

Abstract
Elucidation of the neural substrates underlying complex animal behaviors depends on precise activity control tools, as well as compatible readout methods. Recent developments in optogenetics have addressed this need, opening up new possibilities for systems neuroscience. Interrogation of even deep neural circuits can be conducted by directly probing the necessity and sufficiency of defined circuit elements with millisecond-scale, cell type-specific optical perturbations, coupled with suitable readouts such as electrophysiology, optical circuit dynamics measures and freely moving behavior in mammals. Here we collect in detail our strategies for delivering microbial opsin genes to deep mammalian brain structures in vivo, along with protocols for integrating the resulting optical control with compatible readouts (electrophysiological, optical and behavioral). The procedures described here, from initial virus preparation to systems-level functional readout, can be completed within 4–5 weeks. Together, these methods may help in providing circuit-level insight into the dynamics underlying complex mammalian behaviors in health and disease.
INTRODUCTION
Understanding the circuit-level functional organization of the brain will have important implications for both basic and clinical neuroscience. In particular, we have found that optical manipulation of activity in neural circuits with light-sensitive rhodopsins, such as the Chlamydomonas channelrhodopsin-2 (ChR2)1,2, Volvox channel-rhodopsin-1 (VChR1)3, Natronomonas halorhodopsin (NpHR)4 (Fig. 1a) and synthetic rhodopsin/GPCR chimeras5 (optoXRs; Fig. 1b), may help in illuminating both the normal circuit function and major disease mechanisms6–19. Here we illustrate the critical steps we have developed for the implementation of this optogenetic6,7 approach to probe the function of deep brain circuit elements in mammals. We first review the basic optogenetic control tools we have described, followed by discussion of optimization of expression, targeting and readout technology. We then present the necessary materials and outline a detailed series of our experimental protocols for integrating optogenetic neuronal manipulation with in vivo and ex vivo readout methods, including electrophysiological and optical measures.
Figure 1.

Optogenetic tools. (a) Naturally occurring light-responsive effectors and their microbial sources: ChR2 from Chlamydomonas reinhardtii, VChR1 from Volvox carteri and NpHR from Natronomonas pharaonis; useful light wavelengths for each are indicated. ChR2 and VChR1 are cation-conducting channels and NpHR is a chloride pump. (b) Engineered synthetic rhodopsins for optical control of well-defined intracellular biochemical signaling. The intracellular loops of bovine rhodopsin have been replaced with the intracellular loops of G protein-coupled receptors (GPCRs) to yield light-activated chimeric GPCRs. Green light illumination leads to activation of the downstream Gq and Gs signaling pathways. (c) Action spectra. The absorbance wavelength of the voltage-sensitive dye (VSD) RH 155 is sufficiently separated from the light-sensitive range of all rhodopsins, therefore making it possible to integrate VSD imaging with optogenetic modulation. (d) Viral vectors for introducing microbial opsin genes into the brain. Top and middle: Lentiviral and AAV vectors can be used to deliver a cell-specific promoter along with the opsin gene and its fluorescent marker. Bottom: Cre-dependent adeno-associated virus (AAV) vector carries a doubly floxed inverted opsin (DIO) fusion gene. Upon transduction into Cre recombinase-expressing cells, the opsin fusion gene will be irreversibly inverted and enable cell-specific gene expression. Part a was modified with permission from Nature4 (Nature © 2007; Macmillan Publishers Ltd.).
Experimental design considerations
Temporally precise genetically encoded control
Classical neuronal manipulation techniques (electrical, pharmacological and genetic) either simultaneously affect surrounding cells and processes in addition to the target population or have slow kinetics and poor reversibility, thereby severely limiting the strength of conclusions that can be drawn. To overcome these spatial and temporal limitations, microbial and chimeric-vertebrate opsin genes (Table 1) have been developed recently to control highly-defined electrical (Fig. 1a) and biochemical (Fig. 1b) activity with cell-type selectivity, high temporal precision and rapid reversibility. As most neurons in the brain are not naturally light-sensitive, selective expression of opsin genes in targeted neural populations makes it possible to specifically control the activity in these populations, and the resulting fast on–off kinetics make it possible to evoke or inhibit neural activity within milliseconds, on a timescale relevant to the physiological brain functions.
TABLE 1.
Comparison table of optogenetic tools suitable for fast in vivo use in mammals and other animals.
| Opsin | Host organism | Wavelength sensitivity | Mode of control | Modulatory capabilities | Experimental systems tested |
|---|---|---|---|---|---|
| ChR2, ChR2(H134R), ChR1/2 chimeras, fast (ChETA) mutants | Chlamydomonas reinhardtii | 470 nm (maximum activation) | Depolarizing | Rapid on/off, best used for precise activation of neurons on the millisecond timescale. Can be used to evoke single spikes or defined trains of action potentials over a range of frequencies. The H134R mutation yields larger photocurrents relative to wild-type ChR2, but with slower kinetics. koff ChR1/ChR2 chimera mutants78,79 reduce inactivation and ChETA opsins allow for spiking up to 200 Hz while also improving overall performance80 |
In vitro: dissociated neuron culture1,2,4,80, acute mouse and rat brain sections2,13,18,33,62–64,78,79, 293HEK cells20 In vivo: C. elegans (requires supplementation of all-trans- retinal, ATR)23, D. melanogaster (requires supplementation of ATR)65–67,77, zebrafish68, chicken69, mouse8,9,11,12,14,15,32,49, rat49, primate70 |
| Step function opsin (SFO) genes: ChR2 (C128A), ChR2 (C128S), ChR2 (C128T) | Chlamydomonas reinhardtii | 470 nm (switching on) 546 nm (switching off for C128A and C128S mutants) |
Depolarizing | Point-mutants of ChR2 with slow or optically switchable deactivation. C128A and C128S mutants show the most prolonged activation and the highest light sensitivity, while C128T retains more temporal precision of activation SFOs can be switched on and off with blue and green light pulses, respectively | In vitro: dissociated neuron culture25 |
| VChR1 | Volvox carteri | 535 nm (maximum activation) 589 nm (completely separable activation from ChR2) |
Depolarizing | Red-shifted action spectrum relative to ChR2. Similar to ChR2, VChR1 can be used to drive reliable action potential firing over a range of frequencies. With 589 nm light, VChR1 can be activated independently of ChR2 | In vitro: dissociated neuron culture3 |
| NpHR, eNpHR | Natronomonas pharaonis | 589 nm (maximum activation) | Hyper-polarizing | Light-activated chloride pump. Can be used to hyperpolarize neurons with high temporal precision; capable of inhibiting single action potentials within high frequency spike trains (up to 30 Hz). Also can be used to mediate sustained inhibition of neurons over many minutes |
In vitro: dissociated neuron culture4,21,22,73 and mouse brain slice4,22 In vivo: C. elegans (requires supplementation of ATR)4 and mouse21,22 |
| opto-α1AR | Synthetic | 500 nm (maximum activation) | Biochemical | Light-activated GPCR, via the Gq pathway |
In vitro: 293HEK cell line5 In vivo: mouse5 |
| opto-β2AR | Synthetic | 500 nm (maximum activation) | Biochemical | Light-activated GPCR, via the Gs pathway |
In vitro: 293HEK cell line5 In vivo: mouse5 |
At present, light-activated cation channels from two distinct algal species, channelrhodopsin-2 from Chlamydomonas reinhardtii (ChR2)1,2,20 and channelrhodopsin-1 from Volvox carteri (VChR1)3 (Fig. 1a, left and middle), have proven to be a powerful pair of tools for controlling neural activity. ChR2 is maximally activated by blue light at 470 nm, whereas VChR1 remains significantly light sensitive even at 589 nm, a wavelength at which (importantly) ChR2 is no longer responsive (Fig. 1c). Both ChR2 (deactivation time constant ~12 ms) and VChR1 (deactivation time constant ~120 ms) are able to transduce trains of millisecond-duration light flashes into defined spike trains up to 30–50 Hz1,2,8. Conversely, the chloride-pumping halorhodopsin from Natronomonas pharaonis (NpHR) has been shown to hyperpolarize neurons upon illumination with yellow light (Fig. 1c). Because of sufficient spectral separation, NpHR and ChR2 can simultaneously be expressed in the same neurons to enable bidirectional optical control of neural activity. Moreover, both use all-trans retinal as the chromophore, which is abundantly present in mammalian brain tissue, and therefore these optogenetic tools are fully functional without the addition of chemical cofactors in vivo4.
Optimizing expression and function
For all of these proteins, expression issues will determine crucial aspects of performance. For example, because of the inactivation kinetics of ChR2, higher-expressing cells will be able to follow fast spike frequencies for longer periods of time, as these cells will be able to employ larger pools of the remaining non-inactivated ChR2. As another example, without the proper targeting sequences, high levels of microbial protein expression in mammalian cells can result in the aggregation of misfolded proteins in the Golgi, the endoplasmic reticulum and other intracellular compartments. As the conductance of individual rhodopsins is relatively low (picosiemens or less for ChR2, and even lower for NpHR as a pump, due to its 1:1 coupling between photon absorption and ion flux), it is critical to maximize the number of molecules that are properly integrated into the cytoplasmic membrane, in addition to optimizing transcription of the opsin genes. For this reason, we have been constructing enhanced versions of halorhodopsin (eNpHR) by optimizing membrane trafficking sequences to increase the efficiency of membrane targeting21,22. eNpHR exhibits higher levels of membrane expression and more robust photocurrents, and similar modifications could be applied to optimize other microbial light-activated proteins for enhanced photocurrents.
Molecular engineering is also leading to expanded and refined function of these microbial proteins by altering spectral properties, conductance or kinetics. One point mutation (H134R in ChR2 (ref. 23)) has been shown to result in 2–3× enhanced cellular photocurrents24 (but at the expense of slowed deactivation). Channelrhodopsin deactivation can be slowed so much that a novel utility is achieved; several C128 point mutants of ChR2 exhibit profound bistability, converting a brief pulse of light into a stable step in membrane potential25. Transduced cells are (at steady state) responsive to light at >100× lower light levels because of delayed exit of these proteins from the open state25. These C128 mutants are still activated by blue (470 nm) light, but photocurrents elicited by the opening of ChR2(C128A) and ChR2(C128S) can effectively be terminated by a pulse of green (542 nm) light. These engineered step function opsins (SFO) can optically sensitize cells to native patterns of input, by providing a sustained step depolarization of the target cell membrane potential that can increase excitability. In many settings this approach will be preferable to simply driving user-defined trains with possibly inappropriate spike times. Related experimental leverage in terms of more natural control of native spike timing may also result from use of the optoXRs (Fig. 1; Table 1), which achieve temporally precise modulation of intra-cellular biochemical activity rather than direct control of spiking; of course, in some settings direct control of spike timing is the desired effect, in which case ChR2 or VChR1 are employed.
Genetic strategies for targeting expression to specific neural populations
Opsin genes can be selectively expressed in defined subsets of neurons in the brain using a variety of expression targeting strategies26–28. Here we focus on those techniques that have been shown to be effective in achieving functional expression of optogenic proteins in vivo.
Viral expression systems: unlike many other genetic targeting strategies requiring the use of transgenic animal models, viral vectors29 based on lentivirus and adeno-associated virus (AAV) can be used to target opsin gene expression in a wide range of experimental subjects ranging from rodents to primates. Specifically, high titer lentivirus and AAV-based vectors (>109 transducing units (TU) ml−1 for lentivirus and >1012 genome copies (gc) ml−1 for AAV vectors) can be easily produced in Biosafety Level 2-certified tissue-culture facilities in 1–2 weeks, or alternatively obtained through a number of virus production facilities (e.g., Viral Vector Core, University of North Carolina, Chapel Hill, North Carolina, USA, and Virapur, LLC., San Diego, California, USA). These transduction methods have been shown to achieve high levels of functional opsin gene expression in neurons for several months.
