Relationship to vitamin D deficiency. Am J Med Sci. 2007;333:58–62. doi: 10.1097/00000441-200701000-00009. [DOI] [PubMed] [Google Scholar]
- 77.Fleseriu M, Licata AA. Failure of successful renal transplant to produce appropriate levels of 1,25-dihydroxyvitamin D. Osteoporos Int. 2007;18:363–8. doi: 10.1007/s00198-006-0238-3. [DOI] [PubMed] [Google Scholar]
- 78.Lippuner K, Casez JP, Horber FF, Jaeger P. Effects of deflazacort versus prednisone on bone mass, body composition, and lipid profile: a randomized, double blind study in kidney transplant patients. J Clin Endocrinol Metab. 1998;83:3795–802. doi: 10.1210/jcem.83.11.5235. [DOI] [PubMed] [Google Scholar]
- 79.Mikuls TR, Julian BA, Bartolucci A, Saag KG. Bone mineral density changes within six months of renal transplantation. Transplantation. 2003;75:49–54. doi: 10.1097/00007890-200301150-00009. [DOI] [PubMed] [Google Scholar]
- 80.Nam JH, et al. Pamidronate and calcitriol trial for the prevention of early bone loss after renal transplantation. Transplant Proc. 2000;32:1876. doi: 10.1016/s0041-1345(00)01898-4. [DOI] [PubMed] [Google Scholar]
- 81.Brandenburg VM, et al. Early rapid loss followed by long-term consolidation characterizes the development of lumbar bone mineral density after kidney transplantation. Transplantation. 2004;77:1566–71. doi: 10.1097/01.tp.0000131990.13277.28. [DOI] [PubMed] [Google Scholar]
- 82.Pichette V, et al. Long-term bone loss in kidney transplant recipients: a cross-sectional and longitudinal study. Am J Kidney Dis. 1996;28:105–14. doi: 10.1016/s0272-6386(96)90138-9. [DOI] [PubMed] [Google Scholar]
- 83.Cruz DN, et al. Parameters of high bone-turnover predict bone loss in renal transplant patients: a longitudinal study. Transplantation. 2001;72:83–8. doi: 10.1097/00007890-200107150-00017. [DOI] [PubMed] [Google Scholar]
- 84.Josephson MA, et al. Calcium and calcitriol prophylaxis attenuates posttransplant bone loss. Transplantation. 2004;78:1233–6. doi: 10.1097/01.tp.0000137937.44703.42. [DOI] [PubMed] [Google Scholar]
- 85.Wissing KM, et al. A controlled study of vitamin D3 to prevent bone loss in renal-transplant patients receiving low doses of steroids. Transplantation. 2005;79:108–15. doi: 10.1097/01.tp.0000149322.70295.a5. [DOI] [PubMed] [Google Scholar]
- 86.El-Agroudy AE, El-Husseini AA, El-Sayed M, Mohsen T, Ghoneim MA. A prospective randomized study for prevention of postrenal transplantation bone loss. Kidney Int. 2005;67:2039–45. doi: 10.1111/j.1523-1755.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- 87.Grotz WH, et al. Bone fracture and osteodensitometry with dual energy X-ray absorptiometry in kidney transplant recipients. Transplantation. 1994;58:912–5. doi: 10.1097/00007890-199410270-00009. [DOI] [PubMed] [Google Scholar]
- 88.Abbott KC, et al. Hospitalizations for fractures after renal transplantation in the United States. Ann Epidemiol. 2001;11:450–7. doi: 10.1016/s1047-2797(01)00226-5. [DOI] [PubMed] [Google Scholar]
- 89.Ball AM, et al. Risk of hip fracture among dialysis and renal transplant recipients. Jama. 2002;288:3014–8. doi: 10.1001/jama.288.23.3014. [DOI] [PubMed] [Google Scholar]
- 90.Nisbeth U, Lindh E, Ljunghall S, Backman U, Fellstrom B. Fracture frequency after kidney transplantation. Transplant Proc. 1994;26:1764. [PubMed] [Google Scholar]
- 91.Chiu MY, et al. Analysis of fracture prevalence in kidney-pancreas allograft recipients. J Am Soc Nephrol. 1998;9:677–83. doi: 10.1681/ASN.V94677. [DOI] [PubMed] [Google Scholar]
- 92.Nisbeth U, Lindh E, Ljunghall S, Backman U, Fellstrom B. Increased fracture rate in diabetes mellitus and females after renal transplantation. Transplantation. 1999;67:1218–22. doi: 10.1097/00007890-199905150-00004. [DOI] [PubMed] [Google Scholar]
- 93.Foley RN, Parfrey PS, Sarnak MJ. Epidemiology of cardiovascular disease in chronic renal disease. J Am Soc Nephrol. 1998;9:S16–23. [PubMed] [Google Scholar]
- 94.Ojo AO, et al. Long-term survival in renal transplant recipients with graft function. Kidney Int. 2000;57:307–13. doi: 10.1046/j.1523-1755.2000.00816.x. [DOI] [PubMed] [Google Scholar]
