

Abstract
Cyclin D proteins are elevated in many cancer cells and targeted deletion of Cyclin D1 gene in the mammary tissues protects mice from breast cancer. Accordingly, there is an increasing awareness of this novel non-enzymatic target for cancer therapeutics. We have developed novel, non-alkylating styryl benzyl sulfones that induce cell death in wide variety of cancer cells without affecting the proliferation and survival of normal cells. The development of derivatized Styryl Benzyl Sulfones followed logically from a tumor cell cytotoxicity screen performed in our laboratory that did not have an a priori target profile. Modifications of some of the precursor molecules led to lead optimization with regard to tumor cell cytotoxicity. In this report we describe the synthesis and structure-activity relationships of novel, non-alkylating (E) styryl benzyl sulfones, and the development of the novel anti-cancer agent sodium (E)-2-{2-methoxy-5-[(2′,4′,6′-trimethoxystyrylsulfonyl)methyl]phenylamino}-acetate (ON 01910.Na), which is in Phase III trials for myelodysplastic syndromes (MDS) associated with aberrant expression of cyclin D proteins.
Introduction
In a recent paper,1 we described the synthesis of a group of styryl benzyl sulfones that induce apoptotic death of a wide variety of human tumor cell lines at nanomolar concentrations while exhibiting relatively low toxicity to normal human cells. Our studies showed that the cytotoxic ‘activity of styryl benzyl sulfones is completely dependent on the nature and position of the substituents on the two aromatic rings. Structure–function studies showed that position of functional groups on the styryl aromatic ring play a critical role in determining the biochemical and biological activity of these molecules. Biological evaluation of the activity of these compounds showed that these compounds are highly active against a wide variety of human tumor cell lines including those that are resistant to the activity of many of the currently used chemotherapeutic agents. The low toxicity profile, both in vitro and in vivo, and their potent tumor inhibitory activity as seen in soft agar and nude mouse xenograft assays pointed to the potential value of these compounds as safe therapies for cancer, lacking many of the side effects normally associated with current chemotherapeutic agents. Many of the compounds described in this study were found to act as allosteric inhibitors of serine/theronine and tyrosine kinases providing a rationale for further expansion of this chemotype for applications related to cancer therapy.
The present report describes the synthesis and structure-activity relationships of novel, non-alkylating (E)-styryl benzyl sulfones, and the development of the novel anti-cancer agent sodium (E)-2-{2-methoxy-5-[(2′, 4′, 6′-trimethoxystyrylsulfonyl)methyl]phenylamino}acetate (ON 01910.Na, 28)2. The development of derivatized styryl benzyl sulfones followed logically from a tumor cell cytotoxicity screen performed in our laboratory. Precursors of 28 were identified specifically based upon their ability to target cancer cells while leaving non-malignant cell cultures virtually unaffected. Modifications of some of the precursor molecules led to lead optimization with regard to tumor cell cytotoxicity. Structure-activity studies confirmed that the nature, number, and position of substituents on the two aromatic rings of the parent molecule are the determining factor in the tumor cell cytotoxicity of these compounds.
In previous studies in our laboratory, 28 displayed desirable pharmacokinetic and pharmacodynamic properties, and was able to reduce tumor size and increase survival in mice carrying tumor cell xenografts.2 28 Received orphan drug status for the myelodysplastic syndrome, a heterogeneous hematopoietic stem cell disorder that affects cell proliferation, differentiation, and function. MDS is characterized by dyspoiesis, hyperproliferative bone marrow, and peripheral blood cytopenias involving one or more lineages.3–5 Majority of untreated patients with high risk MDS die from progressive bone marrow failure within one year due to hemorrhage and/or infection. In vitro studies with 28 showed that incubation of human leukemic cells with this compound results in the inhibition of PI3K/AKT pathway, down regulation of cyclin D1, induction of NOXA and BIM and activation of JNK pathway.6 Treatment of MDS patients with 28 results in a dramatic reduction of cytogenetically abnormal cells with a minimal inhibition of normal hematopoiesis. This drug is currently in Phase III clinical trials.
Chemistry
Multiple synthetic routes for synthesis of the (E)-styryl benzyl sulfone scaffold were explored. The initial method (Method A) involved the synthesis of substituted (E)-styryl benzyl sulfones by the reaction of benzyl bromides (2) with mercaptoacetic acid in presence of a strong base, sodium hydroxide in methanol, to obtain benzylthioacetic acids (5) in quantitative yields (Scheme 1). Oxidation of 5 with 30% hydrogen peroxide (H2O2) in glacial acetic acid afforded benzylsulfonylacetic acids 6.7 Knovenagel condensation of 6 with aromatic aldehydes 7 either in benzylamine/acetic acid7 or piperidine/benzoic acid8 in toluene, afforded (E)-styryl benzyl sulfones 8 in good yields.
Scheme 1.

Synthesis of (E)-styryl benzyl sulfones from benzylsulfonylacetic acidsa
a Reagents and conditions: (a) HSCH2COOH, MeOH, NaOH, rt, 3 h; (b) 30% H2O2, ACOH, rt, 24 h; (c) benzylamine, AcOH, reflux, 2–8 h; (d) piperidine, benzoic acid, toluene, 2–4 h.
Some of the substituted benzyl bromides 2 that are not commercially available were synthesized as shown in the scheme 2. Substituted toluenes 1 were brominated with N-bromosuccinimide (NBS) in presence of a catalytic amount of benzoyl peroxide in carbon tetrachloride (CCl4) to obtain 2.9 Some of these nitro substituted benzyl bromides 2 were also made starting from substituted nitro benzaldehydes 3 which were reduced with sodium borohydride (NaBH4)10 and on subsequent bromination of the resulting alcohol 4 with phosphorous tribromide (PBr3) (scheme 2).11
Scheme 2.

Synthesis of benzyl bromidesa
a Reagents and conditions: (a) NBS, benzoyl peroxide, CCl4, 18 h; (b) NaBH4, MeOH, 0 °C–5 °C, 1 h; (c) PBr3, Toluene, 100 °C, 30 min.
In method B, 3-Nitro-4-methoxybenzyl mercaptan (9) was treated with 4-methoxyphenacyl bromide (10) to obtain 4-methoxyphenacyl-3-nitro-4-methoxybenzyl sulfide 11 which on oxidation with 30% H2O2 gave 4-methoxyphenacyl-3-nitro-4-methoxybenzyl sulfone 13.7
Sequential reduction of 13 with sodium borohydride1 and subsequent dehydration with p-toluenesulfonic acid (p-TSA) afforded the desired 8 in moderate yields (Scheme 3).12 Method A is superior to method B for the synthesis of 8 as it involves steps with relatively higher yields. The 3-nitro-4-methoxy benzyl mercaptan 9 was in turn synthesized from 2a as outlined in Scheme 4. 2a on treatment with thiourea in water gave an intermediate isothiouronium salt (12) which on reduction with ammonia yielded 9 in low to moderate yields.13
Scheme 3.

Synthesis of (E)-styryl benzyl sulfones from phenacy benzyl sulfonesa
a Reagents and conditions: (a) NaOH, MeOH, rt, 2 h; (b) 30% H2O2, ACOH, rt, 24 h; (c) NaBH4, tetrahydrofuran, 0 °C, 1 h; (d) p-toluenesulfonic acid, toluene, 2–4 h.
Scheme 4.
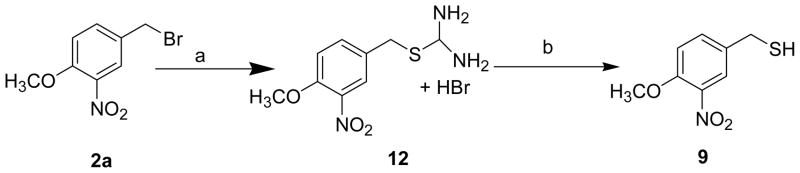
Synthesis of 4-methoxy-3-nitrobenzylmercaptana
a Reagents and conditions: (a) Thiourea, H2O, reflux, 2 h; (b) Ammonia, hexane, reflux, 30 min.
To see the effect of trans sulfide and sulfoxide on the biological activity of these (E)-styryl benzyl sulfones, we made (Z) (16) and (E) (17) 4-methoxy-3-nitrobenzyl-2′,4′,6′-trimethoxystyryl sulfides by the reaction of 3-nitro-4-methoxybenzyl mercaptan (9) and 2,4,6-trimethoxyphenyl acetylene (15) in the presence of triethylborane-hexane (Et3B) in benzene (Scheme 5).14 In this reaction, (Z) to (E) ratio of 40:60 resulting in mainly trans isomers. 17 on controlled oxidation with 1,1,1,3,3,3-hexafluoro-2-propanol and 30% H2O2 at room temperature resulted in sulfoxide 20,15 which on reduction with sodium hydrosulfite in acetone-water mixture at 50 °C afforded the corresponding amine 21.9
Scheme 5.

Synthesis of (Z) and (E)-styryl benzyl sulfIdes from 4-methoxy-3-nitro benzylmercaptan and 2,4,6-trimethoxyphenyl acetylene and (E)-styryl benzyl sulfoxide and sulfonea
a Reagents and conditions: (a) Et3B-hexane, benzene, 25 °C, 2 h; (b) 1,1,1,3,3,3-hexafluoro-2-propanol, 30% H2O2, 25 °C, 2 h; (c) Acetone: water (2:1), sodium hydrosulfite, 50 °C, 30 min; (d) 30% H2O2, AcOH, rt, 24 h; (e) m-CPBA, CH2Cl2, 0 °C - rt, 3 h.
The sulfoxide 20 was oxidized to sulfone 22 with 30% hydrogen peroxide in glacial acetic acid7 which was later reduced with sodium hydrosulfite to corresponding sulfone 23.9 The nitro sulfide 17 was also converted to corresponding amino sulfide 24 which on further oxidation with m-chloroperoxybenzoic acid (m-CPBA) afforded sulfone 23. The acetylene 15 was synthesized starting from 2,4,6-trimethoxybenzaldehyde 18 and tetrabromomethane (CBr4) in the presence of triphenyphosphine (Ph3P) in dichloromethane.16 The resulting 2′,2′-dibromovinyl-1,3,5-trimethoxybenzene 19 on treatment with n-BuLi in THF at −78 °C gave 1516 in high yields. The synthesis of nitro (E)-styryl benzyl sulfoxide 20 and amino (E)-styryl benzyl sulfoxide 21 was also achieved from 4-methoxy-3-nitrobenzylthioacetic acid 5a by oxidation to sulfoxide 2517 and then condensation with 2,4,6-trimethoxybenzaldehyde 18 in the presence of benzylamine and acetic acid (Scheme 7).7 The resulted 20 was reduced to 21 as described earlier in Scheme 5.
Scheme 7.

Synthesis of (E)-styryl 4-methoxy-3-nitrobenzyl sulfoxide from 4-methoxy-3-nitro benzyl sulfoxideacetic acida
a Reagents and conditions: (a)NaOH/deionized H2O, NaHCO3, Acetone, Oxone solution, sodium bisulfite, 6N HCl; (b) benzylamine, AcOH, reflux, 2–8 h; (c) Acetone: water (2:1), sodium hydrosulfite, 50 °C, 30 min.
To enhance the solubility and bioavailability of these (E)-styryl benzyl sulfones, several 3-amino substituted esters and acids were made from (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone (8p) and (E)-3′,4′,5′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone (8q) (Scheme 8). 8p and 8q were treated with different α-bromo esters in the presence of mild base sodium acetate in ethanol for 48 h to give amine esters (26) which on subsequent hydrolysis with sodium hydroxide in ethanol afforded the corresponding acids 27.
Scheme 8.