Although most common AAV (Fig. 1d, top) and lentivirus (Fig. 1d, middle) vectors carry strong ubiquitous or pan-neuronal promoters, some more specific promoter fragments retain cell type-specificity, allowing selective targeting in animals where transgenic technology is not accessible. In addition, viruses are capable of mediating high levels of opsin gene expression by introducing multiple gene copies into each target cell, an important function for overcoming the low transcriptional activity of some cell-specific promoters. In general for rodent brains, opsin gene expression reaches functional levels within 3 weeks after AAV injection and within 2 weeks after lentivirus injection. We have found that to reach the high steady-state levels of expression in distal axonal processes, longer periods of expression (>6 weeks) may be necessary.
Electroporation: specific cell types can also be targeted developmentally with in utero electroporation24,30, e.g. at precisely timed embryonic days in mouse to target cortical layers II and III (E15.5), layer IV (E13.5) or layers V and VI (E12.5). In utero electroporation also can be used to express opsin genes in the inhibitory neurons of the striatum or in the hippocampus30,31. In addition, unlike viral delivery methods, in utero electroporation can be used to deliver DNA of any size, therefore permitting the use of larger promoter segments to achieve higher cell-type specificity (Table 2). Electroporation also allows high copy number of genes to be introduced into the target cells.
TABLE 2.
List of cell type-specific expression strategies proven compatible with optogenetics in mammalian tissues.
| Method | Cell-types targeted | Timing | Pros | Cons | Comments |
|---|---|---|---|---|---|
| Transgenic technology | In principle, any cell type identifiable via a molecular marker. A transgenic construct can be made by fusing the opsin gene to the promoter region of the cell type-specific marker | 6 months to 1 year for obtaining a stable transgenic (mouse or rat) line; marmoset transgenics also now possible | Consistent spatial distri- bution of targeted cells within each animal in a given transgenic line Higher specificity can be obtained by using bacterial artificial chromosomes (BACs) to introduce a large fragment of the promoter sequence Relatively uniform levels of opsin gene expression across targeted cells in the targeted population |
Endogenous promoters tend to drive low levels of transgene expression, often leading to insufficient levels of opsin gene expression to mediate robust optical control Generation of a transgenic line is slow and labor intensive Transgenic technology is more challenging in non-murine species |
Two transgenic mouse lines expressing a microbial opsin gene have been described thus far: Thy1::ChR2–EYFP32,33 and Thy1::NpHR–EYFP22 |
| Lentivirus | Cell type-specific promoters: CaMKIIα49, excitatory glutamatergic neurons SynapsinI4, neuron specific GFAP9, astrocytes ppHcrt8, hypocretin neurons |
2 weeks for construction and production of the recombinant viral vector, 2 weeks after injection to achieve a high level of opsin gene expression | Short testing cycle enables rapid screening of recombinant promoters for targeting specific cell types Can be pseudotyped with rabies glycoprotein to gain retrograde-transduction properties Can achieve high levels of opsin gene expression by increasing the copy number of transgene Expression persists for years |
Limited packaging capacity (< 10 kb total length between long terminal repeats, LTRs) prevents the use of large promoter fragments, therefore compromising specificity Precision of stereotactic injection is limited Variable levels of opsin gene expression across transduced cells Small volume of transduction Some cell type-specific promoters have weak expression levels |
Lentiviral vectors have been successfully applied in a range of mammalian hosts, ranging from mice and rats to birds and monkeys. When pseudotyped with VSVg, lentiviral vectors can effectively transduce all mammalian neural tissues |
| Adeno-associated virus (AAV) | Cell type-specific promoters: SynapsinI71, neuron specific ppSST56, SST neurons ppMCH72, melanin concentrating hormone neurons |
2 weeks for construction and production of the recombinant viral vector, 3 weeks after injection to achieve a high level of opsin gene expression | Short testing cycle enables rapid screen of recombinant promoters for targeting specific cell types Retrograde-transduction properties possible in certain serotypes Can achieve high levels of opsin gene expression by increasing the copy number of transgene Expression persists for years Low immunogenicity |
Limited packaging capacity (< 5 kb total length between LTRs) prevents the use of large promoter fragments, therefore compromising specificity Precision of stereotactic injection is limited Variable levels of opsin gene expression across transduced cells Moderate volume of transduction Some cell type-specific promoters have weak expression levels |
Different AAV serotypes have slightly different tropism and transduction efficiency. For neural tissue, AAV1, 2, 5, 8, and 9 have been shown to transduce neurons in the brain, but with varying distribution and efficiency. The cell type-specific tropisms of each serotype remain to be fully explored and compared |
| Cre-dependent AAV expression system | Cell type specificity is determined by the choice of Cre transgenic lines. | 3 weeks for construction and production of the viral vector, 3 weeks after injection to achieve expression | As with conventional AAV vectors High level of cell type specificity when used with Cre drivers Overcomes the low transcriptional strength of some cell type-specific promoters by amplifying opsin gene expression via a Cre-dependent strong promoter74 Leak in absence of Cre prevented by use of the doubly floxed inverted opsin (DIO) system34,10,11,36,74 |
As with the AAV vector system | |
| Herpes simplex virus 1 (HSV-1) | Cells can be labeled based on their projection targets | ~2 months for the construction of the expression vector and production of recombinant viral vectors 1 to 2 weeks to achieve sufficient level of opsin gene expression | Strong and rapid expression Robust retrograde transporting property75 Can target specific cell populations based on projection target Can be applied in all mammalian models Larger (~150 kb) packaging capacity than lentivirus and AAV |
More difficult to produce than lentivirus More stringent biosafety precautions than AAV |
Commercial HSV vectors may be obtained from Neurovex Other retrograde possibilities include pseudorabies virus and rabies virus vectors |
| Selective control of light-sensitive neural afferents | Axonal projections coming from a brain region of interest can be targeted via focal injection of virus into the axon tract origin. Light-sensitive axon processes coming from the site of injection can be activated at the target region. | For virus-mediated expression, wait at least 4 weeks after injection to allow accumulation in the axonal membrane in the target region | Can used to study specific neural projections Not limited to transgenic mice |
Must wait >4 weeks after viral injection Cannot discriminate among fibers with different local cellular targets, which will require additional emerging targeting technologies |
Transgenic mice: transgenic technologies can be used to restrict gene expression to specific subsets of neurons in mice or rats. Using either short transgene cassettes carrying recombinant promoters or bacterial artificial chromosomes (BACs)-based transgenic constructs, microbial opsin genes can be functionally expressed in subsets of neurons in intact circuits. Several transgenic mouse lines carrying ChR2 under the Thy-1 promoter, that have proven useful for a wide array of experiments, have been generated without any noticeable behavioral or reproductive defects22,32,33.
Conditional expression systems: although cell-specific promoters are effective at restricting gene expression to subsets of genetically defined neurons, some promoters have weak transcriptional activity. Therefore when used to direct the expression of microbial opsin genes, many cell-specific promoters are unable to achieve the level of opsin gene expression necessary to mediate effective action potential firing or blockade. To amplify the transcriptional activity in a cell-specific manner, conditional AAV vectors34–36 (Fig. 1d, bottom) have been developed recently to capitalize on the numerous cell-specific Cre-driver transgenic mouse lines that have been made available by individual labs and collective projects such as GENSAT. These conditional AAV expression vectors carry transgene cassettes that are activated only in the presence of Cre, and the use of strong ubiquitous promoters to drive the Cre-activated transgene selectively amplifies opsin gene expression level only in the cells of interest.
Circuit-specific cell targeting based on neuronal projection patterns: neurons identified by a given genetic marker can still be quite diverse, either receiving innervations from or sending axonal projections to distinct brain regions. For example, some of the tyrosine hydroxylase-expressing dopaminergic (DA) neurons in the midbrain innervate reward-related brain structures such as the nucleus accumbens, whereas other DA neurons project to motor control centers such as the striatum, and spatial separation between different DA neuron populations is not complete. However, as rhodopsins are trafficked to cellular processes including axons, the axonal processes may become light sensitive. Therefore, it may be possible to selectively control a connection-defined neural pathway through focal injection of viral vectors followed by photostimulation of axon terminals in the target downstream brain structure. For example, to study the medial prefrontal cortical afferents in the amygdala, it is possible to inject a viral vector carrying ChR2–enhanced yellow fluorescent protein (EYFP) into the medial prefron-tal cortex and implant the stimulation optical fiber into the amygdala. It is even possible to combinatorially interrogate the role of multiple subsets of afferent fiber bundles using multiple optogenetic proteins with distinct spectral sensitivities (e.g., using yellow light to activate VChR1-expressing afferents independent of ChR2-expressing afferents within the same neural tissue).
A number of plant and microbial proteins and several viral vectors with unique anterograde- or retrograde-transporting properties28,37,38 may be engineered with recombinases to activate gene expression in sub-populations of neurons with cell type- and circuit specificity. For example, expression of fusion proteins containing Cre and either wheat germ agglutinin or tetanus toxin fragment C in the cell bodies of one brain region will allow the recombinase to be trans-neuronally delivered to up- or down-stream neurons in another brain region. Similarly, viral vectors39,40, such as rabies virus28 or herpes simplex virus 1 (HSV-1) vectors, can be used for retrograde gene delivery, and the H129 strain76 of HSV might be developed for anterograde gene delivery. When combined with conditional expression systems, either Cre-dependent transgenic mice or viral vectors, this strategy may allow circuit-specific gene expression in a variety of mammalian animal models not limited to mice. Moreover, microbial protein expression can also be restricted to specific intra-cellular compartments and locations by fusing to targeting motifs and protein domains24,41.
Readouts compatible with optogenetic control of intact circuits
Several classes of circuit readout can be made compatible with optogenetic control. First, voltage-sensitive dye (VSD) imaging is an effective approach for monitoring the electrical activity of large populations of neurons ex vivo and in vivo with high temporal resolution. VSDs are lipophilic molecules whose optical absorbance or emission properties vary depending on the electrical potential across the membrane42. Along with high-speed cameras to capture changes in the optical signal, VSD imaging allows researchers to measure electrical activity changes in neurons on the millisecond timescale and with micro-scale spatial resolution43–46. Previously, we have utilized VSDs to image ex vivo and quantify the activity of neural circuits involved in depression-related behaviors47. VSD imaging can also be combined with optogenetic techniques to conduct all-optical stimulation and imaging of evoked circuit responses in brain slices. Optically compatible VSDs such as RH-155 (absorption ~700 nm) are sufficiently separated from the excitation peaks of ChR2, VChR1 and NpHR48 (Fig. 1c) to permit multimodal all-optical stimulation and imaging. The protocol described below for fast all-optical interrogation has been optimized for brain slices from Thy1::ChR2–YFP transgenic mice32,33.
A second class of readout involves in vivo control integrated with either or both of electrical recording and behavioral measures. Targeting light-stimulation and electrical-recording devices to reach the site of opsin gene delivery poses an experimental challenge that can readily be overcome with the appropriate protocol. We have developed a fiber optic-based optical neural interface (ONI) that meets this challenge49. The ONI uses stereotactically implanted (Fig. 2a–i) cannula guides for targeted virus infusion and light delivery into desired brain structures (Fig. 3a).
Figure 2.