- 95.Braun J, et al. Electron beam computed tomography in the evaluation of cardiac calcification in chronic dialysis patients. Am J Kidney Dis. 1996;27:394–401. doi: 10.1016/s0272-6386(96)90363-7. [DOI] [PubMed] [Google Scholar]
- 96.Blacher J, Guerin AP, Pannier B, Marchais SJ, London GM. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension. 2001;38:938–42. doi: 10.1161/hy1001.096358. [DOI] [PubMed] [Google Scholar]
- 97.Nguyen PT, et al. Prevalence and determinants of coronary and aortic calcifications assessed by chest CT in renal transplant recipients. Am J Nephrol. 2007;27:329–35. doi: 10.1159/000102978. [DOI] [PubMed] [Google Scholar]
- 98.Kasiske BL. Cardiovascular disease after renal transplantation. Semin Nephrol. 2000;20:176–87. [PubMed] [Google Scholar]
- 99.Moe SM, et al. Natural history of vascular calcification in dialysis and transplant patients. Nephrol Dial Transplant. 2004;19:2387–93. doi: 10.1093/ndt/gfh303. [DOI] [PubMed] [Google Scholar]
- 100.Moe SM. Vascular calcification and renal osteodystrophy relationship in chronic kidney disease. Eur J Clin Invest. 2006;36(Suppl 2):51–62. doi: 10.1111/j.1365-2362.2006.01665.x. [DOI] [PubMed] [Google Scholar]
- 101.Goodman WG, et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med. 2000;342:1478–83. doi: 10.1056/NEJM200005183422003. [DOI] [PubMed] [Google Scholar]
- 102.Adragao T, et al. Vascular Calcifications and bone turnover in hemodialysis patients. Nephrol Dial Transplant. 2006;21(suppl iv):292. [Google Scholar]
- 103.Barreto DV, et al. Association of changes in bone remodeling and coronary calcification in hemodialysis patients: a prospective study. Am J Kidney Dis. 2008;52:1139–50. doi: 10.1053/j.ajkd.2008.06.024. [DOI] [PubMed] [Google Scholar]
- 104.Weisinger JR, Carlini RG, Rojas E, Bellorin-Font E. Bone disease after renal transplantation. Clin J Am Soc Nephrol. 2006;1:1300–13. doi: 10.2215/CJN.01510506. [DOI] [PubMed] [Google Scholar]
- 105.Palmer SC, McGregor DO, Strippoli GF. Interventions for preventing bone disease in kidney transplant recipients. Cochrane Database Syst Rev. 2007:CD005015. doi: 10.1002/14651858.CD005015.pub3. [DOI] [PubMed] [Google Scholar]
- 106.Triponez F, Clark OH, Vanrenthergem Y, Evenepoel P. Surgical treatment of persistent hyperparathyroidism after renal transplantation. Ann Surg. 2008;248:18–30. doi: 10.1097/SLA.0b013e3181728a2d. [DOI] [PubMed] [Google Scholar]
- 107.Ebeling PR. Approach to the patient with transplantation-related bone loss. J Clin Endocrinol Metab. 2009;94:1483–90. doi: 10.1210/jc.2009-0205. [DOI] [PubMed] [Google Scholar]
- 108.Bergua C, et al. Effect of cinacalcet on hypercalcemia and bone mineral density in renal transplanted patients with secondary hyperparathyroidism. Transplantation. 2008;86:413–7. doi: 10.1097/TP.0b013e31817c13e1. [DOI] [PubMed] [Google Scholar]
- 109.Cho, M.E. et al. in American Society of Nephrology (Philadelphia, 2008).
- 110.Coco M, et al. Prevention of bone loss in renal transplant recipients: a prospective, randomized trial of intravenous pamidronate. J Am Soc Nephrol. 2003;14:2669–76. doi: 10.1097/01.asn.0000087092.53894.80. [DOI] [PubMed] [Google Scholar]
- 111.Malluche HH, Goldstein DA, Massry SG. Effects of 6 months therapy with 1,25 (OH) 2D3 on bone disease of dialysis patients. Contrib Nephrol. 1980;18:98–104. doi: 10.1159/000403277. [DOI] [PubMed] [Google Scholar]
- 112.Reeve J, et al. Anabolic effect of human parathyroid hormone fragment on trabecular bone in involutional osteoporosis: a multicentre trial. Br Med J. 1980;280:1340–4. doi: 10.1136/bmj.280.6228.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Neer RM, et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434–41. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]
- 114.Cejka D, et al. Effect of teriparatide on early bone loss after kidney transplantation. Am J Transplant. 2008;8:1864–70. doi: 10.1111/j.1600-6143.2008.02327.x. [DOI] [PubMed] [Google Scholar]