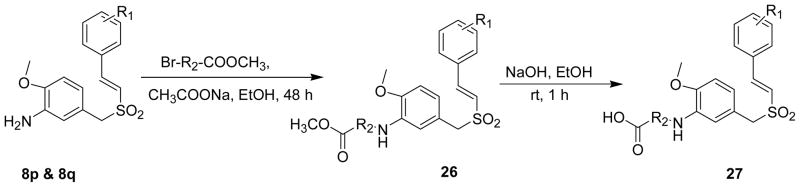
Synthesis of amine esters and acids of 4-methoxy-3-amino styryl benzyl sulfones (8p and 8q)
The lead compound of our investigation, 28, was synthesized from 8p by its reaction with methyl 2-bromo acetate in the presence of a mild base sodium acetate in methanol for 4–6 h to give amino substituted methyl ester 26a which on subsequent hydrolysis with sodium hydroxide in aqueous ethanol and dichloromethane followed by methyl ethyl ketone washing afforded crystalline 28 in high yields with 2 molecules of water of hydration as determined by Karl-Fisher analysis (scheme 9).
Scheme 9.

Synthesis of 28
Structure-Activity Relationships (SAR)
After the synthesis of these compounds, their in vitro cytotoxicity was assessed using two different human tumor cell lines derived from human prostate (DU145) and leukemic (K562) cancers. The results of the study are presented in Table 1. These studies reveal that the cytotoxicity of the (E)-styryl benzyl sulfones is totally dependent on the nature and position of the substituents present on the two aromatic rings. For the purpose of structure activity relationship, we have selected a few compounds from a library of two thousand (E)-styryl benzyl sulfones synthesized from our laboratory. In most of the selected compounds described here, we have kept a methoxy group at the fourth position and an amino group at the third position of benzylsulfonyl aromatic ring and one or more methoxy groups on styryl aromatic ring at different positions. A moderate cytotoxicity in tumor cells was seen in a compound when an amino and methoxy groups are at 3rd and 4th position of the benzyl ring and a methoxy group at the 4th position of the styryl ring (8d). By introduction of second methoxy group on the styryl ring of 8d, the cytotoxicity of the resulting compounds (8g) (8h) and (8i) was further enhanced by several folds. Whereas the results are quite surprising for the molecules that are disubstituted with methoxy groups on the styryl aromatic ring compared to 8d, the results obtained in cytotoxicity assays using these compounds (8g, 8h, 8i, 8j and 8k) clearly shows that the methoxy group, when present at 2, 6 positions (8i), enhances the activity of the molecule by greater than 10 fold when compared to other disubstituted methoxy groups (8g and 8h). It is also clear from the cytotoxicity profile of the compounds 8j and 8k that when amino and methoxy groups are placed at other than 3rd and 4th position, the compounds exhibit decreased cell killing activity: 15 to 500 fold. Because the introduction of two methoxy groups on the styryl aromatic ring enhanced the biological activity, we have synthesized some trimethoxystyryl analogs to determine if this further enhances their cytotoxic properties. Analysis of these compounds (8n, 8o, 8p, 8q, and 8r) in cell-killing assays showed that (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone (8p) exhibited best activity in the entire series. This compound, 8p, is almost 11 folds more active than 8i in both cell lines. These results also show that when 2, 4 and 6 positions of the styryl ring are occupied by methoxy groups (8p) the molecule attains optimum biological activity compared to other trimethoxy substituted styryl sulfones (8n, 8o, 8q and 8r). To validate the critical requirement of the methoxy group at 4th position of 2′, 4′, 6′-trimethoxystyryl moiety, we have replaced the methoxy group at 4th postion by a hydroxy (8t) and a carboxypropoxy group (8u). Both the replacements in 8p caused severe loss of cytotoxicity in the molecules indicating the indispensability of a methoxy group at 4th position of the ring. Further replacing the methoxy group with halogen atoms or changing the position of the amino group in benzyl aromatic ring resulted in molecules (8w, 8x, 8y and 8aa) that suffered substantial loss of cytotoxicity. Also the compounds formed by replacing all three methoxy groups of the styryl ring by fluorine atoms (8ab and 8ac) in 8p have reduced level of cytotoxicity. The SAR analysis clearly shows that all the compounds with nitro substitutions on the benzylic ring (8a, 8c, 8e, 8f, 8l, 8m, 8s, 8v and 8z) are far less active than the corresponding amino compounds indicating that the amino group in that position is critical for the interaction of the compounds with their target and activity.
Table 1.
In vitro cytotoxicity of (E)-styryl benzyl sulfones 8
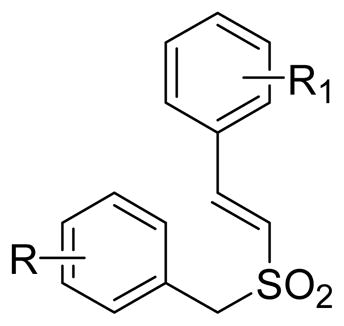
| ||||
|---|---|---|---|---|
| Compd | R | R1 | IC50 (μM)
|
|
| DU145 | K562 | |||
| 8a | 3-NO2, 4-CH3O | 2-CH3O | 20 | 20 |
| 8b | 3-NH2, 4-CH3O | 2-CH3O | 7.5 | 7.5 |
| 8c | 3-NO2, 4-CH3O | 4-CH3O | 25 | 25 |
| 8d | 3-NH2, 4-CH3O | 4-CH3O | 2.5 | 2.5 |
| 8e | 3-NO2, 4-CH3O | 2, 6-(CH3O)2 | 20 | 20 |
| 8f | 2-NO2, 4-CH3O | 2, 6-(CH3O)2 | 50 | 50 |
| 8g | 3-NH2, 4-CH3O | 2, 4-(CH3O)2 | 0.5 | 0.5 |
| 8h | 3-NH2, 4-CH3O | 2, 5-(CH3O)2 | 0.75 | 0.75 |
| 8i | 3-NH2, 4-CH3O | 2, 6-(CH3O)2 | 0.05 | 0.05 |
| 8j | 2-NH2, 4-CH3O | 2, 6-(CH3O)2 | 0.75 | 0.75 |
| 8k | 3-NH2, 6-CH3O | 2, 6-(CH3O)2 | 25 | 25 |
| 8l | 3-NO2, 4-CH3O | 2, 4, 6-(CH3O)3 | 2.5 | 2.5 |
| 8m | 2-NO2, 4-CH3O | 2, 4, 6-(CH3O)3 | 20 | 20 |
| 8n | 2-NH2, 4-CH3O | 2, 4, 6-(CH3O)3 | 0.025 | 0.025 |
| 8o | 3-NH2, 4-CH3O | 2, 4, 5-(CH3O)3 | 0.25 | 0.25 |
| 8p | 3-NH2, 4-CH3O | 2, 4, 6-(CH3O)3 | 0.004 | 0.003 |
| 8q | 3-NH2, 4-CH3O | 3, 4, 5-(CH3O)3 | 3.0 | 0.25 |
| 8r | 3-NH2, 6-CH3O | 2, 4, 6-(CH3O)3 | 7.5 | 7.5 |
| 8s | 3-NO2, 4-CH3O | 2, 6-(CH3O)2, 4-OH | 5.0 | 5.0 |
| 8t | 3-NH2, 4-CH3O | 2, 6-(CH3O)2, 4-OH | 0.2 | 0.3 |
| 8u | 3-NH2, 4-CH3O | 2, 6-(CH3O)2, 4-O(CH2)3COOH | 25 | 20 |
| 8v | 3-NO2, 4-Br | 2, 4, 6-(CH3O)3 | 25 | 25 |
| 8w | 3-NH2, 4-Br | 2, 4, 6-(CH3O)3 | 0.75 | 0.75 |
| 8x | 3-NH2, 4-Cl | 2, 4, 6-(CH3O)3 | 0.01 | 0.01 |
| 8y | 2-NH2, 4-Cl | 2, 4, 6-(CH3O)3 | 2.5 | 2.5 |
| 8z | 3-NO2, 4-F | 2, 4, 6-(CH3O)3 | 25 | 25 |
| 8aa | 3-NH2, 4-F | 2, 4, 6-(CH3O)3 | 2.5 | 2.5 |
| 8ab | 3-NH2, 4-CH3O | 2, 4, 6-F3 | 2.0 | 0.8 |
| 8ac | 3-NH2, 4-CH3O | 2, 4, 5-F3 | 5.0 | 15 |
Once we established that 3-amino-4-methoxy groups on benzyl aromatic ring and 2, 4, 6-trimethoxy groups on styryl ring produced a compound (8p) with best cytotoxic activity, we then analyzed the role of sulfone in the activity of the molecule (Table 2). To understand the oxidative state of sulfur in the molecule, we have made a sulfide (24) and sulfoxide (21) analogs of 8p. Both sulfide (24) and sulfoxide (21) analogs are 10 fold less active than sulfone (8p) indicating complete oxidative state of sulfur in the compound is required for optimum activity.
Table 2.
In vitro cytotoxicity of (E)-styryl benzyl sulfides (24) and sulfoxides (21)

| |||||
|---|---|---|---|---|---|
| Compd | R | R1 | n | IC50 (μM)
|
|
| DU145 | K562 | ||||
| 17 | 3-NO2, 4-OCH3 | 2, 4, 6-(OCH3)3 | 0 | 5.0 | 5.0 |
| 20 | 3-NO2, 4-OCH3 | 2, 4, 6-(OCH3)3 | 1 | 15 | 15 |
| 21 | 3-NH2, 4-OCH3 | 2, 4, 6-(OCH3)3 | 1 | 0.04 | 0.02 |
| 24 | 3-NH2, 4-OCH3 | 2, 4, 6-(OCH3)3 | 0 | 0.05 | 0.03 |
The compound 8p, which has a high potency, has very low solubility in aqueous buffers and solutions. In order to enhance its water solubility and bioavailability the amino group in third position was converted to amino acids (27a–27l). From Table 3, it is clear that all 3-substituted amino acids (27a–27l) are more active than the corresponding esters (26a–26l). As the 3-glycine substituted compound 27a has superior activity over other molecules (27b–27l), and as it doesn’t have a chiral center at glycine carbon, we have made a water-soluble sodium analogue of it (28) to carry out our preclinical and clinical studies. Compound 28 has 28mg/ml solubility in water and other aqueous buffers which makes it suitable for intravenous, intraperitonial and oral administration of the compound.
Table 3.