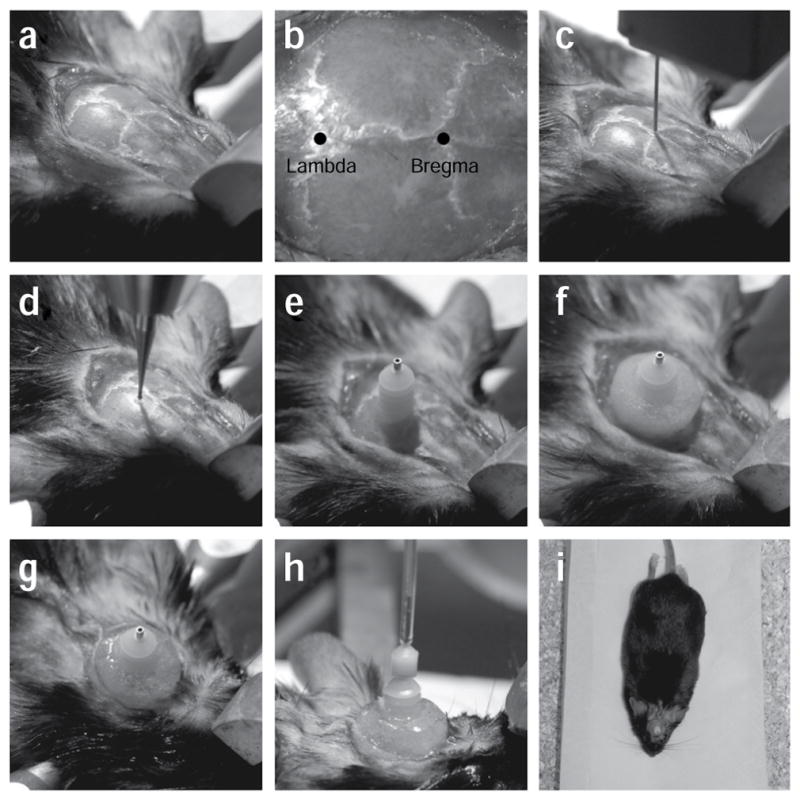
Stereotactic implantation of the cannula guide. (a) After mounting the animal into the stereotactic frame, a first incision is made to open the skin above the skull. The skin is gently pulled to the side to reveal the cranial sutures. (b) After quickly wiping the skull with hydrogen peroxide, the bregma and the lambda can be easily identified (marked spots). (c) A thin needle is used to align the skull. (d) A dental drill is used to create a small craniotomy at the desired location on the skull, without puncturing the dura. The dura is later removed using fine forceps to minimize damage to the cortex. (e) A cannula guide is implanted on the skull through the craniotomy. (f) Metabond and dental cement are used to secure the cannula guide to the skull. (g) Vetbond and sutures are used to close the incision around the cannulation site. (h) An internal cannula guide connected to a pump is inserted through the cannula guide and is used to infuse virus into the target area in the brain. (i) The animal is allowed to rest in a recovery cage after surgical implantation. The surgery was conducted according to established animal care guidelines and protocols at Stanford University.
Figure 3.

Preparation of the optical fiber for in vivo neural control in mammals. (a) Diagram of the optical neural interface (ONI), consisting of a stereotactically implanted cannula, an optical fiber, and a solid state laser controlled by a signal generator. The fiber is prepared with the appropriate length to illuminate the target brain region. (b) The tools and parts (fiber with FC connection, dummy cannula, internal cannula, cannula guide, dental drill, super glue, diamond scribe and fiber stripping tool) needed to manufacture ONI fibers. (c,d) A drill is used to produce a small bore on the center of a cannula cap. (e,f) The steel tubing is removed from an internal cannula. The plastic adapter is used as the fiber guide. (g) A fiber stripping tool is used to remove the plastic cladding from a fiber to reveal the fiber core. (h) The bare fiber core is threaded through the bore on the cannula cap. (i,j) The plastic adapter from the internal cannula is also placed on the fiber. (k,l) Superglue is applied to the fiber to secure the plastic adapter against the plastic cladding on the fiber. The plastic adapter is held tightly against the fiber cladding for several minutes to allow the superglue to harden. (m) After superglue has hardened, the fiber is inserted through a cannula guide with the right projection length for the target brain region. (n) The cannula guide is securely clipped into the plastic adapter. (o) A diamond scribe is used to remove the excess fiber from the tip of the cannula guide. (p) The finished ONI fiber is allowed to protrude from the tip of the cannula guide by ~0.5 mm. Part a was modified with permission7 (Nature © 2007; Macmillan Publishers Ltd.).
Stereotactic targeting of brain structures: stereotactic surgeries can be used to target opsin gene expression to genetically defined and spatially restricted neuronal populations in the brain and can also be used to place a light delivery device to target the transduced cells using widely available brain atlas coordinates50 as described below. There are two major methods for stereotactically delivering lentiviral or AAV vectors to the brain. In the first approach, primarily applicable for in vitro acute slice electrophysiology or imaging studies, the viral vectors can be injected through a glass needle, targeted to the brain region of interest via a stereotactic frame; the use of fine glass needles fabricated from glass capillaries minimizes the extent of tissue damage. A small diameter Hamilton syringe can also be used in place of the glass needle; the Hamilton syringe can be mounted onto a micro-pump such as the WPI UltraMicroPump III, which is directly attached to a stereotactic frame.
The second approach utilizes a stereotactically implanted cannula guide not only to deliver virus but also to direct an optical fiber to the same brain area of interest (Fig. 3a). In this approach, the use of a single cannula guide for both viral vector delivery and optical fiber targeting ensures the co-registration of transduced brain area and light illumination. The use of cannula guides and infusion cannulae is a well-suited method for infusion of compounds into specific brain areas. Cannula systems consist of three major components: cannula guide, injection (or internal) cannula and dummy cannula. The projection length of each component can be customized for each component depending on the brain region of interest. Depending on the experimental application, the material composition and physical dimensions of each component can also be varied; e.g., silica instead of steel cannulae are more suitable for integration with magnetic resonance imaging, and larger diameter cannula guides can be used with bigger rodents such as rats to accommodate the insertion of a larger diameter optical fiber to increase the volume of illumination. The cannula guide is chronically implanted onto the skull of each experimental subject (Fig. 2h). A dummy cannula (stylet and screw cap) is temporarily inserted into the cannula guide between experiments to prevent clogging and infection. During behavioral experiments, an optical fiber can be inserted through the cannula guide to illuminate the light-sensitive neurons. In addition, bilateral cannula systems are suitable for targeting multiple brain regions within the same experimental subject.
Choice of light sources: there are several factors to be considered, including the intensity of light needed to reliably activate a sufficient volume of tissue and the ability to deliver high frequencies of light flashes to the experimental sample. For in vivo setups, it is also important to consider the mechanics and co-registration of the light delivery with the opsin-expressing brain region. For in vitro experiments where the tissue sample is manipulated under a microscope, most conventional light sources such as halogen/xenon arc lamps, LEDs and lasers can be directly coupled to the microscope’s light path. Light flashes can be generated either by a lamp coupled to a fast shutter (e.g., Lambda DG-4 or Uniblitz shutter) or by directly modulating the intensity of light output using a waveform generator (lasers and LEDs; for optimal excitation wavelengths see Table 1).
For in vivo light delivery in freely moving animals, high power lasers (~10–15 mW output at the tip of a 100 μm fiber) and LEDs are most suitable. For superficial stimulation of cortical layers, commercially available small LEDs can also be directly mounted above the brain over a thinned skull area or cranial glass window15,24. To target deep brain structures, we have found that thin optical fibers can be used to efficiently transmit sufficient powers of light to the target area8,49. Optical fibers are versatile elements that can be fused to recording electrodes (creating an optrode24 as described below for electrophysiological readout) and coupled to lasers and LEDs by FC/PC connectors. For mice, up to 300 μm fibers (bare fiber without plastic cladding) can be used without compromising animal movement, whereas we find that rats can tolerate up to 400 μm fibers. The length of the bare optical fiber can be customized based on the depth of the brain area by removing the precisely suitable length of plastic cladding. Mammalian brain tissue scatters light heavily, but ~10% of initial light power density remains at a distance from the fiber tip of ~500 μm8,49.
To avoid fiber breakage resulting from repeated insertion through the cannula guide system, a short fiber segment can be permanently implanted in the target region. During experiments, this short indwelling fiber segment can be coupled to a longer fiber connected to the laser by a custom fiber-to-fiber connector (Doric Lenses, Quebec, Canada). This strategy avoids fiber breakage, minimizes damage to brain tissue compared with implanted cannulas and reduces the likelihood of infection due to environmental exposure through the cannula. However, the fiber connector can lead to up to 50% loss of transmitted light, which may be overcome with more powerful laser diodes.
Choice of behavioral subjects: it is important to choose the appropriate species and genetic background according to the behavioral assay. For example, BALB/C animals are more suitable than C57BL6/J animals for anxiety and depression studies, whereas C57BL6/J are preferred over BALB/C for locomotor and cognitive studies51. When using transgenic mouse lines, it is often important to make sure that the mice are backcrossed (at least for six generations)52 to the appropriate genetic background.
Mechanical design considerations for behavioral equipments: during optogenetic behavioral experiments, each animal subject will have an optical fiber tethered to the skull throughout the experiment. Therefore, each subject should undergo sufficient habituation for the behavioral setup. Certain kinds of experimental apparatus containing obstacles such as doors, tunnels or closed compartments may need to be modified to allow free passage of the optical fiber. In some experiments, the acute insertion and removal of an optical fiber before and after the behavior test may be stressful for the experimental subject; therefore, previous handling will help alleviate this stress. A fiber commutator (Doric Lenses) may be used to relieve tension in the fiber and provide more rotational freedom for the experimental animals.
Summary
We have provided a compact collection of our key technical insights and protocols for optogenetic targeting of deep brain structures in mammals, as well as our protocols for major readout strategies, both in vivo and ex vivo. These methods promise to permit systematic exploration of brain circuits properties that would otherwise be intractable for this kind of work. Together these protocols are intended to facilitate the widespread and versatile interrogation of circuit dynamics underlying mammalian behaviors in health and disease.
MATERIALS
REAGENTS
Virus production: lentiviral plasmids
-
Channelrhodopsin vectors
pLenti-CaMKIIa-hChR2(H134R)-EYFP-WPRE
pLenti-Synapsin-hChR2(H134R)-EYFP-WPRE
pLenti-CaMKIIa-VChR1-EYFP-WPRE
pLenti-EF1a-hChR2(H134R)-EYFP-WPRE
pLenti-CaMKIIa-hChR2(H134R)-mCherry-WPRE
-
Halorhodopsin vectors
pLenti-CaMKIIa-eNpHR-EYFP-WPRE
-
SFO channelrhodopsin vectors
pLenti-CaMKIIa-hChR2(C128A)-EYFP-WPRE
pLenti-CaMKIIa-hChR2(C128S)-EYFP-WPRE
pLenti-CaMKIIa-hChR2(C128T)-EYFP-WPRE
-
Lentivirus support plasmids
pCMVdeltaR8.74
pVSVg ▲ CRITICAL These plasmids can be obtained from the Deisseroth laboratory directly (http://www.optogenetics.org) or from the Deisseroth plasmid list at Addgene. University-based virus production services, such as the virus vector cores at the University of North Carolina and University of Pennsylvania, will accept plasmid DNA and produce ready-for-injection virus for a fee.
Virus production: mammalian expression vector plasmids for transient transfection
-
Channelrhodopsin vectors
pcDNA3.1/VChR1-EYFP
pcDNA3.1/hChR2(H134R)-EYFP
pcDNA3.1/hChR2(H134R)-mCherry
-
OptoXR Vectors
pcDNA3.1/opto-α1AR-EYFP
pcDNA3.1/opto-β2AR-EYFP
Virus production: cre-dependent DIO AAV vectors
pAAV-EF1a-double floxed-hChR2(H134R)-EYFP-WPRE-hGHpA
pAAV-EF1a-double floxed-eNpHR-EYFP-WPRE-hGHpA ▲ CRITICAL These plasmids can be obtained directly from the Deisseroth laboratory (http://www.optogenetics.org) or from the Deisseroth plasmid list at Ad-dgene. University-based virus production services, such as the virus vector cores at the University of North Carolina and University of Pennsylvania, will accept plasmid DNA and produce ready-for-injection virus for a fee.
293FT cells (Invitrogen, cat. no. R700-07)
Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, cat. no. 12-604Q)
UltraCULTURE serum-free medium (Lonza, cat. no. 12-725F)
Penicillin–streptomycin–L-glutamine mixture (Lonza, cat. no. 17-718R)
Sodium pyruvate solution (Lonza, cat. no. 13-115E)
Sodium bicarbonate solution (Lonza, cat. no. 17-613E)
Phosphate buffered saline (PBS) without Ca2 + and Mg2 + (Lonza, cat. no. 17-516F)
Defined fetal bovine serum (HyClone, cat. no. SH30070.03)
Sodium butyrate (Sigma, cat. no. 19364-1G)
HEPES (Sigma, cat. no. 54457-50G-F)
Sodium phosphate dibasic (Sigma, cat. no. 71636-250G)
Sodium chloride (Sigma, cat. no. 71376-1KG)
Hexadimethrine bromide (Sigma, cat. no. 107689-10G)
Sodium hydroxide (Sigma, cat. no. 71689-500G) ! CAUTION Strong base, handle and store according to hazardous material protocol.
Distilled H2O (Lonza, cat. no. 17-724F)
Calcium chloride 2 M stock solution (Quality Biological, cat. no. 351-130-061)
Stereotactic injection and cannula implantation
Anesthetics (Mice: Ketamine 80 mg kg−1 and Xylazine 12 mg kg−1; Sigma, cat. no. K113; Rats: 80 mg kg−1 and Xylazine 6 mg kg−1, Sigma, cat. no. K4138). Alternatively, isoflurane may also be used. ! CAUTION Ketamine is a controlled substance and should be handled according to relevant rules of the host institutions. Isoflurane is a highly volatile substance and a proper fume hood and ventilation device should be used.
Analgesics (Rats: Buprenorphine 0.01–0.05 mg kg−1; Mice: Buprenorphine 0.05–0.1 mg kg−1 subcutaneous, Sigma, cat. no. B9275) ! CAUTION Buprenorphine is a controlled substance and should be handled according to relevant rules of the host institutions.
Lubricant eye ointment (Pharmaderm, cat. no. NDC 0462-0211-38)
Sterile PBS (PBS 10×, Gibco, cat. no. 70011)
Ethanol (Sigma, cat. no. 459836) ! CAUTION Ethanol is flammable. Avoid exposure to ignition.
Hydrogen peroxide 30% (Sigma, cat. no. 31642)
C&B metabond (Parkell, cat. no. IS380)
Dental cement (Stoelting, cat. no. 51459)
Tissue adhesive (Fisher, cat. no. NC9259532)
Paraffin oil (Fisher, cat. no. BP26291)
Betadine (PurduePharma, cat. no. a8658)
-
8-week old C57BL/6J mice (The Jackson Laboratory, cat. no. 000664)
! CAUTION All experiments using animals should be carried out under institutional and national guidelines.
VSD imaging
D(+)-Sucrose (Fluka, cat. no. 8410026)
D(+)-Glucose (Sigma, cat. no. S6297)
NaCl (EMD Chemicals, cat. no. SX0420-1)
KCl (Fluka, cat. no. 60128)
NaH2PO4 (Fluka, cat. no. 74196)
NaHCO3 (Sigma, cat. no. S6297)
2 M CaCl2 stock solution (Quality Biological, cat. no. 351-130-061)
2 M MgCl2 stock solution (Quality Biological, cat. no. 340-034-061)
RH 155 (AnaSpec, cat. no. 84726)
Agarose (Invitrogen, cat. no. 16500100)
Super Glue (Duro, cat. no. SUP-1)
EQUIPMENT
Virus production
0.45 μm Low-protein binding filter flask (Millipore, cat. no. SCHVU02RE)
T-75 and T-225 tissue culture flasks (Nunc, cat. nos. 156499 and 159934)
Four-layer cell factory culture flask (Nunc, cat. no. 140004)
Ultracentrifuge tubes (Beckman Coulter, cat. no. 344058)
Beckman Coulter Allegra X-12 bench-top centrifuge (Beckman Coulter)
Beckman Coulter L-100 K preparative ultracentrifuge (Beckman Coulter)
Beckman Coulter SW-28 ultracentrifuge rotor (Beckman Coulter)
Stereotactic injection and cannula implantation
Surgical tools including scissors, forceps and scalpel blades (Fine Science Tools)
Small animal stereotactic frame (Kopf Instruments, cat. no. 922 for mice, cat. no. 955 for rat) with cannula holder (Kopf Instruments cat. no. 1766-AP)
Programmable microsyringe pump (WPI, cat. no. SP220I)
10 μl Hamilton microsyringe (WPI, cat. no. nanofil) and tubing (Intramedic, cat. no. 4705)
Cannula guide for 200 μm fiber in mice (PlasticsOne, cat. no. C313GS-5/SPC) internal/injector cannula (PlasticsOne, cat. no. C313IS-5/SPC) and dummy cannula (PlasticsOne, cat. no. C313DCS-5/SPC). The length of the cannula guide and internal cannula can be customized for desired target depth (recommended length for the cannula guide is 0.5 mm above the target region; internal cannula should have a projection length of 0.5 mm beyond the cannula guide tip; dummy cannula should be flush with the tip of the cannula guide).
Dissection stereomicroscope (1–×10) with stand (Leica MZ6, cat. no. 10445614)
Cotton swabs (Puritan Medical Products, cat. no. 25-806 10WC)
High-speed micro drill with charger (Fine Science Tools, cat. no. 18000-17)
0.5 mm Micro drill stainless steel burrs (Fine Science Tools, cat. no. 19007-05)
0.9 mm Micro drill stainless steel burrs (Fine Science Tools, cat. no. 19007-05)
1 ml Syringes with subcutaneous needles (VWR, cat. no. 82002-326)
Surgical suture (Myco Medical, cat. no. SK7772)
Heating blanket (Fine Science Tools, cat. no. 21061 or 21060)
Epoxy glue (Fisher, cat. no. NC9863515)
Optrode (integrated fiber-electrode) recording
473 nm diode laser (Cryst Laser, cat. no. BCL-473-050; 50mW, 473nm, TEMoo CW laser with TTL on/off, analog output intensity modulation, PC connector at laser output)
Optical fiber (ThorLabs, cat. no. BFL37-200 for 200 μm fiber; cat. no. BFL37-300 for 300 μm fiber; fibers need to have a FC end to connect to the PC adapter on the output of the laser; length can be specified depending on the experimental setup; recommended to have at least 1 m of slack fiber from the head of the animal to the first fiber fixture).
Fiber stripping tool (300 μm fiber: ThorLabs, cat. no. T18S31; 200 μm fiber: ThorLabs, cat. no. T12S25)
Electrodes, tungsten, 1 mΩ, ~125 μm (AM systems, cat. no. 573220)
Electrode holder (Kopf Instruments, cat. no. 1774)
Amplifier (AM systems, model 1800 2-channel microelectrode AC amplifier, cat. no. 700,000)
Data recording system (Axon Instruments/Molecular Devices Digidata 1440A or previous Digidata 1320 series, with pCLAMP 10 software)
In vivo optical stimulation for behavioral studies
Arbitrary waveform generator, lasers and optical fiber (Agilent, cat. no. 33220A)
Light power meter (ThorLabs, sensor, cat. no. S130A, and digital console, cat. no. PM100D)
Dust cap (PlasticsOne, cat. no. 303DC/1)
Custom-made aluminum rotating optical commutator (Doric Lenses)
Thy1::ChR2–EYFP line 18 transgenic mice (The Jackson Laboratory, cat. no. 007612) ! CAUTION all experiments using animals should be carried out under institutional and national guidelines.
VSD imaging
Olympus BX51W upright microscope (Olympus)
×4 Objective (Olympus XL Fluoro ×4/340 objective, 0.28 NA, Olympus)
Stable light source for VSD illumination (Olympus, cat. no. TH4-100)
VSD imaging filter cube in Olympus filter housing (Chroma Technologies, emission filter, cat. no. D710/40; dichroic mirror, cat. no. 665DCXXR)
Slice perfusion chamber (Siskiyou, cat. no. PC-V)
High-speed CCD camera and processor with preconfigured Dell Optiplex 755 (SciMedia, cat. no. MiCAM02)
Ultra-fast optical filter switcher and light source for providing optical stimulation (Sutter Instruments, cat. no. Lambda DG-4)
Excitation filters (Semrock, ChR2, cat. no. FF01-470/22-25, NpHR, cat. no. FF01-593/40-25)
Cell strainers (BD Falcon, cat. no. 352360)
Illumination filter for RH155 imaging (Chroma Technologies, cat. no. D710/40)
Slice anchor (Warner Instruments, cat. no. SHD-26H/2)
Waveform generator (Agilent Technologies, cat. no. 3320A)
2-Channel peristaltic pump (Rainin, RP-1 pump, cat. no. 7103-052)
Tygon Tubing 1/16″ I.D. 1/8″ O.D. and 1/32″ wall (US Plastics, cat. no. 57102)
Connectors for tubing (Harvard Apparatus, cat. no. 721410)
Coaxial cables
Dissection tools (Fine Science Tools)
Vacuum centrifuge for evaporation of solvents used in preparation of dye aliquots (Savant DNA Speed Vacuum)
Vibrating slice cutter (Leica Microsystems, cat. no. VT 1000S)
Table-top laboratory animal anesthesia system (VetEquip, cat. no. 901806)
Blades (Fine Science Tools, cat. no. 10050-00)
95% Oxygen/5% CO2 (Praxair, cat. no. OXM)
Shutter (Uniblitz, cat. no. Vmm-D1)
Air table (Newport)
Microscope-fixed stage (Sutter Instruments, cat. no. MT-1000/Y51)
Crystallizing dishes for slice recovery (VWR, cat. no. 89000/290)
50 and 1,000 ml beakers (Corning, cat. nos. 1000-50 and 1000-1L)
1.5 ml Microcentrifuge tubes (Eppendorf, cat. no. 022364111)
10 cm Petri dish (BD, cat. no. 351005)
Parafilm M (VWR, cat. no. 52858-000)
REAGENT SETUP
Virus injection setup
Fill the syringe, the tubing and the glass needle or injection cannula with paraffin oil. Load the virus solution in the needle or injection cannula using the programmable microsyringe pump.
D-10 cell maintenance medium
Prepare 500 ml of D-10 medium by supplementing 500 ml of DMEM with 50 ml of fetal bovine serum, 5 ml of penicillin–streptomycin–L-glutamine mixture, 5 ml of sodium pyruvate and 5 ml of sodium bicarbonate. The final mixture can be stored at 4 °C for 1 month.
Virus production medium
Prepare serum-free virus production medium by supplementing 500 ml of UltraCULTURE with 5 ml of penicillin–streptomycin–L-glutamine mixture, 5 ml of sodium pyruvate and 5 ml of sodium bicarbonate. The final mixture can be stored at 4 °C for 1 month.
20% Sucrose solution
Prepare 10 g of sucrose and then add PBS to a final volume of 50 ml. Filter using 0.22 μm filter and then store at 4 °C for up to 1 month.
2 × HBS buffer
To 450 ml of distilled H2O, add 5.96 g of HEPES (50 mM), 0.106 g of Na2HPO4 (1.5 mM) and 8.18 g of NaCl (280 mM). The pH should initially be ~5.8. Titrate with NaOH to 7.05 (use 5 M NaOH first and then switch to 1 M NaOH). Bring the final volume to 500 ml and then filter with 0.22 μm filter. The solution is stable at room temperature (RT, 22 °C) for 6 months.
Cutting solution
In ddH2O add 120 mM sucrose, 64 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3 and 10 mM glucose. Store at 15 °C. Remains fresh for 2 weeks.
Artificial cerebral spinal fluid (ACSF), 10× stock solution
In ddH2O add 124 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 26 mM NaHCO3, 10 mM glucose, 2.4 mM CaCl2 and 1.3 mM MgCl2. ▲ CRITICAL The solution must be freshly prepared.