In vitro cytotoxicity of 3-amino substituted esters 26 and acids 27 and sodium salt 28

| ||||
|---|---|---|---|---|
| Compd | R1 | R2 | IC50 (μM)
|
|
| DU145 | K562 | |||
| 26a | 2, 4, 6-(CH3O)3 | CH2 | 0.1 | 0.1 |
| 26b | 3, 4, 5-(CH3O)3 | CH2 | 75 | 30 |
| 26c | 2, 4, 6-(CH3O)3 | CH2CH2 | 1.5 | 1.0 |
| 26d | 2, 4, 6-(CH3O)3 | CH(CH3) | 0.75 | 0.2 |
| 26e | 2, 4, 6-(CH3O)3 | CF2 | 0.25 | 0.25 |
| 26f | 2, 4, 6-(CH3O)3 | CH(CF3) | 0.25 | 0.25 |
| 26g | 2, 4, 6-(CH3O)3 | C(CH3)2 | 2.5 | 0.80 |
| 26h | 2, 4, 6-(CH3O)3 | CH(C6H5) | 0.075 | 0.03 |
| 26i | 2, 4, 6-(CH3O)3 | CH(4-FC6H4) | 0.3 | 0.8 |
| 26j | 2, 4, 6-(CH3O)3 | CH(4-ClC6H4) | 0.4 | 0.2 |
| 26k | 2, 4, 6-(CH3O)3 | CH(4-BrC6H4) | 0.3 | 0.2 |
| 26l | 2, 4, 6-(CH3O)3 | CH(4-MeOC6H4) | 0.25 | 0.15 |
| 27a | 2, 4, 6-(CH3O)3 | CH2 | 0.075 | 0.0075 |
| 27b | 3, 4, 5-(CH3O)3 | CH2 | 5.0 | 3.0 |
| 27c | 2, 4, 6-(CH3O)3 | CH2CH2 | 5.0 | 5.0 |
| 27d | 2, 4, 6-(CH3O)3 | CH(CH3) | 0.025 | 0.015 |
| 27e | 2, 4, 6-(CH3O)3 | CF2 | 0.15 | 0.15 |
| 27f | 2, 4, 6-(CH3O)3 | CH(CF3) | 0.02 | 0.075 |
| 27g | 2, 4, 6-(CH3O)3 | C(CH3)2 | 0.15 | 0.0075 |
| 27h | 2, 4, 6-(CH3O)3 | CH(C6H5) | 0.02 | 0.015 |
| 27i | 2, 4, 6-(CH3O)3 | CH(4-FC6H4) | 0.15 | 0.08 |
| 27j | 2, 4, 6-(CH3O)3 | CH(4-ClC6H4) | 0.4 | 0.25 |
| 27k | 2, 4, 6-(CH3O)3 | CH(4-BrC6H4) | 0.3 | 0.2 |
| 27l | 2, 4, 6-(CH3O)3 | CH(4-OMeC6H4) | 0.1 | 0.015 |
| 28 | ON 01910 Na | 0.1 | 0.015 | |
Biological Results and Discussion
In vitro anti-tumor effects of 8p and 28 compounds
We next tested the activity of two of the most active compounds (8p and 28) listed in Tables 1–3 against 94 different human tumor cell lines and surprisingly, they were found to induce apoptosis of all of these cell lines with very similar GI50 values (selected data shown in Table 4). These compounds (such as 8p, 28) were also tested by the National Cancer Institute, USA, through its Developmental Therapeutics Program (DTP) against their panel of 60 human cancer cell-lines.18 Their results showed that these compounds exhibited broad-spectrum activity and inhibited the growth of all of the tested cell lines, including drug-resistant cell-lines, at nanomolar concentrations.. The finding that such a large number of tumor cell lines are sensitive to these compounds suggests that the target is essential for the proliferation and survival of cancer cells. Statistical comparison (using the NCI algorithm COMPARE) revealed that these drugs are mitotic blockers of tumor cells. This statistical observation was further substantiated by flow cytometry analysis shown in Figure 2.
Table 4.
Evaluation of 8p and 28 against a panel of Human Tumor Cell lines and normal fibroblasts.
| Cell Line | Tumor Type | IC50 (μM) | |
|---|---|---|---|
| 8p | 28 | ||
| BT20 | Breast (ER−) | 0.03 | 0.08 |
| T47D | Breast (ER+) | 0.003 | 0.17 |
| MCF-7 | Breast (ER+) | 0.001 | 0.075 |
| SK-BR-3 | Breast (ER−) | 0.003 | 0.075 |
| BT474 | Breast (ER+) | 0.002 | 0.05 |
| MDA-MB-231 | Breast (Triple Neg) | ND | 0.025 |
| MDA-MB-157 | Breast (Triple Neg) | ND | 0.075 |
| Hcc70 | Breast | ND | 0.075 |
| HCC1428 | Breast | ND | 0.06 |
| DU145 | Prostate (AR−) | 0.005 | 0.1 |
| PC-3 | Prostate (AR+) | 0.006 | 0.15 |
| OV-CAR-3 | Ovarian | 0.003 | 0.075 |
| MIA-Paca2 | Pancreatic | 0.003 | 0.05 |
| BxPC-3 | Pancreatic | ND | 0.075 |
| PANC-1 | Pancreatic | ND | 0.04 |
| U87 | Glioblastoma | 0.003 | 0.08 |
| H157 | NSCLC | 0.004 | 0.08 |
| A549 | NSCLC | 0.003 | 0.09 |
| H1975 | NSCLC | 0.003 | 0.09 |
| H187 | SCLC | 0.004 | 0.08 |
| N417 | SCLC | 0.003 | 0.08 |
| AGS | Gastric | 0.003 | 0.08 |
| RF1 | Gastric | 0.002 | 0.05 |
| RF48 | Gastric | 0.001 | 0.05 |
| HELA | Cervical | ND | 0.1 |
| COLO-205 | Colo-rectal | 0.005 | 0.15 |
| DLD-1 | Colo-rectal | 0.005 | 0.15 |
| HCT-116 | Colo-rectal | 0.003 | 0.075 |
| HCT-15 | Colo-rectal | 0.003 | 0.09 |
| COLO-320 | Colo-rectal | 0.003 | 0.06 |
| SW480 | Colo-rectal | 0.005 | 0.06 |
| RPMI-7951 | Melanoma | ND | 0.025 |
| WM983A | Melanoma | ND | 0.04 |
| WM3211 | Melanoma | ND | 0.075 |
| WM1341D | Melanoma | ND | 0.1 |
| WM278 | Melanoma | ND | 0.15 |
| WM239A | Melanoma | ND | 0.075 |
| WM-793 | Melanoma | ND | 0.075 |
| 451-LU | Melanoma | ND | 0.025 |
| DND-1A | Melanoma | ND | 0.075 |
| K562 | CML | 0.0025 | 0.015 |
| MOLT-4 | T-Lymphoblastic: ALL | 0.004 | 0.04 |
| Z138C | Mantle Cell Lymphoma | 0.003 | 0.075 |
| GRANTA-519 | Mantle Cell Lymphoma | 0.003 | 0.075 |
| Bel-7402 | Hepatoma | ND | 0.1 |
| KB | Nasopharyngeal | ND | 0.07 |
| HELA | Cervical | ND | 0.1 |
| U937 | Lymphoma | ND | 0.01 |
| LP-1 | Multiple Myeloma | 0.003 | 0.03 |
| U266 | Multiple Myeloma | 0.003 | 0.025 |
| OPM-2 | Multiple Myeloma | 0.003 | 0.015 |
| RPMI-8266 | Multiple Myeloma | 0.003 | 0.01 |
| HL-60 | AML M3 | ND | 0.02 |
| KG1a | AML M1 | ND | 0.03 |
| HEL | AML M6 | ND | 0.05 |
| Daudi | Burkitt’s Lymphoma (B-cell) | 0.003 | 0.15 |
| Raji | Burkitt’s Lymphoma (B-cell) | 0.002 | 0.075 |
| Namalwa | Burkitt’s Lymphoma (B-cell) | 0.005 | 0.075 |
| Fibroblasts | PS-41 | ND | >100 |
| Endothelial | Hu-VEC | ND | >100 |
Figure 2.

28 selectively induces mitotic G2/M arrest and apoptosis in cancer cells. (A) Normal diploid fibroblasts HFL-1 cells or (B) human prostate cells (DU145) were treated with increasing concentrations of 28 and incubated in medium containing 10% fetal bovine serum for 24 h and the cells were fixed, stained with propidium iodide and then subjected to FACS analysis and analyzed for their DNA content. 28 induces a mitotic arrest and induction of a subG1 population (blue) indicative of apoptosis in the cancer cell line while only inducing a small mitotic block in the normal cells.
8p and 28 compounds are highly active against drug resistant tumor cell lines
Development of multidrug resistance (MDR) to classical chemotherapeutic agents is a clinical problem Oncologists face as patients fail first round treatment or become resistant during or following recurrent tumor growth. The MDR phenotype is caused by the overexpression of ATP-binding cassette (ABC) transporters divided into seven subfamily members. The overexpression of various members of this family enables the tumor cells to actively pump out a wide range of amphipathic drugs such that the intracellular concentrations are no longer cytotoxic. To further investigate the activity of these compounds against MDR positive tumor types, we determined the IC50 values of 8p and 28 using two classical MDR positive cell lines (Table 5). The results shown in Figure 1A show a 96 h dose response of the uterine sarcoma cell line MES-SA and the multidrug resistant sub line MES-SA/DX519 treated with 28 compared to a dose response against paclitaxel a known substrate for P-glycoprotein. This cell line has been shown to express high levels of P-glycoprotein (ABCB1) and is resistant to a number of hydrophobic compounds including doxorubicin, paclitaxel, vincristine, vinblastine, etoposide, mitoxantrone, dactinomycin, and daunorubucin. Our results show that the parental cell line was very sensitive to Paclitaxel (IC50 4 nM) but the MDR positive sub line was greater than 100 fold resistant (IC50 750 nM). When the two cell lines were treated with 28, both the parental and the MDR positive cell lines were equally sensitive to the cell killing activity of the compound with an IC50 value of 0.1 μM (Table 5). A second MDR cell line, derived from an ovarian tumor, was also tested. Once again, both the parental and the resistant cell lines were equally sensitive to compound number 28 (Table 5). We also investigated as to whether atypical multidrug resistant cell are sensitive to 28. For these studies, we employed the parental leukemic cell line CEM and its MDR sub line CEM/C2 (Figure 1B).20 CEM/C2 was selected and sub cloned for resistance to camptothecin and has cross resistance to etoposide, dactinimycin, bleomycin, mitoxantrone, doxorubicin, and daunorubicin. As shown in figure 1B and Table 5, compound 28 was active against both the parental and the camptothecin resistant sub clone, with the CEM/C2 clone being 2 fold more sensitive than the parental. These studies clearly demonstrate that this chemotype will not be a substrate for ABC transport proteins and therefore be a suitable treatment option for tumors expressing both classical and atypical MDR resistant markers.
Table 5.
Evaluation of 8p and 28 against a panel of Multidrug-resistance Human Tumor Cell lines
Figure 1.

28 is active against multidrug resistant cells (MDR). (A) The MDR-1 positive MES-SA/DX5 clone as well as an atypical MDR resistant to topoisomerase inhibitors CEM/C2 (B) as well as their respective sensitive parental controls were plated into six-well dishes and treated with increasing concentrations of 28 or a representative chemotherapeutic agent, paclitaxel (PTX) or camptothecin (CPT), respectively for 96 h. The number of viable cells from duplicate plates was determined by trypan blue exclusion. As expected, the parentals are sensitive to both 28 and the chemotherapeutic agent, but while the resistant clones are over 500 fold resistant to PTX or CPT, they are remaining sensitive to the cytotoxic action of 28.
Effects of 28 on cell cycle progression of normal and tumor cells
We next examined the effect of these compounds on normal and tumor cell cycle progression using FACS analysis. Figure 2A show the effect of 28 on the cell cycle progression of normal diploid human fetal lung (HFL-1) and DU145 (prostate cancer) cells (Figure 2B). The results of this study show that the addition of the 28 to DU145 cells resulted in a block of their cell cycle progression in G2/M phase of the cell cycle in a dose dependent manner, In addition, treatment of the tumor cells resulted in an accumulation of cells containing subG1 content of DNA which is an indication of cell death. On the other hand, treatment of normal diploid fibroblasts cells (HFL-1) with 28 resulted in a block of their cell cycle progression in the G1 and G2/M phases of the cell cycle, without the corresponding induction of cell death. We further investigated the selective nature of cell killing by running studies aimed at studying the activation of the apoptotic pathway. Tumor and normal cell lines were then treated with 28 and activation of apoptotic pathways, as judged by PARP [Poly(ADP-ribose) polymerase-1] cleavage,21 is shown in (Figure 3). Treatment of 28 selectively induced PARP cleavage in the tumor cell line while there was no PARP cleavage in the treated normal cell line. Since it is well known that PARP cleavage is the result of activation of caspase-3 we further investigated the activation of the apoptotic pathway by looking at cellular viability together with the activity of caspases 3/7. A dual viability and caspase activation assay was performed in A549 cells treated with 28 which showed the concentrations at which 28 activated the apoptotic machinery to induce a cytotoxic effect (Figure 4). This data shows that treatment with 0.25 μM induced loss of viability with the concomitant induction of caspase 3/7 within 24 h of tumor cell treatment. Taken together, this data shows that 28 selectively induces G2/M cell cycle block with the induction of apoptosis in tumor cells.