Preparation of dye aliquots
Dilute 25 mg powder (MW: 950.3 g mol−1) in 5 ml ethanol and aliquot into ten 0.5 ml aliquots (5 mg ml−1) in 1.5 ml microcentrifuge tubes. Evaporate the solvent from aliquots by centrifuging it in a vacuum centrifuge until dry (~30 min, drying rate: medium, no heat). Each aliquot will contain 2.5 mg of RH 155 dye. Dried aliquots may be stored at 0 °C for up to 1 year.
Preparation of slice staining solution
On the day of experiment, transfer 2.5 mg of RH 155 dye into 25 ml of ACSF to yield a final concentration of 0.1 mg ml−1. Minimize the exposure to light with aluminum foil wrapping and store at RT. Equilibrate the solution with 95% CO2/5% O2.
Agarose blocks for slicing
Prepare 1% agarose solution by adding 1 g of agarose into 100 ml of distilled water. Heat the agarose/water mixture in a microwave until agarose has completely dissolved (~1 min at high heat). Cool to 50 °C and then pour the agarose solution into 10 cm petri dishes (~50 ml per petri dish). After the agarose has solidified, wrap the plates with parafilm to avoid evaporation and store at 4 °C for up to 6 months. On the day of experiment, cut the block large enough to support brain samples (1 cm × 0.5 cm × 0.5 cm).
PROCEDURE
Production of recombinant lentivectors for optogenetic gene delivery in mammals
-
1|
Cell preparation Aspirate the growth medium from one 95% confluent T-225 flask of 293FT cells. Gently rock the flask to ensure complete coverage of the cell monolayer and incubate at room temperature for 5 min or until all of the cells have detached. Add 5 ml of D-10 to the flask to neutralize trypsin present in the medium and pipette three times to break up any cell clumps.
▲ CRITICAL STEP It is important to use low passage 293FT cells for the production of viruses. To make sure that the cells are always in the fastest growth phase, never let the cells approach full confluence.
-
2|
Split the 10 ml of cell suspension into four new T-225 flasks. Add 30 ml of fresh room temperature D-10 to each flask. Gently rock the flasks to ensure that the cells are evenly distributed in the flask. Put all four flasks into a 37 °C CO2 incubator for 2 d or until 95% confluent.
-
3|
Harvest the cells from each of four 95% confluent T-225 flasks of 293FT cells as described in Step 1. Transfer all the cells into 500 ml of room temperature D-10 medium, mix, and place the cell and medium mixture into one four-layer cell factory. Transfer the cell factory to a 37 °C CO2 incubator for 24 h.
-
4|
Transfection of cell factory In a 50 ml conical tube, prepare the following mixture: 690 μg of lentivirus plasmid (e.g., pLECYT and pFCK-hChR2-mCherry), 690 μg of pCMVdeltaR8.74, 460 μg of pVSVg and 5.7 ml of 2 M CaCl2 solution. Mix gently and bring the total volume to 23.75 ml with distilled H2O. Mix thoroughly.
-
5|
Add 23.75 ml of 2× HBS to the DNA–CaCl2 mixture. Mix thoroughly and quickly. Then transfer directly into 500 ml of room temperature D-10.
▲ CRITICAL STEP Mix the DNA–CaCl2 with 2× HBS quickly and then transfer to room temperature D-10 immediately.
Short but thorough mixing ensures efficient formation of small calcium phosphate precipitates.
-
6|
Remove the old medium from the four-layer cell factory and replace with the D-10 containing the calcium phosphate transfection mix. Transfer the plate back to the incubator for 15 h
-
7|
Remove the transfection medium from the cell factory and gently wash using 200 ml of fresh D-10. Add 400 ml of fresh room temperature D-10 to the cell factory. Put cells back into incubator for 8 h
! CAUTION The medium from the cell factory may contain recombinant viral vectors. Apply bleach to the medium to decontaminate.
-
8|
At 24 h post transfection, remove medium from the cell factory and replace with 200 ml of Virus Production Medium supplemented with 5 mM Na butyrate. Return the cell factory into the incubator.
▲ CRITICAL STEP Cells are easily detached at this stage and should be handled gently.
! CAUTION The medium from the cell factory may contain recombinant viral vectors. Apply bleach to the medium to decontaminate.
-
9|
Virus harvest Treat six ultracentrifuge tubes by spraying with ethanol and air-drying in the tissue culture hood.
-
10|
At 48 h post transfection, collect the virus containing supernatant into four 50 ml conical tubes and centrifuge for 5 min at 500×g.
-
11|
Prewash a low-protein binding 0.45 μm filter flask with 30 ml of D10 medium, then filter the virus-containing supernatant
-
12|
Divide the filtered virus-containing supernatant among the six centrifuge tubes
-
13|
Pipette 2 ml of 20% sucrose solution to the bottom of the centrifuge tube.
▲ CRITICAL STEP It is important to pipette slowly so that the sucrose solution forms a discrete layer.
-
14|
Centrifuge in a Beckman SW-28 rotor for 2 h at 82,700g at 4 °C.
-
15|
Gently carry the centrifuge tubes back to the tissue culture hood and pour out the supernatant. There should be a tiny semi-transparent pellet at the bottom of each tube that resembles a soft contact lens. Dry the side of each tube with Kimwipe.
! CAUTION Supernatant may contain trace virus. Apply bleach to the medium to decontaminate.
-
16|
Add 100 μl of cold PBS to the first tube and resuspend the pellet by swirling and gentle pipetting.
▲ CRITICAL STEP Excessive pipetting will degrade the virus.
-
17|
Transfer the medium from the first centrifuge tube into the next tube to resuspend the second pellet. Repeat for the four additional tubes
-
18|
Pipette the virus solution into an Eppendorf tube and spin the solution at 1,000g for 5 min to remove unsuspended virus debris. Centrifugation can be carried out at either room temperature or 4 °C.
-
19|
Aliquot the supernatant and store at − 80 °C for up to 1 year.
▪ PAUSE POINT The supernatant can be stored at − 80 °C for up to 1 year.
Stereotactic injection and cannula placement into the rodent brain (Fig. 2)
-
20|
Animal preparation. C57BL/6 mice (15–30 g) or Fisher rats (~200–300 g) should be housed and handled according to institutional and national guidelines.
-
21|
Ensure that all surgical tools are clean and sterile. Disinfect the area with 70% ethanol and sterilize all tools by autoclaving or immersion in a disinfectant.
▲ CRITICAL STEP Perform all the surgeries under aseptic conditions.
-
22|
Anesthetize rodents using 1.5% isoflurane (for surgeries longer than 1 h) or i.p. injection (90 mg kg−1 ketamine and 5 mg kg−1 xylazine for rats; 80 mg kg−1 and 15–20 mg kg−1, respectively, for mice).
▲ CRITICAL STEP Check for the absence of toe-pinch reflex. Dispose of excess anesthesia according to institutional regulations.
? TROUBLESHOOTING
-
23|
Apply ophthalmic ointment to prevent eye drying
-
24|
Use a heating pad to maintain the body temperature at 35 °C
-
25|
Place the animal in a stereotactic frame. Fix the first ear (or head) bar and appose the animal’s ear canal to the ear bar by applying moderate pressure; place the second ear bar in the opposite ear while gently holding the animal; insert the mouth holder between the jaws of the animal; and finally, attach the nose holder to the animal using low pressure.
▲ CRITICAL STEP Correct head position in the stereotactic frame allows tilt, but no lateral movement of the head.
-
26|
Shave and clean the head, wipe with 70% ethanol and betadine
-
27|
Inject saline solution subcutaneously to prevent dehydration during surgery (30 ml kg−1 of body weight: ~0.5 ml for adult mouse and ~5.0 ml for adult rat).
-
28|
Make a midline scalp incision of ~1 cm using scalpel. Gently pull the skin aside to expose the skull (Fig. 2a). Clean the skull with cotton swabs immersed in hydrogen peroxide.
▲ CRITICAL STEP The thin membranes covering the skull will be disrupted immediately upon exposure to hydrogen peroxide and will appear as a layer of white foam. Stop the reaction with PBS immediately after the application of hydrogen peroxide by wiping with a cotton swab soaked in PBS and remove debris with a scalpel blade.
-
29|
After cleaning the skull with cotton swabs, identify the bregma (the point of intersection of the sagittal suture with the curve of best fit along the coronal suture) and the lambda (midpoint of the curve of best fit along the lambdoid suture)53 (Fig. 2b).
▲ CRITICAL STEP Align the bregma and the lambda to the same dorsal–ventral (or z) coordinates by adjusting the height of the nose clamp on the stereotactic frame until the head is flat. Align the x and y tilt of the animal’s head by measuring the x and y coordinates of the bregma and the lambda.
-
30|
Use a brain atlas to identify the coordinates for the target structure
-
31|
Move to the targeted area defined by the coordinates (Fig. 2c). Slowly make a small craniotomy (slightly larger diameter than the cannula guide) using a drill mounted on the stereotactic frame (Fig. 2d).
▲ CRITICAL STEP To minimize damage to the brain, do not drill through the dura and instead use fine forceps to remove the dura. This is crucial for cortical injections.
? TROUBLESHOOTING
-
32|
Thaw the virus on ice. Lentivirus can be kept on ice for 6 h without significantly affecting the titer, whereas AAV can be stored at temperatures as warm as 4 °C for at least 1 month without the loss of significant titer
-
33|
Place the cannula in the cannula holder on stereotactic frame and slowly lower the cannula guide into the brain.
? TROUBLESHOOTING
-
34|
Dry the skull with cotton swabs. Apply a thin layer of C&B Metabond where the dental cement will be in contact with the skull, including around the cannula pedestal
-
35|
Once the C&B Metabond hardens, release the cannula from the holder and withdraw the cannula holder without moving the cannula guide (Fig. 2e). Secure the cannula pedestal with dental cement and ensure that at least two rounds of threads in the cannula pedestal are exposed (Fig. 2f).
-
36|
Glue the skin back with Vetbond surgical adhesive (Fig. 2g).
-
37|
Perform virus injection during surgery (A) or after surgery (B). It may be necessary to titrate the virus (Box 1).
-
Virus injection during surgery (We describe here our protocol using the micropump from WPI and with special attention on the surgical procedure and cannula implantation for virus injection into rodent brain. Virus can also be delivered using glass capillaries to minimize tissue damage54.)
Inject 0.2–0.5 μl of virus into the brain using a 10 μl microsyringe and a thin 34 G metal needle.
Control the injection volume and flow rate (0.1 μl min−1) with a high precision injection pump (Fig. 2h).
After injection leave the needle in place for 10 additional min to allow the virus to diffuse in the brain. Withdraw the needle slowly afterwards.
After completely withdrawing the needle check that it is not clogged by pumping out a small droplet (0.1 μl).
-
Virus injection after surgery
Carry out this procedure after the dental cement is dry, while the animal is still anesthetized.
Use an internal cannula fitted to the cannula guide’s length (Fig. 2i).
Connect the internal/injector cannula to the tubing filled with oil (which is connected to the microsyringe), load it with virus solution and place it into the cannula guide.
-
Infuse the virus at a very low rate (0.1 μl min−1) as described above.
? TROUBLESHOOTING
-
-
38|
Give buprenorphine (0.03 mg kg−1) subcutaneously following the surgical procedure to minimize pain/discomfort.
-
39|
Let the animal fully recover in a clean cage placed over a heating blanket (Fig. 2i). Monitor the recovery of the animal every 10 min.
-
40|
Simultaneous optical stimulation and electrical recording in living mice with the optrode method
Prepare the optrode by firmly attaching an extracellular tungsten electrode (1 mΩ, ~125 μm) to an optical fiber (~200 μm) (TIMING 10 min).
▲ CRITICAL STEP The tip of the electrode must be deeper (>0.3mm) than the tip of the fiber to ensure illumination of the recorded neurons and prevent artifacts24.
▲ CRITICAL STEP Do not contact the tip of the electrode.