Figure 3.

DU145 and HFL-1 (normal human fibroblasts) cells were treated with increasing concentrations of 28 or DMSO (Vehicle) for 48 h. Cells were harvested and total protein was resolved by 10% SDS-PAGE, western blotted and hybridized to anti-PARP (Cell Signaling # 542) anti body. The blot was then washed and treated with anti-rabbit secondary labeled with infrared dye (700) and scanned using the Odyssey scanner (LiCor).
Figure 4.

Cellular viability together with the activity of caspases 3/7 were assayed concomitantly in A549 cells treated with 28 for 24 h (n=3). Viability was measured after addition of CellTiter-Blue® reagent (Promega Corporation). Caspase 3/7 activity was measured after addition of Caspase-Glo® 3/7 reagent (Promega Corporation). Flourescence and luminescence were plotted together to reveal the concentrations at which 28 activated the apoptotic machinery to induce a cytotoxic effect.
In vivo anti-tumor effect of 28
In order to determine in vivo efficacy, we utilized the nude mouse model system. A highly aggressive human estrogen negative breast carcinoma cell line (BT20) was xenografted into athymic nude mice. The animals were treated with 200 mg/kg of 28 using a Q2D × 20 schedules. The animals were treated when the tumors were approximately 70 mm3 in size. Figure 5A shows that an intraperitonial (IP) injection of 28 was able to significantly inhibit the growth of the tumors. The tumors of vehicle treated mice, on average, increased in volume over the 22 day period by 5 fold (70 mm3–480 mm3), while the Q2D 28 treated tumors increased in volume by only 2.5 fold (70 mm3–180 mm3). 28 were well tolerated at these doses as determined by body weights and physical observations (Figure 5B). These studies show that 28 is efficacious against human tumor xenografts while showing no signs of toxicity at the schedules tested under this study.
Figure 5.

Human breast cancer cells (BT20) were injected subcutaneously into female nude mice. The mice were grouped and then treated with 200 mg/kg 28 by intraperitoneal injections Q2D x 20 formulated in phosphate buffered saline (PBS) or placebo (PBS) alone. Tumor volumes (A) and body weights (B) were determined and the average (+/−SEM) for each group (N=9) were determined and plotted. 28 significantly inhibited the in vivo growth of tumors. Placebo tumors doubled in 6 days while it took over 18 days for the 28 treated tumors to double in size. There was no sign of toxicity or body weight loss.
Conclusion
In this communication, we describe the synthesis of a group of (E)-styryl benzyl sulfones which induce apoptotic death of a wide variety of human tumor cell lines at sub nanomolar concentrations while exhibiting relatively low toxicity to normal human cells. Our studies show that the cytoxic activity of the (E)-styryl benzyl sulfones is completely dependent on the nature and position of the substituents on the two aromatic rings. These structure function studies show that a molecule with a benzyl moiety having 3-amino, 4-methoxy groups and a styryl ring with methoxy groups at 2, 4 and 6-positions showed optimum biological activity (8p). Biological evaluation of the activity of these compounds show that these compounds are highly active against a wide variety of human tumor cell lines including those that are resistant to the activity of many of the currently used chemotherapeutic agents.
The low toxicity profile, both in vitro and in vivo and their potent tumor inhibitory activity as seen in nude mouse xenograft assays point to the potential value of these compounds as safe therapies for cancer, lacking many of the side effects normally associated with current chemotherapeutic agents. Recent studies with 28 show that this compound altered the growth and cell cycle status of mantle cell lymphoma cell lines and potently inhibited the expression of several important proteins, including cyclin D1, p-mTOR, and PI3-K.6 Since 28 is highly effective in various combinations with conventional chemotherapy,6 the lack of overt hematotoxicity of this compound is beneficial for testing novel combinations for advanced cancers, including tumors resistant to conventional chemotherapy. In addition, its safety profile seen with normal hematopoietic cells suggest that these compounds have a potential use in in vitro purging of tumor cells from patient bone marrow for use in autologous bone marrow transplantation. Clinical studies in MDS (Phase III) and pancreatic (Phase II/III) patients currently underway will reveal the best way to utilize this compound in cancer therapy.
Experimental Section
Chemistry: General Experimental Procedures
All reactions requiring anhydrous conditions were run under an atmosphere of dry nitrogen and solvents were dried using standard procedures. Reagents and solvents were obtained in the highest available purity and used without further purification unless indicated. Reactions were monitored by thin layer chromatography (TLC) on preloaded silica gel 60 glass–backed plates with F254 plates as the indicator (Sigma – Aldrich), developed using mobile phases of varying compositions of ethyl acetate/hexane, methanol/chloroform and the spots were visualized by a UV indicator. Column chromatography was performed with Merck 70 – 320 mesh silica gel. Melting points were determined using an Electro thermal MEL – Temp 3.0 micro melting point apparatus with a FLUKA 51 K/J electronic thermometer and are uncorrected. Nuclear magnetic resonance spectra for proton (1H NMR) were recorded on a Bruker Avance III (300 MHz), Varian INOVA (400 MHz) and GE (500 MHz) spectrometer. 13 C NMR spectra (75 MHz) were obtained on a Bruker Avance III 300 MHz spectrometer. The chemical shift values are expressed in ppm (parts per million) relative to tetramethylsilane as an internal standard: s, singlet; d, doublet; dd, doublet of a doublet; t, triplet; m, multiplet; br s, broad singlet. Coupling constants (J values) were measured in hertz (Hz). Compounds purity was determined by elemental analyses (0.4%) or LC/MS analysis and was confirmed to be > 95% for all compounds. All LC/MS data were gathered on an Agilent 1200 LC with Agilent 6410 triple quadrupole mass spectrometer detectors. The compound solution was infused into the electron spray ionization source operating in positive and negative mode.
General Procedure for the Preparation of benzyl bromides (2). Method A (Scheme 2)
To a well stirred solution of toluene 1 (40 mmol) in carbon tetrachloride (150 mL) were added benzoyl peroxide (4.0 mmol), and N-bromosuccinimide (48 mmol). The reaction mixture was heated at reflux for 18 h. After completion of the reaction (TLC monitoring, hexane/ethyl acetate, 9:1 on silica gel plate), the contents cooled to room temperature, water was added, and the product was isolated by extraction with dichloromethane. The organic phase was washed with water, brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to obtain the desired crude product 2. The pure compound 2 was obtained following purification by silica gel flash column chromatography (hexane/ethyl acetate, 9:1). The following benzyl bromides 2 were prepared using the above procedure.
4-Methoxy-3-nitrobenzyl bromide (2a)
Radical benzylic bromination of 4–methyl-2-nitroanisole yielded the corresponding 4-methoxy-3-nitrobenzyl bromide. The yield of this reaction was 68%, giving light yellow solid with a melting point 106–108 °C. 1H NMR (CDCl3, 300 MHz): δ 3.97 (s, 3H, OCH3), 4.47 (s, 2H, CH2), 7.08 (d, J = 8.7 Hz, 1H, Ar-H), 7.58 (dd, J = 8.7, 2.1 Hz, 1H, Ar-H), 7.89 (d, J = 2.4 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 245.9688; found 245.9682. Anal. (C8H8BrNO3) C, H, N.
4-Methoxy-2-nitrobenzyl bromide (2b)
Radical benzylic bromination of 4-methyl-3-nitroanisole yielded the corresponding 4-methoxy-2-nitrobenzyl bromide. The yield of this reaction was 65%, giving yellow solid with a melting point 60–62 °C. 1H NMR (CDCl3, 300 MHz): δ 3.89 (s, 3H, OCH3), 4.81 (s, 2H, CH2), 7.14 (dd, J = 8.4, 2.7 Hz, 1H, Ar-H), 7.48 (d, J = 8.7 Hz, 1H, Ar-H), 7.56 (d, J = 2.7 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 245.9688; found 245.9681. Anal. (C8H8BrNO3) C, H, N.
4-Bromo-3-nitrobenzyl bromide (2c)
Radical benzylic bromination of 4-bromo-3-nitrotoluene yielded the corresponding 4-bromo-3-nitrobenzyl bromide. The yield of this reaction was 78%, giving yellow solid with a melting point 62–63 °C. 1H NMR (CDCl3, 500 MHz): δ 4.45 (s, 2H, CH2), 7.47 (dd, J = 8.4, 2.4 Hz, 1H, Ar-H), 7.73 (d, J = 8.4 Hz, 1H, Ar-H), 7.89 (d, J = 2.0 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 295.8667; found 295.8662. Anal. (C7H5Br2NO2) C, H, N.
4-Chloro-3-nitrobenzyl bromide (2d)
Radical benzylic bromination of 4-chloro-3-nitrotoluene yielded the corresponding 4-chloro-3-nitrobenzyl bromide. The yield of this reaction was 87%, giving pale yellow liquid with a boiling point 115–120 °C (0.25 mm Hg). 1H NMR (CDCl3, 300 MHz): δ 4.60 (s, 2H, CH2), 7.42 (dd, J = 8.4, 2.4 Hz, 1H, Ar-H), 7.68 (d, J = 8.4 Hz, 1H, Ar-H), 7.86 (d, J = 2.4 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 249.9192; found 249.9186. Anal. (C7H5BrClNO2) C, H, N.
4-Chloro-2-nitrobenzyl bromide (2e)
Radical benzylic bromination of 4-chloro-2-nitrotoluene yielded the corresponding 4-chloro-2-nitrobenzyl bromide. The yield of this reaction was 69%, giving pale yellow solid with a melting point 40–42 °C. 1H NMR (CDCl3, 500MHz): δ 4.80 (s, 2H, CH2), 7.54 (d, J = 8.3 Hz, 1H, Ar-H), 7.61 (dd, J = 8.3, 2.1 Hz, Ar-H), 8.07 (d, J = 2.1Hz, Ar-H). HRMS: m/z calcd [M + H] 249.9192; found 249.9189. Anal. (C7H5BrClNO2) C, H, N.
4-Fluoro-3-nitrobenzyl bromide (2f)
Radical benzylic bromination of 4-fluoro-3-nitrotoluene yielded the corresponding 4-fluoro-3-nitrobenzyl bromide. The yield of this reaction was 84%, giving yellow solid with a melting point 50–52 °C. 1H NMR (CDCl3, 300 MHz): δ 4.49 (s, 2H, CH2), 7.32(dd, J = 8.7, 1.8 Hz, 1H, Ar-H), 7.66–7.71 (m, 1H, Ar-H), 8.11 (dd, J = 6.9, 2.4 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 233.9488; found 233.9481. Anal. (C7H5BrFNO2) C, H, N.
General Procedure for the Preparation of benzyl alcohols (4). Method B (Scheme 2)
Sodium borohydride (20 mmol) was added in small portions to an ice-cold solution of aldehyde 3 (20 mmol) in dry methanol (100 mL) with stirring. The reaction mixture was left at 0–5 °C for 1 h. After completion of the reaction (TLC monitoring, hexane/ethyl acetate, 9:1 on silica gel plate), the solvent was evaporated, then the chloroform (100 mL) was added to the residue obtained. The organic layer was washed with 5% sodium bicarbonate (50 mL) and water (75 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to obtain the desired crude product 4. The pure compound 4 was obtained after recrystallization from benzene/hexane. The following benzyl alcohols 4 were prepared using the above procedure.