-
41|
Prepare animals by performing craniotomies (A) or use animals that have a cannula implanted in the desired region (B).
! CAUTION All experiments using animals should be carried out under institutional and national guidelines.
-
New craniotomy above target region
Prepare the animal as described in Steps 20–31, in a stereotactic frame on an optics table. ▲ CRITICAL STEP Use a Faraday cage to shield out electrical noise.
-
Existing cannula above target region
Anesthetize the animal as described in Steps 22–24.
Place animal in stereotactic frame (Step 25) so that the cannula is vertically aligned.
-
-
42|
Use an electrode holder to mount the optrode (or optrodes for more than one location) on the stereotactic frame.
-
43|
Lower the optrode to the desired depth through the freshly made craniotomies or through the cannula.
▲ CRITICAL STEP Take all precautions not to damage the electrode tip. Avoid touching the skull or the cannula directly with the electrode tip; once the tip is safely inside the cannula it will not get damaged as it is lowered further.
-
44|
Attach a ground wire to the scalp as a reference signal.
-
45|
Connect the optical fiber to a laser diode of desired wavelength and the electrode to an amplifier. For single unit recordings the recorded signal is band pass filtered between 300 Hz and 5 kHz. For field potentials, low-pass filter the signal at 300 Hz.
-
46|
Use an ADC board (e.g., Digidata) and control software (e.g., pClamp) to collect data and generate light pulses through the fiber.
? TROUBLESHOOTING
-
47|
If desired, sacrifice the animal immediately after recording. The brain may be extracted and sectioned to verify opsin gene expression and accurate placement of the optrode or acute slices can be prepared for electrophysiology or VSDI.
BOX 1. CRITICAL STEP VIRUS INJECTION AND VALIDATION.
It is important to quantitatively validate the targeting of virus injection.
It is recommended to perform several injections with increasing volumes of virus at the same concentration.
Start at 0.2 μl per injection site with 0.2 μl increments up to 1 μl final injection volume.
Assess spatial extent, efficacy and specificity of transduction by histological analysis.
Setting up optical stimulation for behavioral assays
-
48|
Preparation of ONI fibers (Fig. 3). Prepare the ONI fiber using cannula parts from PlasticsOne. These consist of a screwing cap for securing the fiber to the animal’s head, a fiber guard from the internal cannula adapter and a bare fiber with length customized based on the depth of the target region. Prepare the screwing cap by drilling a ~2 mm diameter hole centrally on the top of a dust cap to allow the optical fiber to pass through (Fig. 3c,d).
-
49|
Remove the metal tubing from the plastic pedestal of an internal cannula (Fig. 3e,f).
-
50|
Use a fiber stripping tool to remove ~40 mm of plastic coating from the bare end of an optical fiber (Fig. 3g).
-
51|
Thread the fiber through the cap with the cap opening facing away from the FC connector on the fiber (Fig. 3h). Thread the fiber through the plastic pedestal so that the snap-on part faces away from the FC connector on the fiber (Fig. 3i).
-
52|
Apply a very thin layer of glue to the interface of coated/uncoated fiber and slide the plastic pedestal onto the fiber so it is tight against the plastic coating on the fiber (Fig. 3j–l). Allow the glue to dry for 10 min.
▲ CRITICAL STEP Do not apply excess glue. Too much glue will set slowly, prevent the cap from fitting over the internal cannula and also prevent the internal cannula from snapping onto the cannula guide.
-
53|
Insert the ONI fiber through a cannula guide (same dimensions as the ones implanted on the experimental animals) and ensure that the components snap tightly (Fig. 3m,n). Use a diamond knife to cut the fiber so that it projects 0.5 mm from the tip of the cannula guide (Fig. 3o,p). It is not necessary to polish the fiber end.
▲ CRITICAL STEP It is important to use a diamond knife so the fiber tip is flat to ensure consistent illumination. After cutting, check the fiber tip under a stereomicroscope.
-
54|
Connect the FC end of the fiber to the PC connector on the output port of the laser. Connect the laser control box to a waveform generator.
-
55|
Program the waveform generator with the desired stimulation protocol (i.e., pulse frequency, duration and intensity).
▲ CRITICAL STEP Use a power meter to measure the light output at the end of the fiber before and after behavioral testing.
-
56|
In vivo stimulation for behavior. Insert the ONI fiber through the cannula guide on the animal’s head and tighten the screw cap to secure the fiber.
▲ CRITICAL STEP Extra care should be taken during fiber insertion to prevent breakage.
▲ CRITICAL STEP For most behaviors, it is necessary to habituate the animals to the experimental setup by practicing fiber insertion/removal several times. The animals should be placed in the behavioral testing room for at least 3 h before behavioral testing.
? TROUBLESHOOTING
-
57|
Begin behavioral testing. Stimulation can be applied by triggering the waveform generator.
-
58|
Verify the cannula placement and opsin gene expression by post-behavioral histological analysis.
All-optical stimulation and activity imaging in slices using VSD
-
59|
Optics setup On an upright microscope, set up the stimulation light source (e.g., DG-4) to interfere with the epifluorescence port. The light source or a shutter needs to be in place to allow precisely timed flashes of stimulation light.
-
60|
Set up the VSD filters by placing the imaging filter cube carrying both the dichroic (reflect < 665 nm light to the tissue sample) and emission filter (710 nm bandpass).
-
61|
Connect the halogen lamp to the diascopic lamphouse port and use this light source for illumination of RH 155-stained slices. Place the illumination filter (710 nm) into a slot in the condenser. This filter isolates 710 nm light from the diascopic lamphouse for illumination of RH 155-stained slices. RH 155 is an absorbance dye; the filter placed in the condenser filters the light coming from the bright field light source below the tissue sample, and the amount of transmittance reflects the changes in membrane voltage.
-
62|
Staining in RH 155 dye Prepare acute brain sections from the brain region of interest55. While the slices are recovering in cutting solution with perfusion of CO2/O2, prepare fresh ACSF solution.
▲ CRITICAL STEP Make sure that the pH of the ACSF is between 7.3 and 7.4. Perfuse the ACSF with 95% CO2/5% O2 for 10 min before using.
-
63|
Dilute one aliquot of RH 155 dye with 25 ml ACSF to prepare staining solution. The solution should appear dark blue in color.
▲ CRITICAL STEP It is important to minimize light exposure to the dye. Wrap aluminum foil around all dye containers and keep all the dye solutions in the dark.
-
64|
Equilibrate the staining solution in 95%O2/5%CO2 at room temperature (25 °C) for 10 min.
▲ CRITICAL STEP Wrap foil around staining container and keep in dark.
-
65|
Quickly transfer the slices from the water bath to the staining solution and incubate the slices in the dark for three hours while bubbling with 95%O2/5%CO2.
▲ CRITICAL STEP Place slices flat and spaced evenly, completely immersed in solution. Minimize light exposure by wrapping containers in foil and staining in darkness. Consistent timing, dye concentration and staining area are crucial.
-
66|
Setting up the sample and optimizing the image Transfer the slices in the cell strainer from the staining solution to fresh, ACSF equilibrated with 95% O2/5% CO2 to wash away excess dye. Incubate the slices in the fresh ACSF for 15 min. While the slices are recovering, perfuse the slice chamber on the microscope with equilibrated ACSF. The slices should be evenly stained dark blue.
-
67|
Transfer a single stained slice from the cell strainer into the slice chamber on the microscope stage. Continue to perfuse the chamber with 95% O2/5% CO2 ACSF.
-
68|
Ensure slice stability by placing a slice anchor around the edges of the slice. Ensure Lycra strings from the slice anchor do not cover the region of interest.
-
69|
Adjust the slice to place it in the center of the field of the view and adjust intensity of excitation light (from halogen light source) to enhance contrast while minimizing camera saturation.
▲ CRITICAL STEP Ensure 100% of the illumination light is directed to the camera. After positioning the slice, close the shutter for the halogen light source to minimize photobleaching. All imaging sessions should be carried out in the dark to minimize noise and photobleaching.
-
70|
Stimulation and image acquisition Bear in mind that perfusion of ACSF during image acquisition can introduce motion artifacts. The camera can be set to trigger the function generator and DG-4, if desired. It is also recommended to simultaneously record light-evoked field potentials and to validate the effectiveness of optical stimulation, slice health and proper configuration of the VSDI setup. Process images using MATLAB.
● TIMING
Steps 1–3, Preparation of cells for transfection: 2 d
Steps 4–8, Transfection and culturing of cells to produce lentivirus: 39 h
Steps 9–19, Harvesting, concentration and aliquoting of lentivirus: 4 h
Steps 20–39, Stereotactic injection of virus in rodents: 1–2 h. At least 2 weeks are required after injection before sufficient levels of opsin gene products are expressed in the brain.
Steps 40–47, Simultaneous optical stimulation and electrical recording in vivo: 2–3 h
Steps 48–58, Behavioral testing with in vivo optical stimulation: 2 or more hours depending on the specific behavioral experiment
Steps 59–61, Setting up optics for VSD imaging: 1 h
Steps 62–65, Staining brain sections with VSD: 3 h
Steps 66–69, Setting up samples for imaging and optimizing the settings: 30 min
Step 70, Acquiring VSDI images: 30 min
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 3.
TABLE 3.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 22 | Incomplete anesthesia: persistent response to toe pinch | High tolerance or clearance | Administer additional 10% volume ketamine/xylazine (or corresponding isoflurane increment) |
| 31 | Poor cannula positioning observed during histological validation Excess bleeding after craniotomy |
Stereotactic coordinates based on brain atlas are not accurate for the experimental animal age or strain Vessel trauma |
Tune the co-ordinates for age and species Place cotton swab over craniotomy site. When hemostasis is achieved, irrigate craniotomy site with sterile saline |
| 33 | Excess bleeding after cannula placement | Vessel trauma | Place cotton swab over craniotomy site. When hemostasis is achieved, irrigate the site with sterile saline |
| 37 | No solution can be injected | Needles may clog after advancement through intact tissue | Replace the needle |
| 46 | No electrical signal is recorded Electrical noise observed during recording Optical stimulation does not modulate physiology |
Sensitive electrode tip has been damaged by contact Ground loop present in the setup Opsin-expressing cells may be distant from recording electrode |
After confirming integrity of electrical connections, replace electrode Check all converging grounds to the table and the table ground to the amplifier. Confirm that the ground wire is in contact with the tissue and provide ~30–50 μl saline to the ground wire to ensure electrical connectivity Calculate relevant scattering parameters (accounting for light wavelength, intensity and source geometry) in the context of local neuroanatomy. Optrode may need to be redesigned accordingly or optrode positioning may need to be revised by complete retraction and replacement |
| 56 | Fiber breakage during behavior | Rotational behavior results in fiber torsion | Employ fiberoptic commutator to relieve torsion; some light loss may result |
ANTICIPATED RESULTS
Virus-mediated cell-specific expression of microbial opsin genes in neural tissues
Both lentiviral and AAV vectors carrying an appropriate cell-specific promoter and the opsin gene of interest can mediate robust expression in targeted cell types (Fig. 4a–d)49. Injection of 1 μl of high titer VSVg pseudotyped lentivirus (>109 pfu ml−1) or AAV (>1012 gc ml−1) into the cortex or the hippocampus can result in transduction volumes >1 mm3. Prolonged expression of any membrane protein driven by strong ubiquitous promoters, such as CAG, can lead to toxicity in neurons. However, when the transgene is driven by a reliable and strong cell-specific promoter, expression can be very well-tolerated, stable and also specific, with < 10% of transduced cells belonging to non-targeted cell types as identified by immunostaining for the marker protein. Thus far, both the human Synapsin I and murine CaMKIIa promoters have been used in viral systems (lentivirus or AAV) to drive strong pan-neuronal or excitatory neuron-specific expression, respectively, in the mammalian brain in vivo. The glial fibrillary acidic protein (GFAP) promoter can also be used to drive astrocyte-specific expression from a lentiviral vector. Other promising cell-specific promoters include ppHcrt8, Sst56 and CCK57 (see Table 2 for a list of previously reported cell-specific promoters). When using cell-specific Cre transgenic mice, 1 μl of doubly floxed inverted opsin (DIO) Cre-dependent AAV can be delivered to the target brain area to achieve virtually 100% cell-specific expression in the Cre-expressing cell population.