4-Methoxy-3-nitrobenzyl alcohol (4a)
Reduction of 4-methoxy-3-nitrobenzaldehyde yielded the corresponding 4-methoxy-3-nitrobenzyl alcohol. The yield of this reaction was 78%, giving yellow solid with a melting point 69–70 °C. 1H NMR (CDCl3, 300 MHz): δ 2.65 (br s, 1H, OH), 3.96 (s, 3H, OCH3), 4.64 (s, 2H, CH2), 7.12 (d, J = 8.4 Hz, 1H, Ar-H), 7.61 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 7.84 (d, J = 2.1 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H]184.0532; found 184.0522. Anal. (C8H9NO4) C, H, N.
4-Methoxy-2-nitrobenzyl alcohol (4b)
Reduction of 4-metoxy-2-nitrobenzaldehyde yielded the corresponding 4-methoxy-2-nitrobenzyl alcohol. The yield of this reaction was 73%, giving colorless needles with a melting point 80–82 °C. 1H NMR (CDCl3, 400 MHz): δ 2.53 (t, J = 6.8 Hz, 1H, OH), 3.88 (s, 3H, OCH3), 4.86 (d, J = 6.7 Hz, 2H, CH2), 7.19 (dd, J = 8.6, 2.7 Hz, 1H, Ar-H), 7.58 (d, J = 8.6 Hz, 1H, Ar-H), 7.60 (d, J = 2.7 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 184.0532; found 184.0523. Anal. (C8H9NO4) C, H, N.
4-Bromo-3-nitrobenzyl alcohol (4c)
Reduction of 4-bromo-3-nitrobenzaldehyde yielded the corresponding 4-bromo-3-nitrobenzyl alcohol. The yield of this reaction was 89%, giving yellow solid with a melting point 60–62 °C. 1H NMR (CDCl3, 300 MHz): δ 2.61 (br s, 1H, OH), 4.71 (s, 2H, CH2), 7.38 (d, J = 8.5 Hz, 1H, Ar-H), 7.67 (d, J = 8.3 Hz, 1H, Ar-H), 7.81 (s, 1H, Ar-H). HRMS: m/z calcd [M + H] 231.9531; found 231.9533. Anal. (C7H6BrNO3) C, H, N.
4-Chloro-3-nitrobenzyl alcohol (4d)
Reduction of 4-chloro-3-nitrobenzaldehyde yielded the corresponding 4-chloro-3-nitrobenzyl alcohol. The yield of this reaction was 81%, giving yellow solid with a melting point 63–65 °C. 1H NMR (CDCl3, 300 MHz): δ 2.63 (br s, 1H, OH), 4.49 (s, 2H, CH2), 7.48 (dd, J = 8.0, 2.0 Hz, 1H, Ar-H), 7.53 (d, J = 8.0 Hz, 1H, Ar-H), 7.85 (d, J = 2.0 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 188.0036; found 188.0031. Anal. (C7H6ClNO3) C, H, N.
4-Chloro-2-nitrobenzyl alcohol (4e)
Reduction of 4-chloro-2-nitrobenzaldehyde yielded the corresponding 4-chloro-2-nitrobenzyl alcohol. The yield of this reaction was 78%, giving pale yellow solid with a melting point 89–91 °C. 1H NMR (CDCl3, 500MHz): δ 2.91 (br s, 1H, OH), 4.92 (s, 2H, CH2), 7.47(d, J = 8.7 Hz, 1H, Ar-H), 7.72 (dd, J = 8.7, 2.4 Hz, Ar-H), 8.09 (d, J = 2.1 Hz, Ar-H). HRMS: m/z calcd [M + H] 188.0036; found 188.0029. Anal. (C7H6ClNO3) C, H, N.
4-Fluoro-3-nitrobenzyl alcohol (4f)
Reduction of 4-fluoro-3-nitrobenzaldehyde yielded the corresponding 4-fluoro-3-nitrobenzyl alcohol. The yield of this reaction was 91%, giving colorless solid with a melting point 42–44 °C. 1H NMR (CDCl3, 300 MHz): δ 2.69 (br s, 1H, OH), 4.76 (s, 2H, CH2), 7.28 (dd, J = 10.7, 8.6 Hz, 1H, Ar-H), 7.61–7.66 (m, 1H, Ar-H), 8.06 (dd, J = 10.7, 8.6 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 172.0732; found 172.0713. Anal. (C7H6FNO3) C, H, N.
6-Methoxy-3-nitrobenzyl alcohol (4g)
Reduction of 6-methoxy-3-nitrobenzaldehyde yielded the corresponding 6-methoxy-3-nitrobenzyl alcohol. The yield of this reaction was 77%, giving white solid with a melting point 123–125 °C. 1H NMR (CDCl3, 300 MHz): δ 2.31 (br s, 1H, OH), 3.97 (s, 3H, OCH3), 4.73 (d, J = 4.1 Hz, 2H, CH2), 6.93 (d, J = 9.0 Hz, 1H, Ar-H), 8.19 (dd, J = 9.0, 2.8 Hz, 1H, Ar-H), 8.26 (d, J = 2.8 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 184.0532; found 184.0521. Anal. (C8H9NO4) C, H, N.
General Procedure for the Preparation of benzyl bromides (2). Method B (Scheme 2)
Phosphorous tribromide (24 mmol) was added to a stirred solution of alcohol 4 (20 mmol) and toluene (40 mL) at 40 °C. The solution was heated to 100 °C for 30 min, and after completion of the reaction (TLC monitoring, hexane/ethyl acetate, 9:1 on silica gel plate), the contents cooled to ambient temperature. The liquid was decanted and washed with water (2 × 50 mL) and brine (50 mL). The combined aqueous washes were extracted with ether (2 × 75 mL), and the combined organic fractions were dried and evaporated to give crude residue. The residue was dissolved in ether (100 mL) and washed with water (2 × 50 mL) and brine (50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and evaporated to get the crude product 2, which on silica gel flash chromatography (hexane/ethyl acetate, 9:1) resulted in pure benzyl bromide 2. The following benzyl bromides 2 were prepared using the above procedure.
4-Methoxy-3-nitrobenzyl bromide (2a)
Bromination of the alcohol 4a with phosphorous tribromide yielded the corresponding 2a. The yield of this reaction was 52%. The analytical data are in accord with above method A procedure.
4-Methoxy-2-nitrobenzyl bromide (2b)
Bromination of the alcohol 4b with phosphorous tribromide yielded the corresponding 2b. The yield of this reaction was 58%. The analytical data are in accord with above method A procedure.
4-Bromo-3-nitrobenzyl bromide (2c)
Bromination of the alcohol 4c with phosphorous tribromide yielded the corresponding 2c. The yield of this reaction was 62%. The analytical data are in accord with above method A procedure.
4-Chloro-3-nitrobenzyl bromide (2d)
Bromination of the alcohol 4d with phosphorous tribromide yielded the corresponding 2d. The yield of this reaction was 59%. The analytical data are in accord with above method A procedure.
4-Chloro-2-nitrobenzyl bromide (2e)
Bromination of the alcohol 4e with phosphorous tribromide yielded the corresponding 2e. The yield of this reaction was 63%. The analytical data are in accord with above method A procedure.
4-Fluoro-3-nitrobenzyl bromide (2f)
Bromination of the alcohol 4f with phosphorous tribromide yielded the corresponding 2f. The yield of this reaction was 54%. The analytical data are in accord with above method A procedure.
6-Methoxy-3-nitrobenzyl bromide (2g)
Bromination of the 6-methoxy-3-nitrobenzyl alcohol 4g with phosphorous tribromide yielded the corresponding 6-methoxy-3-nitrobenzyl bromide 2g. The yield of this reaction was 55%, giving white solid with a melting point 76–78 °C. 1H NMR (CDCl3, 300 MHz): δ 3.97 (s, 3H, OCH3), 4.73 (s, 2H, CH2), 6.93 (d, J = 9.0 Hz, 1H, Ar-H), 8.19 (dd, J = 9.0, 2.8 Hz, 1H, Ar-H), 8.26 (d, J = 2.8 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 245.9688; found 245.9674. Anal. (C8H8BrNO3) C, H, N.
General Procedure for the Preparation of benzylthioacetic acids (5). Method A (Scheme 1)
The following benzylthioacetic acids were prepared according to the procedure reported in the literature.7
4-Methoxy-3-nitrobenzylthioacetic acid (5a)
Condensation of 4-methoxy-3-nitrobenzyl bromide 2a with mercaptoacetic acid yielded the corresponding 4-methoxy-3-nitrobenzylthio-acetic acid. The yield of this reaction was 96%, giving pale yellow solid with a melting point 129–133 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.12 (s, 2H, -SCH2), 3.82 (s, 2H, CH2S), 3.90 (s, 3H, OCH3), 7.32 (d, J = 8.4 Hz, 1H, Ar-H), 7.59 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.82 (d, J = 2.1 Hz, 1H, Ar-H), 12.60 (br s, 1H, COOH). HRMS: m/z calcd [M - H] 256.0358; found 256.0346. Anal. (C10H11NO5S) C, H, N.
4-Methoxy-2-nitrobenzylthioacetic acid (5b)
Condensation of 4-methoxy-2-nitrobenzyl bromide 2b with mercaptoacetic acid yielded the corresponding 4-methoxy-2-nitrobenzylthioacetic acid. The yield of this reaction was 86%, giving light yellow solid with a melting point 86–88 °C. 1H NMR (CDCl3, 300 MHz): δ 3.05 (s, 2H, -SCH2), 3.81 (s, 3H, OCH3), 4.11 (s, 2H, CH2S), 7.04 (dd, J = 8.4, 2.7 Hz, 1H, Ar-H), 7.33 (d, J = 8.7 Hz, 1H, Ar-H), 7.49 (d, J = 2.7 Hz, 1H, Ar-H), 12.41 (br s, 1H, COOH). HRMS: m/z calcd [M - H] 256.0358; found 256.0349. Anal. (C10H11NO5S) C, H, N.
4-Bromo-3-nitrobenzylthioacetic acid (5c)
Condensation of 4-bromo-3-nitrobenzyl bromide 2c with mercaptoacetic acid yielded the corresponding 4-bromo-3-nitrobenzylthioacetic acid. The yield of this reaction was 92%, giving yellow solid with a melting point 133–135 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.15 (s, 2H, -SCH2), 4.05 (s, 2H, CH2S), 7.51 (d, J = 8.1, Hz, 1H, Ar-H), 7.91 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 8.21 (d, J = 1.8 Hz, 1H, Ar-H), 12.60 (br s, 1H, COOH). HRMS: m/z calcd [M - H] 303.9357; found 303.9348. Anal. (C9H8BrNO4S) C, H, N.
4-Chloro-3-nitrobenzylthioacetic acid (5d)
Condensation of 4-chloro-3-nitrobenzyl bromide 2d with mercaptoacetic acid yielded the corresponding 4-chloro-3-nitrobenzylthioacetic acid. The yield of this reaction was 90%, giving yellow solid 110–114 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.15 (s, 2H, -SCH2), 3.86 (s, 2H, CH2S), 7.42 (d, J = 8.1 Hz, 1H, Ar-H), 7.69 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 7.92 (d, J = 1.8 Hz, 1H, Ar-H), 12.70 (br s, 1H, COOH). HRMS: m/z calcd [M - H] 259.9863; found 259.9856. Anal. (C9H8ClNO4S) C, H, N.
4-Chloro-2-nitrobenzylthioacetic acid (5e)
Condensation of 4-chloro-2-nitrobenzyl bromide 2e with mercaptoacetic acid yielded the corresponding 4-chloro-2-nitrobenzylthioacetic acid. The yield of this reaction was 92%, giving yellow solid with a melting point 103–105 °C. 1H NMR (CDCl3, 300 MHz): δ 3.13 (s, 2H, -SCH2), 4.22 (s, 2H, CH2S), 7.47 (d, J = 8.1 Hz, 1H, Ar-H), 7.57 (dd, J = 8.1, 2.1 Hz, 1H, Ar-H), 8.04 (d, J = 2.1 Hz, 1H, Ar-H), 12.73 (br s, 1H, COOH). HRMS: m/z calcd [M - H] 259.9863; found 259.9851. Anal. (C9H8ClNO4S) C, H, N.
4-Fluoro-3-nitrobenzylthioacetic acid (5f)
Condensation of 4-fluoro-2-nitrobenzyl bromide 2f with mercaptoacetic acid yielded the corresponding 4-fluoro-3-nitrobenzylthioacetic acid. The yield of this reaction was 88%, giving pale yellow solid with a melting point 74–76 °C. 1H NMR (CDCl3, 300 MHz): δ 3.12 (s, 2H, -SCH2), 3.92 (s, 2H, CH2S), 7.28 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 7.61–7.68 (m, 1H, Ar-H), 8.07 (dd, J = 6.9, 2.4 Hz, 1H, Ar-H), 12.61 (br s, 1H, COOH). HRMS: m/z calcd [M - H] 244.0158; found 244.0143. Anal. (C9H8FNO4S) C, H, N.
6-Methoxy-3-nitrobenzylthioacetic acid (5g)
Condensation of 6-methoxy-3-nitrobenzyl bromide 2g with mercaptoacetic acid yielded the corresponding 6-methoxy-3-nitrobenzylthioacetic acid. The yield of this reaction was 86%, giving pale yellow solid with a melting point 76–78 °C. 1H NMR (CDCl3, 300 MHz): δ 3.08 (s, 2H, -SCH2), 3.74 (s, 2H, CH2S), 3.98 (s, 3H, OCH3), 7.29 (d, J = 9.0 Hz, 1H, Ar-H), 8.21 (m, 1H, Ar-H), 8.30 (d, J = 3.0 Hz, 1H, Ar-H). HRMS: m/z calcd [M - H] 256.0358; found 256.0349. Anal. (C10H11NO5S) C, H, N.
General Procedure for the Preparation of benzylsulfonylacetic acids (6). Method A (Scheme 1)
The following benzylsulfonylacetic acids were prepared according to the procedure reported in the literature.7
4-Methoxy-3-nitrobenzylsulfonylacetic acid (6a)
Oxidation of 4-methoxy-3-nitrobenzylthioacetic acid 5a with 30% hydrogen peroxide yielded the corresponding 4-methoxy-3-nitrobenzylsulfonylacetic acid. The yield of this reaction was 51%, giving yellow solid with a melting point 137–139 °C. 1H NMR (DMSO-d6, 500 MHz): δ 3.79 (s, 2H, SCH2), 3.86 (s, 3H, OCH3), 4.48 (s, 2H, CH2S), 7.08 (d, J = 9.0 Hz, 1H, Ar-H), 7.59 (dd, J = 8.5, 2.5 Hz, 1H, Ar-H), 7.86 (d, J = 2.5 Hz, 1H, Ar-H), 13.42 (br s, 1H, COOH). HRMS: m/z calcd [M - H] 288.0256; found 288.0251. Anal. (C10H11NO7S) C, H, N.
4-Methoxy-2-nitrobenzylsulfonylacetic acid (6b)
Oxidation of 4-methoxy-2-nitrobenzylthioacetic acid 5b with 30% hydrogen peroxide yielded the corresponding 4-methoxy-2-nitrobenzylsulfonylacetic acid. The yield of this reaction was 63%, giving pale yellow solid with a melting point 158–161 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.87 (s, 3H, OCH3), 4.27 (s, 2H, - SCH2), 5.02 (s, 2H, CH2S), 7.36 (dd, J = 8.4, 2.7 Hz, 1H, Ar-H), 7.53 (d, J = 8.7 Hz, 1H, Ar-H), 7.57 (d, J = 2.4 Hz, 1H, Ar-H), 13.53 (br s, 1H, COOH). HRMS: m/z calcd [M - H] 288.0256; found 288.0244. Anal. (C10H11NO7S) C, H, N.
4-Bromo-3-nitrobenzylsulfonylacetic acid (6c)
Oxidation of 4-bromo-3-nitrobenzylthioacetic acid 5c with 30% hydrogen peroxide yielded the corresponding 4-bromo-3-nitrobenzylsulfonylacetic acid. The yield of this reaction was 62%, giving a yellow solid with a melting point 172–174 °C. 1H NMR (DMSO-d6, 400 MHz): δ 4.30 (s, 2H, -SCH2), 4.81(s, 2H, CH2S), 7.65 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 8.01 (d, J = 8.4 Hz, 1H, Ar-H), 8.08 (d, J = 1.8 Hz, 1H, Ar-H), 13.58 (br s, 1H, COOH). HRMS: m/z calcd [M - H] 335.9256; found 335.9252. Anal. (C9H8BrNO6S) C, H, N.
4-Chloro-3-nitrobenzylsulfonylacetic acid (6d)
Oxidation of 4-chloro-3-nitrobenzylthioacetic acid 5d with 30% hydrogen peroxide yielded the corresponding 4-chloro-3-nitrobenzylsulfonylacetic acid. The yield of this reaction was 68%, giving yellow solid with a melting point 161–163 °C. 1H NMR (DMSO-d6, 300 MHz): δ 4.27 (s, 2H, -SCH2), 4.77 (s, 2H, CH2S), 7.61 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 7.98 (d, J = 8.4 Hz, 1H, Ar-H), 8.04 (d, J = 1.8 Hz, 1H, Ar-H), 13.60 (br s, 1H, COOH). HRMS: m/z calcd [M - H] 291.9761; found 291.9755. Anal. (C9H8ClNO6S) C, H, N.
4-Chloro-2-nitrobenzylsulfonylacetic acid (6e)
Oxidation of 2-(4-chloro-2-nitrobenzylthioacetic acid 5e with 30% hydrogen peroxide yielded the corresponding 2-(4-chloro-2-nitrobenzylsulfonylacetic acid. The yield of this reaction was 64%, giving yellow solid with a melting point 113–115 °C. 1H NMR (DMSO-d6, 300 MHz): δ 4.36 (s, 2H, -SCH2), 5.00 (s, 2H, CH2S), 7.65 (dd, J = 8.4, 3.0 Hz, 1H, Ar-H), 7.80 (d, J = 8.4 Hz, 1H, Ar-H), 8.18 (d, J = 1.8 Hz, 1H, Ar-H), 13.70 (br s, 1H, COOH). HRMS: m/z calcd [M - H] 291.9761; found 291.9753. Anal. (C9H8ClNO6S) C, H, N.
4-Fluoro-3-nitrobenzylsulfonylacetic acid (6f)
Oxidation of 4-fluoro-3-nitrobenzylthioacetic acid 5f with 30% hydrogen peroxide yielded the corresponding 4-fluoro-3-nitrobenzylsulfonylacetic acid. The yield of this reaction was 69%, giving yellow solid with a melting point 120–122 °C. 1H NMR (DMSO-d6, 300 MHz): δ 4.24 (s, 2H, -SCH2), 4.75 (s, 2H, CH2S), 7.64 (dd, J = 8.4, 2.7 Hz, 1H, Ar-H), 7.79–7.84 (m, 1H, Ar-H), 8.20 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 13.40 (br s, 1H, COOH). HRMS: m/z calcd [M - H] 276.0056; found 276.0046. Anal. (C9H8FNO6S) C, H, N.
6-Methoxy-3-nitrobenzylsulfonylacetic acid (6g)
Oxidation of 6-methoxy-3-nitrobenzylthioacetic acid 5g with 30% hydrogen peroxide yielded the corresponding 6-methoxy-3-nitrobenzylsulfonylacetic acid. The yield of this reaction was 67%, giving pale yellow solid with a melting point 166–168 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.93 (s, 3H, OCH3), 4.28 (s, 2H, - SCH2), 4.79 (s, 2H, CH2S), 7.30 (d, J = 9.0 Hz, 1H, Ar-H), 8.26–8.28 (m, 1H, Ar-H), 8.31 (d, J = 3.0 Hz, 1H, Ar-H), 13.40 (br s, 1H, COOH). HRMS: m/z calcd [M - H] 288.0256; found 288.0248. Anal. (C10H11NO7S) C, H, N.
General Procedure for the Preparation of (E)-Styryl benzyl sulfone (8). Method A (Scheme 1)
A mixture of benzylsulfonylacetic acid 6 (10 mmol), araldehyde 7 (10 mmol), glacial acetic acid (15 mL), and a catalytic amount of benzylamine (200 μL) was refluxed for about 2–8 h. After completion of the reaction (TLC monitoring, chloroform on silica gel plate), with the contents cooled to room temperature, the precipitated product was filtered and washed with 2-propanol. If solid was not formed, the reaction mixture was diluted with ether and washed successively with saturated sodium bicarbonate, dilute hydrochloric acid, and water. The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to obtain the desired crude product 8. The crude product was recrystallized in 2-propanol to yield an analytically pure sample of 8. The following (E)-Styryl benzyl sulfones 8 were prepared using the above procedure.
(E)-2′-Methoxystyryl-4-methoxy-3-nitrobenzylsulfone (8a)
The title compound was obtained from 4-methoxy-3-nitrobenzylsulfonylacetic acid 6a and 2-methoxybenzaldehyde following the procedure as described in method A. Yield, 56%; yellow solid, mp 169–171 °C. 1H NMR (CDCl3, 300 MHz): δ 3.89 (s, 3H, OCH3), 3.99 (s, 3H, OCH3), 4.27 (s, 2H, CH2), 6.92 – 6.99 (m, 2H, Ar-H), 7.01 (d, J = 15.6 Hz, 1H, =CH), 7.12 (d, J = 8.7 Hz, 1H, Ar-H), 7.35–7.45 (m, 2H, Ar-H), 7.62 (d, J = 15.6 Hz, 1H, CH=), 7.63 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.87 (d, J = 2.4 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 364.0777; found 364.0780. Anal. (C17H17NO6S) C, H, N.
(E)-2′-Methoxystyryl-4-methoxy-3-aminobenzylsulfone (8b)
(E)-2′-methoxystyryl-4-methoxy-3-nitrobenzylsulfone 8a (900 mg, 2.5 mmol) was dissolved in acetone:water (40:20 mL) and heated to 50 °C. After 30 min sodium hydrosulfite (8.79 g, 50.0 mmol) was added slowly and maintained temperature at 50 °C for further 30 min. After completion of reaction (TLC monitoring, chloroform on silica gel plate), the contents cooled to room temperature, water was added, and the product was isolated by extraction with ethyl acetate. The organic phase was washed with water (3 X 100 mL), brine (50 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to obtain the desired crude product 8b. The pure compound 8b was obtained following purification by silica gel flash column chromatography (chloroform). Yield, 51%; white solid, mp 140–142 °C. 1H NMR (CDCl3, 300 MHz): δ 3.78 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 4.15 (s, 2H, CH2), 6.95 – 7.02 (m, 2H, Ar-H), 6.98 (d, J = 15.6 Hz, 1H, =CH), 7.12 (d, J = 8.7 Hz, 1H, Ar-H), 7.37–7.45 (m, 2H, Ar-H), 7.62 (d, J = 15.6 Hz, 1H, CH=), 7.63 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.87 (d, J = 2.4 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 334.1035; found 334.1066. Anal. (C17H19NO4S) C, H, N.