Figure 4.
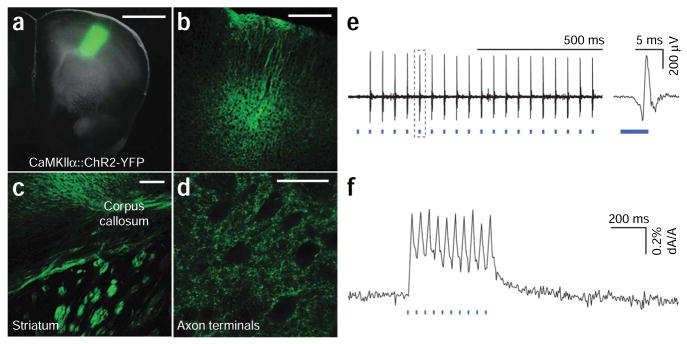
Functional expression of microbial opsin genes in the rodent brain. (a) Fluorescence image superimposed on bright field image of the motor cortex of a wild-type mouse injected with 2 μl lentivirus carrying the CaMKIIα::ChR2–EYFP construct, into layer V of the anterior M1 cortical area (AP 2 mm; ML 2 mm; DV 2 mm); 2 weeks were allowed for expression before preparing 200 μm slices that were examined under confocal (images are z-projection of single planes). (b) The injection produced strong expression in layer V of the cortex (the dendrites projecting from layer V neurons to the surface of the brain are clearly visible). (c) Fibers from layer V of the cortex travel through the corpus callosum (CC) and striatum. (d) Axons terminating in target structures. (e) Optrode recording during optical stimulation of the subthalamic nucleus of a Thy-1::ChR2–EYFP mouse. Example trace shows ten pulses of 5 ms blue light (470 nm) flashes delivered at 20 Hz. At right, zoomed view shows a single 5 ms light flash and resulting evoked spike. (f) Fast all-optical interrogation: typical voltage sensitive dye imaging signal from an acute horizontal hippocampal slice stained with RH 155. Trace shows the voltage changes in the slice resulting from 10 pulses of 10 ms blue light flashes delivered at 20 Hz. Acquisition rate: 200 Hz. The trace is averaged over four acquisition periods and a region of interest of 147 μm × 133 μm was defined in the entorhinal cortex. (Scale bars: a, 1 mm; b, 500 μm; c, 50 μm; d, 25 μm.)
With both lentivirus and AAV, microbial opsin genes will express stably for at least 6 months after injection, and when tagged with a fluorescent protein marker, the transduced cells will exhibit strong membrane-localized fluorescence. Comparatively, ChR2 is better localized to the membrane than NpHR and VChR1, but it may still be difficult to identify the somata of ChR2–EYFP expressing cells in intact tissue. To improve the visualization and identification of expressing cells, the optogenetic protein and the fluorescent protein tag can be expressed as two separated proteins using IRES58 or 2A59,60 bicistronic linkers. Expressing different rhodopsins as separate proteins also ensures that both will integrate into the plasma membrane in the correct orientation. For most reliable optical control of axon terminals (Fig. 4e), it is important to achieve a high level of expression in the axonal membranes; therefore, although physiological effects can be seen earlier, for viral mediated expression, it is optimal to wait until 4 or more weeks after injection before conducting experiments.
Electrophysiological validation of functional expression
Depending on targeted cell type, the maximal evoked firing frequency will vary (for pyramidal neurons in the neocotex, cells expressing ChR2 can be driven to fire spikes up to 30 Hz reliably, whereas other cell types such as fast-spiking interneurons will be able to fire at 100 Hz or more). Spiking properties in targeted cells depend on spike and illumination history as well as on membrane expression level and local illumination intensity; for any given cell type and circuit, a detailed characterization should be carried out to determine the efficacy of light evoked spike trains. To track and validate activity modulation, optrode recordings can be carried out in vivo (Fig. 4e). Artifacts, although much smaller than for electrical stimulation, can be occasionally observed61; when present, such artifacts are correlated with the onset and offset of the light pulse; amplitude depends on light power and can be reduced with proper grounding and use of electrodes with coating extending to the tip and staggered relative to the optical fiber by 300–500 μm, as is typically important in any case for proper illumination of the recorded area.
Imaging optogenetically controlled signals using VSD
Voltage-sensitive dyes imaging of optically evoked activity in neural circuits expressing light-gated ion channels allows for fast, all-optical interrogation and quantification of the spatiotemporal dynamics of ex vivo circuits. In general, brain sections prepared from younger animals (< 6 weeks) yield optimal signals; however, older animals may still be used but with reduced signal properties. Brain sections should be prepared with sufficient thickness to preserve network connections in the target circuit (e.g., ~300 μm for mice and ~400 μm for rats). Because of the inherently low signal of VSDI (~0.1% change in absorbance or fluorescence) and high levels of optical noise, averaging individual pixels over multiple trials is often necessary. Along with averaging these pixels over time, several pixels can also be averaged over space (with reference to anatomical landmarks) to increase the signal-to-noise ratio. Software provided by Scimedia or other fast camera producers (i.e., Redshirtimaging) can carry out these initial tasks. However, more advanced post-processing and quantitative analysis methods are often carried out in our laboratory using algorithms written in Matlab47, available upon request.
Optical stimulation of brain sections from a Thy1::ChR2–YFP transgenic mouse with blue (470 nm) light reveals quantitatively reproducible optical changes in the brain tissue (Fig. 4f). The signal is typically strongest in the CA1 region, where ChR2 expression is highest. Signals are also seen in the entorhinal cortex, the subiculum and the dentate gyrus as a result of both direct activation of ChR2 and percolating circuit activity.
Acknowledgments
We thank the entire Deisseroth laboratory for their support. F.Z. is supported by an NIH NRSA. V.G. is supported by SGF and SIGF (Stanford Graduate Fellowships). A.R.A. is supported by the Fonds National de la Recherche Scientifique, NARSAD, NIH (K99) and the Fondation Leon Fredericq. L.d.L. is supported by NIDA, DARPA and NARSAD. K.D. is supported by the William M. Keck Foundation, the Snyder Foundation, the Albert Yu and Mary Bechmann Foundation and the Wallace Coulter Foundation, as well as by California Institute for Regenerative Medicine, the McKnight Foundation, the Esther A. and Joseph Klingenstein Fund, NSF, National Institute of Mental Health, National Institute on Drug Abuse and the NIH Pioneer Award.
Footnotes
AUTHOR CONTRIBUTIONS F.Z., V.G., A.R.A., R.D.A., R.D., L.d.L. and K.D. wrote the manuscript.
References
- 1.Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 2.Zhang F, Wang LP, Boyden ES, Deisseroth K. Channelrhodopsin-2 and optical control of excitable cells. Nat Methods. 2006;3:785–792. doi: 10.1038/nmeth936. [DOI] [PubMed] [Google Scholar]
- 3.Zhang F, et al. Red-shifted optogenetic excitation: a tool for fast neural control derived from Volvox carteri. Nat Neurosci. 2008;11:631–633. doi: 10.1038/nn.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang F, et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
- 5.Airan RD, Thompson KR, Fenno LE, Bernstein H, Deisseroth K. Temporally precise in vivo control of intracellular signalling. Nature. 2009;458:1025–1029. doi: 10.1038/nature07926. [DOI] [PubMed] [Google Scholar]
- 6.Deisseroth K, et al. Next-generation optical technologies for illuminating genetically targeted brain circuits. J Neurosci. 2006;26:10380–10386. doi: 10.1523/JNEUROSCI.3863-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang F, Aravanis AM, Adamantidis A, de Lecea L, Deisseroth K. Circuit-breakers: optical technologies for probing neural signals and systems. Nat Rev Neurosci. 2007;8:577–581. doi: 10.1038/nrn2192. [DOI] [PubMed] [Google Scholar]
- 8.Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450:420–424. doi: 10.1038/nature06310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science. 2009;324:354–359. doi: 10.1126/science.1167093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsai HC, et al. Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science. 2009;324:1080–1084. doi: 10.1126/science.1168878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bi A, et al. Ectopic expression of a microbial-type rhodopsin restores visual responses in mice with photoreceptor degeneration. Neuron. 2006;50:23–33. doi: 10.1016/j.neuron.2006.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petreanu L, Huber D, Sobczyk A, Svoboda K. Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nat Neurosci. 2007;10:663–668. doi: 10.1038/nn1891. [DOI] [PubMed] [Google Scholar]
- 14.Alilain WJ, et al. Light-induced rescue of breathing after spinal cord injury. J Neurosci. 2008;28:11862–11870. doi: 10.1523/JNEUROSCI.3378-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber D, et al. Sparse optical microstimulation in barrel cortex drives learned behaviour in freely moving mice. Nature. 2008;451:61–64. doi: 10.1038/nature06445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lima SQ, Hromadka T, Znamenskiy P, Zador AM. PINP: a new method of tagging neuronal populations for identification during in vivo electrophysiological recording. PLoS ONE. 2009;4:e6099. doi: 10.1371/journal.pone.0006099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hira R, et al. Transcranial optogenetic stimulation for functional mapping of the motor cortex. J Neurosci Methods. 2009;179:258–263. doi: 10.1016/j.jneumeth.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 18.Petreanu L, Mao T, Sternson SM, Svoboda K. The subcellular organization of neocortical excitatory connections. Nature. 2009;457:1142–1145. doi: 10.1038/nature07709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arenkiel BR, Peca J. Using light to reinstate respiratory plasticity. J Neurophysiol. 2009;101:1695–1698. doi: 10.1152/jn.00009.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagel G, et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci USA. 2003;100:13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gradinaru V, Thompson KR, Deisseroth K. eNpHR: a Natronomonas halorhodopsin enhanced for optogenetic applications. Brain Cell Biol. 2008;36:129–139. doi: 10.1007/s11068-008-9027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao S, et al. Improved expression of halorhodopsin for light-induced silencing of neuronal activity. Brain Cell Biol. 2008;36:141–154. doi: 10.1007/s11068-008-9034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagel G, et al. Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr Biol. 2005;15:2279–2284. doi: 10.1016/j.cub.2005.11.032. [DOI] [PubMed] [Google Scholar]
- 24.Gradinaru V, et al. Targeting and readout strategies for fast optical neural control in vitro and in vivo. J Neurosci. 2007;27:14231–14238. doi: 10.1523/JNEUROSCI.3578-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berndt A, Yizhar O, Gunaydin LA, Hegemann P, Deisseroth K. Bi-stable neural state switches. Nat Neurosci. 2009;12:229–234. doi: 10.1038/nn.2247. [DOI] [PubMed] [Google Scholar]