(E)-4′-Methoxystyryl-4-methoxy-3-nitrobenzylsulfone (8c)
The title compound was obtained from 4′-methoxy-3-nitrobenzylsulfonylacetic acid 6a and 4-methoxybenzaldehyde following the procedure as described in method A. Yield, 58%; yellow solid, mp 172–174 °C. 1H NMR (CDCl3, 500 MHz): δ 3.88 (s, 3H, OCH3), 3.98 (s, 3H, OCH3), 4.27 (s, 2H, CH2), 6.93 – 6.98 (m, 2H, Ar-H), 6.95 (d, J = 15.5 Hz, 1H, =CH), 7.12 (d, J = 9.0 Hz, 1H, Ar-H), 7.36 (dd, J = 7.5, 1.5 Hz, 1H, Ar-H), 7.42–7.49 (m, 2H, Ar-H), 7.61 (d, J = 15.5 Hz, 1H, CH=), 7.86 (d, J = 2.0 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 364.0777; found 364.0765. Anal. (C17H17NO6S) C, H, N.
(E)-4′-Methoxystyryl-4-methoxy-3-aminobenzylsulfone (8d)
The title compound was obtained by the reduction of (E)-4-methoxystyryl-4-methoxy-3-nitrobenzylsulfone 8c following the procedure as described in compound 8b. Yield, 49%; yellow solid, mp 152–154 °C. 1H NMR (CDCl3, 300 MHz): δ 3.89 (s, 3H, OCH3), 3.99 (s, 3H, OCH3), 4.27 (s, 2H, CH2), 6.92 – 6.99 (m, 2H, Ar-H), 7.01 (d, J = 15.6 Hz, 1H, =CH), 7.12 (d, J = 8.7 Hz, 1H, Ar-H), 7.35–7.45 (m, 2H, Ar-H), 7.62 (d, J = 15.6 Hz, 1H, CH=), 7.63 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.87 (d, J = 2.4 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 334.1035; found 334.1068. Anal. (C17H19NO4S) C, H, N.
(E)-2′,6′-Dimethoxystyryl-4-methoxy-3-nitrobenzylsulfone (8e)
The title compound was obtained from 4-methoxy-3-nitrobenzylsulfonylacetic acid 6a and 2,6-dimethoxybenzaldehyde following the procedure as described in method A. Yield, 53%; yellow solid, mp 188–190 °C. 1H NMR (CDCl3, 300 MHz): δ 3.78 (s, 6H, 2 X OCH3), 3.90 (s, 3H, OCH3), 4.17 (s, 2H, CH2), 6.48 (d, J = 8.7 Hz, 2H, Ar-H), 7.03 (d, J = 8.7 Hz, 1H, Ar-H), 7.13 (d, J = 15.6 Hz, 1H, =CH), 7.26 (t, J = 8.4 Hz, 1H, Ar-H), 7.56 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.82 (d, J = 2.4 Hz, 1H, Ar-H), 7.84 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 394.0882; found 394.0889. Anal. (C18H19NO7S) C, H, N.
(E)-2′,6′-Dimethoxystyryl-4-methoxy-2-nitrobenzylsulfone (8f)
The title compound was obtained from 4-methoxy-2-nitrobenzylsulfonylacetic acid 6b and 2,6-dimethoxybenzaldehyde following the procedure as described in method A. Yield, 52%; yellow solid, mp 176–178 °C. 1H NMR (CDCl3, 300 MHz): δ 3.78 (s, 6H, 2 X OCH3), 3.80 (s, 3H, OCH3), 4.74 (s, 2H, CH2), 6.47 (d, J = 8.7 Hz, 2H, Ar-H), 7.09 (dd, J = 8.4, 2.7 Hz, 1H, Ar-H), 7.16 (d, J = 15.6 Hz, 1H, =CH), 7.25 (t, J = 8.4 Hz, 1H, Ar-H), 7.45 (m, 2H, Ar-H), 7.72 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 394.0882; found 394.0869. Anal. (C18H19NO7S) C, H, N.
(E)-2′,4′-Dimethoxystyryl-4-methoxy-3-aminobenzylsulfone(8g)
Step1: (E)-2′,4′-Dimethoxy-styryl-4-methoxy-3-nitrobenzylsulfone
The condensation of 4-methoxy-3-nitrobenzylsulfonylacetic acid 6a with 2,4-dimethoxybenzaldehyde following the procedure as described in method A resulted in the desired product (E)-2′,4′-dimethoxystyryl-4-methoxy-3-nitrobenzylsulfone. Yield, 50%; yellow solid, mp 148–150 °C. 1H NMR (CDCl3, 300 MHz): δ 3.78 (s, 6H, 2 X OCH3), 3.90 (s, 3H, OCH3), 4.17 (s, 2H, CH2), 6.48 (d, J = 8.7 Hz, 2H, Ar-H), 7.03 (d, J = 8.7 Hz, 1H, Ar-H), 7.13 (d, J = 15.6 Hz, 1H, =CH), 7.26 (t, J = 8.4 Hz, 1H, Ar-H), 7.56 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.84 (d, J = 2.4 Hz, 1H, Ar-H), 7.82 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 394.0882; found 394.0877. Anal.Calcd. for C18H19NO7S: C, 54.95%; H, 4.87%; N, 3.56%. Found: C, 55.06%; H, 4.82%; N, 3.43%.
Step 2: (E)-2′,4′-Dimethoxystyryl-4-methoxy-3-aminobenzylsulfone (8g)
The title compound was obtained by the reduction of (E)-2′,4′-dimethoxystyryl-4-methoxy-3-nitrobenzylsulfone following the procedure as described in compound 8b. Yield, 47%; yellow solid, mp 136–138 °C. 1H NMR (CDCl3, 300 MHz): δ 3.86 (s, 6H, 2 X OCH3), 3.98 (s, 3H, OCH3), 4.25 (s, 2H, CH2), 6.46 (d, J = 2.4 Hz, 1H, Ar-H), 6.85 (d, J = 15.3 Hz, 1H, =CH), 7.12 (d, J = 8.7 Hz, 1H, Ar-H), 7.29 (d, J = 8.7 Hz, 1H, Ar-H), 7.51 (d, J = 15.3 Hz, 1H, CH=). 7.63 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.86 (d, J = 2.4 Hz, 1H, Ar-H). HRMS: m/z [M + H] 364.1140; found 364.1129. Anal. (C18H21NO5S) C, H, N.
(E)-2′,5′-Dimethoxystyryl-4-methoxy-3-aminobenzylsulfone (8h)
Step1: (E)-2′,5′-Dimethoxystyryl-4-methoxy-3-nitrobenzylsulfone
The condensation of 4-methoxy-3-nitrobenzylsulfonylacetic acid 6a with 2,5-dimethoxybenzaldehyde following the procedure as described in method A resulted in the desired product (E)-2′,5′-dimethoxystyryl-4-methoxy-3-nitrobenzylsulfone. Yield, 52%; yellow solid, mp 174–175 °C. 1H NMR (CDCl3, 300 MHz): δ 3.80 (s, 6H, 2 X OCH3), 3.94 (s, 3H, OCH3), 4.19 (s, 2H, CH2), 6.54 (d, J = 8.7 Hz, 2H, Ar-H), 7.13 (d, J = 8.7 Hz, 1H, Ar-H), 7.17 (d, J = 15.6 Hz, 1H, =CH), 7.30 (t, J = 8.4 Hz, 1H, Ar-H), 7.59 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.78 (d, J = 2.4 Hz, 1H, Ar-H), 7.82 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 394.0882; found 394.0881. Anal.Calcd. for C18H19NO7S: C, 54.95%; H, 4.87%; N, 3.56%. Found: C, 54.86%; H, 4.75%; N, 3.47%.
Step 2: (E)-2′,5′-Dimethoxystyryl-4-methoxy-3-aminobenzylsulfone (8h)
The title compound was obtained by the reduction of (E)-2′,5′-dimethoxystyryl-4-methoxy-3-nitrobenzylsulfone following the procedure as described in compound 8b. Yield, 49%; yellow solid, mp 130–132 °C. 1H NMR (CDCl3, 300 MHz): δ 3.78 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 4.18 (s, 2H, CH2), 6.68–6.77 (m, 3H, Ar-H), 6.86 (d, J = 9.0 Hz, 2H, Ar-H), 6.91 (d, J = 15.6 Hz, 1H, =CH), 6.93–7.02 (m, 1H, Ar-H), 7.64 (d, J = 15.9 Hz, 1H, CH=). HRMS: m/z [M + H] 364.1140; found 364.1131. Anal. (C18H21NO5S) C, H, N.
(E)-2′,6′-Dimethoxystyryl-4-methoxy-3-aminobenzylsulfone (8i)
The title compound was obtained by the reduction of (E)-2′,6′-Dimethoxystyryl-4-methoxy-3-nitrobenzylsulfone 8e following the procedure as described in compound 8b. Yield, 48%; pale yellow solid, mp 106–108 °C. 1H NMR (CDCl3, 300 MHz): δ 3.83 (s, 3H, OCH3), 3.86 (s, 6H, 2 X OCH3), 4.16 (s, 2H, CH2), 6.52–6.59 (m, 2H, Ar-H), 6.71–6.79 (m, 2H, Ar-H), 7.17 (t, J = 8.1 Hz, 1H, Ar-H), 7.23 (d, J = 15.9 Hz, 1H, =CH), 7.32 (t, J = 8.4 Hz, 1H, Ar-H), 7.94 (d, J = 15.9 Hz, 1H, CH=). HRMS: m/z [M + H] 364.1140; found 364.1131. Anal. (C18H21NO5S) C, H, N.
(E)-2′,6′-Dimethoxystyryl-4-methoxy-2-aminobenzylsulfone (8j)
The title compound was obtained by the reduction of (E)-2′,6′-Dimethoxystyryl-4-methoxy-2-nitrobenzylsulfone 8f following the procedure as described in compound 8b. Yield, 49%; yellow solid, mp 160–164 °C. 1H NMR (CDCl3, 300 MHz): δ 3.70 (s, 3H, OCH3), 3.79 (s, 6H, 2 X OCH3), 4.19 (s, 2H, CH2), 6.24–6.29 (m, 2H, Ar-H), 6.49 (d, J = 8.4 Hz, 2H, Ar-H), 6.91 (d, J = 8.1 Hz, 1H, Ar-H), 7.23 (d, J = 15.9 Hz, 1H, =CH), 7.29 (t, J = 6.3 Hz, 1H, Ar-H), 7.94 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z [M + H] 364.1140; found 364.1151. Anal. (C18H21NO5S) C, H, N.
(E)-2′,6′-Dimethoxystyryl-6-methoxy-3-aminobenzylsulfone (8k)
Step1: (E)-2′,6′-Dimethoxystyryl-6-methoxy-3-nitrobenzylsulfone
The condensation of 6-methoxy-3-nitrobenzylsulfonylacetic acid 6g with 2,6-dimethoxybenzaldehyde following the procedure as described in method A resulted in the desired product (E)-2′,6′-dimethoxystyryl-6-methoxy-3-nitrobenzylsulfone. Yield, 55%; pale yellow solid, mp 189–191 °C. 1H NMR (CDCl3, 500 MHz): δ 3.85 (s, 6H, 2 X OCH3), 3.88 (s, 3H, OCH3), 4.44 (s, 2H, CH2), 6.55 (d, J = 8.5 Hz, 2H, Ar-H), 6.93 (d, J = 9.0 Hz, 1H, Ar-H), 7.27 (d, J = 16.0 Hz, 1H, =CH), 7.33 (t, J = 8.5 Hz, 1H, Ar-H), 7.79 (d, J = 16.0 Hz, 1H, CH=), 8.21 (dd, J = 9.0, 3.0 Hz, 1H, Ar-H), 8.31 (d, J = 3.0 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 394.0882; found 394.0891. Anal.Calcd. for C18H19NO7S: C, 54.95%; H, 4.87%; N, 3.56%. Found: C, 55.10%; H, 4.92%; N, 3.63%.