- 26.Luo L, Callaway EM, Svoboda K. Genetic dissection of neural circuits. Neuron. 2008;57:634–660. doi: 10.1016/j.neuron.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luan H, White BH. Combinatorial methods for refined neuronal gene targeting. Curr Opin Neurobiol. 2007;17:572–580. doi: 10.1016/j.conb.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 28.Wickersham IR, et al. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron. 2007;53:639–647. doi: 10.1016/j.neuron.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dittgen T, et al. Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc Natl Acad Sci USA. 2004;101:18206–18211. doi: 10.1073/pnas.0407976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Navarro-Quiroga I, Chittajallu R, Gallo V, Haydar TF. Long-term, selective gene expression in developing and adult hippocampal pyramidal neurons using focal in utero electroporation. J Neurosci. 2007;27:5007–5011. doi: 10.1523/JNEUROSCI.0867-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borrell V, Yoshimura Y, Callaway EM. Targeted gene delivery to telencephalic inhibitory neurons by directional in utero electroporation. J Neurosci Methods. 2005;143:151–158. doi: 10.1016/j.jneumeth.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 32.Arenkiel BR, et al. In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron. 2007;54:205–218. doi: 10.1016/j.neuron.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H, et al. High-speed mapping of synaptic connectivity using photostimulation in Channelrhodopsin-2 transgenic mice. Proc Natl Acad Sci USA. 2007;104:8143–8148. doi: 10.1073/pnas.0700384104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang F. Larry M. Katz Memorial Lecture, Cold Spring Harbor Laboratory Meeting on Neuronal Circuits. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 2008. Fast optical neural circuit interrogation technology: development and applications. [Google Scholar]
- 35.Kuhlman SJ, Huang ZJ. High-resolution labeling and functional manipulation of specific neuron types in mouse brain by Cre-activated viral gene expression. PLoS ONE. 2008;3:e2005. doi: 10.1371/journal.pone.0002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atasoy D, Aponte Y, Su HH, Sternson SM. A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J Neurosci. 2008;28:7025–7030. doi: 10.1523/JNEUROSCI.1954-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maskos U, Kissa K, St Cloment C, Brulet P. Retrograde trans-synaptic transfer of green fluorescent protein allows the genetic mapping of neuronal circuits in transgenic mice. Proc Natl Acad Sci USA. 2002;99:10120–10125. doi: 10.1073/pnas.152266799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sugita M, Shiba Y. Genetic tracing shows segregation of taste neuronal circuitries for bitter and sweet. Science. 2005;309:781–785. doi: 10.1126/science.1110787. [DOI] [PubMed] [Google Scholar]
- 39.Callaway EM. Transneuronal circuit tracing with neurotropic viruses. Curr Opin Neurobiol. 2008;18:617–623. doi: 10.1016/j.conb.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boldogkoi Z, et al. Genetically timed, activity-sensor and rainbow transsynaptic viral tools. Nat Methods. 2009;6:127–130. doi: 10.1038/nmeth.1292. [DOI] [PubMed] [Google Scholar]
- 41.Lewis TL, Jr, Mao T, Svoboda K, Arnold DB. Myosin-dependent targeting of transmembrane proteins to neuronal dendrites. Nat Neurosci. 2009;12:568–576. doi: 10.1038/nn.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ebner TJ, Chen G. Use of voltage-sensitive dyes and optical recordings in the central nervous system. Prog Neurobiol. 1995;46:463–506. doi: 10.1016/0301-0082(95)00010-s. [DOI] [PubMed] [Google Scholar]
- 43.Kauer JS. Real-time imaging of evoked activity in local circuits of the salamander olfactory bulb. Nature. 1988;331:166–168. doi: 10.1038/331166a0. [DOI] [PubMed] [Google Scholar]
- 44.Tominaga T, Tominaga Y, Ichikawa M. Optical imaging of long-lasting depolarization on burst stimulation in area CA1 of rat hippocampal slices. J Neurophysiol. 2002;88:1523–1532. doi: 10.1152/jn.2002.88.3.1523. [DOI] [PubMed] [Google Scholar]
- 45.Muschol M, Kosterin P, Ichikawa M, Salzberg BM. Activity-dependent depression of excitability and calcium transients in the neurohypophysis suggests a model of ‘stuttering conduction’. J Neurosci. 2003;23:11352–11362. doi: 10.1523/JNEUROSCI.23-36-11352.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grinvald A, Hildesheim R. VSDI: a new era in functional imaging of cortical dynamics. Nat Rev. 2004;5:874–885. doi: 10.1038/nrn1536. [DOI] [PubMed] [Google Scholar]
- 47.Airan RD, et al. High-speed imaging reveals neurophysiological links to behavior in an animal model of depression. Science. 2007;317:819–823. doi: 10.1126/science.1144400. [DOI] [PubMed] [Google Scholar]
- 48.Airan RD, et al. Integration of light-controlled neuronal firing and fast circuit imaging. Curr Opin Neurobiol. 2007;17:587–592. doi: 10.1016/j.conb.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aravanis AM, et al. An optical neural interface: in vivo control of rodent motor cortex with integrated fiberoptic and optogenetic technology. J Neural Eng. 2007;4:S143–156. doi: 10.1088/1741-2560/4/3/S02. [DOI] [PubMed] [Google Scholar]
- 50.Paxinos G, Franklin K. The Mouse Brain in Stereotaxic Coordinates. 2. Academic Press; New York: 2001. [Google Scholar]
- 51.Crawley NJ. What’s Wrong With My Mouse?: Behavioral Phenotyping of Transgenic and Knockout Mice. 2. Wiley; Hoboken, NJ: 2007. [Google Scholar]
- 52.Crawley JN, et al. Behavioral phenotypes of inbred mouse strains: implications and recommendations for molecular studies. Psychopharmacology. 1997;132:107–124. doi: 10.1007/s002130050327. [DOI] [PubMed] [Google Scholar]
- 53.Paxinos G, Watson C, Pennisi M, Topple A. Bregma, lambda and the interaural midpoint in stereotaxic surgery with rats of different sex, strain and weight. J Neurosci Methods. 1985;13:139–143. doi: 10.1016/0165-0270(85)90026-3. [DOI] [PubMed] [Google Scholar]
- 54.Cetin A, Komai S, Eliava M, Seeburg PH, Osten P. Stereotaxic gene delivery in the rodent brain. Nat Protoc. 2006;1:3166–3173. doi: 10.1038/nprot.2006.450. [DOI] [PubMed] [Google Scholar]
- 55.Debanne D, et al. Paired-recordings from synaptically coupled cortical and hippocampal neurons in acute and cultured brain slices. Nat Protoc. 2008;3:1559–1568. doi: 10.1038/nprot.2008.147. [DOI] [PubMed] [Google Scholar]
- 56.Tan W, et al. Silencing preBotzinger complex somatostatin-expressing neurons induces persistent apnea in awake rat. Nat Neurosci. 2008;11:538–540. doi: 10.1038/nn.2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jasnow AM, Ressler KJ, Hammack SE, Chhatwal JP, Rainnie DG. Distinct subtypes of cholecystokinin (CCK)-containing interneurons of the basolateral amygdala identified using a CCK promoter-specific lentivirus. J Neurophysiol. 2009;101:1494–1506. doi: 10.1152/jn.91149.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bochkov YA, Palmenberg AC. Translational efficiency of EMCV IRES in bicistronic vectors is dependent upon IRES sequence and gene location. Biotechniques. 2006;41:283–284. 286, 288. doi: 10.2144/000112243. passim. [DOI] [PubMed] [Google Scholar]
- 59.Holst J, et al. Generation of T-cell receptor retrogenic mice. Nat Protoc. 2006;1:406–417. doi: 10.1038/nprot.2006.61. [DOI] [PubMed] [Google Scholar]
- 60.Tang W, et al. Faithful expression of multiple proteins via 2A-peptide self-processing: a versatile and reliable method for manipulating brain circuits. J Neurosci. 2009;29:8621–8629. doi: 10.1523/JNEUROSCI.0359-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J, et al. Integrated device for optical stimulation and spatiotemporal electrical recording of neural activity in light-sensitized brain tissue. J Neural Eng. 2009;6:55007. doi: 10.1088/1741-2560/6/5/055007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang YP, Oertner TG. Optical induction of synaptic plasticity using a light-sensitive channel. Nat Methods. 2007;4:139–141. doi: 10.1038/nmeth988. [DOI] [PubMed] [Google Scholar]
- 63.Ishizuka T, Kakuda M, Araki R, Yawo H. Kinetic evaluation of photosensitivity in genetically engineered neurons expressing green algae light-gated channels. Neurosci Res. 2006;54:85–94. doi: 10.1016/j.neures.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 64.Zhang YP, Holbro N, Oertner TG. Optical induction of plasticity at single synapses reveals input-specific accumulation of alphaCaMKII. Proc Natl Acad Sci USA. 2008;105:12039–12044. doi: 10.1073/pnas.0802940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hwang RY, et al. Nociceptive neurons protect Drosophila larvae from parasitoid wasps. Curr Biol. 2007;17:2105–2116. doi: 10.1016/j.cub.2007.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pulver SR, Pashkovski SL, Hornstein NJ, Garrity PA, Griffith LC. Temporal dynamics of neuronal activation by Channelrhodopsin-2 and TRPA1 determine behavioral output in Drosophila larvae. J Neurophysiol. 2009;101:3075–3088. doi: 10.1152/jn.00071.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schroll C, et al. Light-induced activation of distinct modulatory neurons triggers appetitive or aversive learning in Drosophila larvae. Curr Biol. 2006;16:1741–1747. doi: 10.1016/j.cub.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 68.Douglass AD, Kraves S, Deisseroth K, Schier AF, Engert F. Escape behavior elicited by single, channelrhodopsin-2-evoked spikes in zebrafish somatosensory neurons. Curr Biol. 2008;18:1133–1137. doi: 10.1016/j.cub.2008.06.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li X, et al. Fast noninvasive activation and inhibition of neural and network activity by vertebrate rhodopsin and green algae channelrhodopsin. Proc Natl Acad Sci USA. 2005;102:17816–17821. doi: 10.1073/pnas.0509030102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Han X, et al. Millisecond-timescale optical control of neural dynamics in the nonhuman primate brain. Neuron. 2009;62:191–198. doi: 10.1016/j.neuron.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kugler S, Kilic E, Bahr M. Human synapsin 1 gene promoter confers highly neuron-specific long-term transgene expression from an adenoviral vector in the adult rat brain depending on the transduced area. Gene Ther. 2003;10:337–347. doi: 10.1038/sj.gt.3301905. [DOI] [PubMed] [Google Scholar]
- 72.van den Pol AN, Acuna-Goycolea C, Clark KR, Ghosh PK. Physiological properties of hypothalamic MCH neurons identified with selective expression of reporter gene after recombinant virus infection. Neuron. 2004;42:635–652. doi: 10.1016/s0896-6273(04)00251-x. [DOI] [PubMed] [Google Scholar]
- 73.Han X, Boyden ES. Multiple-color optical activation, silencing, and desynchronization of neural activity, with single-spike temporal resolution. PLoS One. 2007;2:e299. doi: 10.1371/journal.pone.0000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Petreanu L, Mao T, Sternson SM, Svoboda K. The subcellular organization of neocortical excitatory connections. Nature. 2009;457:1142–1145. doi: 10.1038/nature07709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lima SQ, Hromadka T, Znamenskiy P, Zador AM. PINP: a new method of tagging neuronal populations for identification during in vivo electrophysiological recording. PLoS One. 2009;4:e6099. doi: 10.1371/journal.pone.0006099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rinaman L, Schwartz G. Anterograde transneuronal viral tracing of central viscerosensory pathways in rats. J Neurosci. 2004;24:2782–2786. doi: 10.1523/JNEUROSCI.5329-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Suh GS, et al. Light activation of an innate olfactory avoidance response in Drosophila. Curr Biol. 2007;17:905–908. doi: 10.1016/j.cub.2007.04.046. [DOI] [PubMed] [Google Scholar]
- 78.Lin JY, Lin MZ, Steinback P, Tsien RY. Characterization of engineered channelrhodopsin variants with improved properties and kinetics. J Biophys. 2009;96:1803–1814. doi: 10.1016/j.bpj.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang H, et al. Molecular determinants differentiating photocurrent properties of two channelrhodopsins from chlamydomonas. J Biol Chem. 2009;284:5685–5696. doi: 10.1074/jbc.M807632200. [DOI] [PubMed] [Google Scholar]
- 80.Gunaydin LA, et al. Ultrafast optogenetic control. Nat Neurosci. 2010 Jan 17; doi: 10.1038/nn.2495. advance online publication. [DOI] [PubMed] [Google Scholar]