Step 2: (E)-2′,6′-Dimethoxystyryl-6-methoxy-3-aminobenzylsulfone (8k)
The title compound was obtained by the reduction of (E)-2′,6′-dimethoxystyryl-6-methoxy-3-nitrobenzylsulfone following the procedure as described in compound 8b. Yield, 47%; light yellow solid, mp 172–174 °C. 1H NMR (CDCl3, 500 MHz): δ 3.63 (s, 3H, OCH3), 3.84 (s, 6H, 2 X OCH3), 4.33 (s, 2H, CH2), 6.53 (d, J = 8.5 Hz, 2H, Ar-H), 6.93 (d, J = 9.0 Hz, 1H, Ar-H), 7.27 (d, J = 16.0 Hz, 1H, =CH), 7.33 (t, J = 8.5 Hz, 1H, Ar-H), 7.85 (d, J = 16.0 Hz, 1H, CH=), 8.21 (dd, J = 9.0, 3.0 Hz, 1H, Ar-H), 8.31 (d, J = 3.0 Hz, 1H, Ar-H). HRMS: m/z [M + H] 364.1140; found 364.1134. Anal. (C18H21NO5S) C, H, N.
(E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-nitrobenzylsulfone (8l)
The title compound was obtained from 4-methoxy-3-nitrobenzylsulfonylacetic acid 6a and 2,4,6-trimethoxybenzaldehyde following the procedure as described in method A. Yield, 56%; yellow solid, mp 184–186 °C. 1H NMR (CDCl3, 300 MHz): δ 3.84 (s, 6H, 2 X OCH3), 3.86 (s, 3H, OCH3), 3.98 (s, 3H, OCH3), 4.23 (s, 2H, CH2), 6.09 (s, 2H, Ar-H), 7.03 (d, J = 15.6 Hz, 1H, =CH), 7.10 (d, J = 8.7 Hz, 1H, Ar-H), 7.63 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.80 (d, J = 15.6 Hz, 1H, CH=), 7.85 (d, J = 2.1 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 424.0988; found 424.0982. Anal. (C19H21NO8S) C, H, N.
(E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-2-nitrobenzylsulfone (8m)
The title compound was obtained from 4-methoxy-2-nitrobenzylsulfonylacetic acid 6b and 2,4,6-trimethoxybenzaldehyde following the procedure as described in method A. Yield, 54%; yellow solid, mp 158–160 °C. 1H NMR (CDCl3, 300 MHz): δ 3.84 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 4.80 (s, 2H, CH2), 6.10 (s, 2H, Ar-H), 7.05 (d, J = 15.6 Hz, 1H, =CH), 7.16 (dd, J = 8.7, 2.7 Hz, 1H, Ar-H), 7.50 – 7.53 (m, 2H, Ar-H), 7.70 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 424.0988; found 424.0972. Anal. (C19H21NO8S) C, H, N.
(E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-2-aminobenzylsulfone (8n)
The title compound was obtained by the reduction of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-2-nitrobenzylsulfone 8m following the procedure as described in compound 8b. Yield, 50%; pale yellow solid, mp 147–149 °C. 1H NMR (CDCl3, 300 MHz): δ 3.78 (s, 3H, OCH3), 3.85 (s, 6H, 2 X OCH3), 3.86 (s, 3H, OCH3), 4.25 (s, 2H, CH2), 6.11 (s, 2H, Ar-H), 6.32–6.36 (m, 2H, Ar-H), 6.98 (d, J = 8.1 Hz, 1H, Ar-H), 7.13 (d, J = 15.6 Hz, 1H, =CH), 7.93 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 394.1246; found 394.1261. Anal. (C19H23NO6S) C, H, N.
(E)-2′,4′,5′-Trimethoxystyryl-4-methoxy-3-aminobenzylsulfone (8o)
Step 1: (E)-2′,4′,5′-Trimethoxystyryl-4-methoxy-3-nitrobenzylsulfone
The condensation of 4-methoxy-3-nitrobenzylsulfonylacetic acid 6a with 2,4,5-trimethoxybenzaldehyde following the procedure as described in method A resulted in the desired product (E)-2′,4′,5′-trimethoxystyryl-4-methoxy-3-nitrobenzylsulfone. Yield, 56%; pale yellow solid, mp 200–202 °C. 1H NMR (CDCl3, 300 MHz): δ 3.85 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 3.95 (s, 3H, OCH3), 3.99 (s, 3H, OCH3), 4.26 (s, 2H, CH2), 6.49 (s, 1H, Ar-H), 6.79 (d, J = 15.6 Hz, 1H, =CH) 6.84 (s, 1H, Ar-H), 7.14 (d, J = 8.7 Hz, 1H, Ar-H), 7.56 (d, J = 15.6 Hz, 1H, CH=), 7.63 (dd, J = 8.7, 2.1 Hz, 1H, Ar-H), 7.86 (d, J = 2.1 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 424.0988; found 424.1001. Anal.Calcd. for C19H21NO8S: C, 53.89%; H, 5.00%; N, 3.31%. Found: C, 53.76%; H, 4.91%; N, 3.25%.
Step 2: (E)-2′,4′,5′-Trimethoxystyryl-4-methoxy-3-aminobenzylsulfone (8o)
The title compound was obtained by the reduction of (E)-2′,4′,5′-trimethoxystyryl-4-methoxy-3-nitrobenzylsulfone following the procedure as described in compound 8b. Yield, 48%; pale yellow solid, mp 112–114 °C. 1H NMR (CDCl3, 300 MHz): δ 3.84 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 3.94 (s, 3H, OCH3), 4.17 (s, 2H, CH2), 6.50 (s, 1H, Ar-H), 6.69 (d, J = 2.1 Hz, 1H, Ar-H), 6.73 (dd, J = 8.4, 2.1 Hz, 2H, Ar-H), 6.77 (d, J = 2.1 Hz, 1H, Ar-H), 6.81 (d, J = 15.0 Hz, 1H, =CH), 7.61 (d, J = 15.6 Hz, 1H, =CH). HRMS: m/z calcd [M + H] 394.1246; found 394.1268. Anal. (C19H23NO6S) C, H, N.
(E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-aminobenzylsulfone (8p)
The title compound was obtained by the reduction of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-nitrobenzylsulfone 8l following the procedure as described in compound 8b. Yield, 48%; light yellow solid, mp 146–148 °C. 1H NMR (CDCl3, 400 MHz): δ 3.77 (s, 3H, OCH3), 3.84 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 4.24 (s, 2H, CH2), 4.33 (br s, 2H, NH2), 6.10 (s, 2H, Ar-H), 6.31 – 6.35 (m, 2H, Ar-H), 6.97 (d, J = 8.3 Hz, 1H, Ar-H), 7.12 (d, J = 15.6 Hz, 1H, =CH), 7.93 (d, J = 15.6 Hz, 1H, CH=). 13 C NMR (CDCl3, 75 MHz):δ 163.7, 161.4, 147.6, 136.4, 135.1, 122.8, 121.1, 121.0, 117.2, 110.2, 103.8, 90.3, 61.7, 55.7, 55.5, 55.4. HRMS: m/z calcd [M + H] 394.1246; found 394.1246. Anal. (C19H23NO6S) C, H, N.
(E)-3′,4′,5′-Trimethoxystyryl-4-methoxy-3-aminobenzylsulfone (8q)
Step 1: (E)-3′,4′,5′-Trimethoxystyryl-4-methoxy-3-nitrobenzylsulfone
The condensation of 4-methoxy-3-nitrobenzylsulfonylacetic acid 6a with 3,4,5-trimethoxybenzaldehyde following the procedure as described in method A resulted in the desired product (E)-3′,4′,5′-trimethoxystyryl-4-methoxy-3-nitrobenzylsulfone. Yield, 52%; pale yellow solid, mp 170–172 °C. 1H NMR (CDCl3, 300 MHz): δ 3.88 (s, 6H, 2 X OCH3), 3.90 (s, 3H, OCH3), 3.99 (s, 3H, OCH3), 4.29 (s, 2H, CH2), 6.63 (d, J = 15.3 Hz, 1H, =CH), 6.68 (s, 2H, Ar-H), 7.13 (d, J = 8.7 Hz, 1H, Ar-H), 7.39 (d, J = 15.3 Hz, 1H, CH=), 7.62 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.86 (d, J = 2.4 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 424.0988; found 424.0998. Anal.Calcd. for C19H21NO8S: C, 53.89%; H, 5.00%; N, 3.31%. Found: C, 53.71%; H, 4.93%; N, 3.37%.
Step 2: (E)-3′,4′,5′-Trimethoxystyryl-4-methoxy-3-aminobenzylsulfone (8q)
The title compound was obtained by the reduction of (E)-3′,4′,5′-trimethoxystyryl-4-methoxy-3-nitrobenzylsulfone following the procedure as described in compound 8b. Yield, 48%; light orange solid, mp 108–110 °C. 1H NMR (CDCl3, 400 MHz): δ 3.77 (s, 3H, OCH3), 3.79 (s, 6H, 2 X OCH3), 3.82 (s, 3H, OCH3), 4.11 (s, 2H, CH2), 6.55 (d, J = 15.4 Hz, 1H, =CH), 6.58 (s, 2H, Ar-H), 6.61 (dd, J = 8.2, 2.1 Hz, 1H, Ar-H), 6.67 (d, J = 8.2 Hz, 1H, Ar-H), 6.70 (d, J = 2.1 Hz, 1H, Ar-H), 7.29 (d, J = 15.4 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 394.1246; found 394.1246. Anal. (C19H23NO6S) C, H, N.
(E)-2′,4′,6′-Trimethoxystyryl-6-methoxy-3-aminobenzylsulfone (8r)
Step 1: (E)-2′,4′,6′-Trimethoxystyryl-6-methoxy-3-nitrobenzylsulfone
The condensation of 6-methoxy-3-nitrobenzylsulfonylacetic acid 6g with 2,4,6-trimethoxybenzaldehyde following the procedure as described in method A resulted in the desired product (E)-2′,4′,6′-trimethoxystyryl-6-methoxy-3-nitrobenzylsulfone. Yield, 55%; yellow solid, mp 144–146 °C. 1H NMR (CDCl3, 500 MHz): δ 3.83 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 4.42 (s, 2H, CH2), 6.08 (s, 2H, Ar-H), 6.92 (d, J = 9.0 Hz, 1H, Ar-H), 7.09 (d, J = 15.5 Hz, 1H, =CH), 7.69 (d, J = 16.0 Hz, 1H, CH=), 8.11 (dd, J = 9.0, 2.5 Hz, 1H, Ar-H), 8.30 (d, J = 2.5 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 424.0988; found 424.0998. Anal.Calcd. for C19H21NO8S: C, 53.89%; H, 5.00%; N, 3.31%. Found: C, 53.76%; H, 4.94%; N, 3.37%.
Step 2: (E)-2′,4′,6′-Trimethoxystyryl-6-methoxy-3-aminobenzylsulfone (8r)
The title compound was obtained by the reduction of (E)-2′,4′,6′-trimethoxystyryl-6-methoxy-3-nitrobenzylsulfone following the procedure as described in compound 8b. Yield, 49%; orange solid, mp 124–128 °C. 1H NMR (CDCl3, 300 MHz): δ 3.57 (s, 3H, OCH3), 3.76 (s, 6H, 2 X OCH3), 3.77 (s, 3H, OCH3), 4.25 (s, 2H, CH2), 6.01 (s, 2H, Ar-H), 6.59 (dd, J = 8.2, 2.1 Hz, 2H, Ar-H), 6.77 (d, J = 8.2 Hz, 1H, Ar-H), 7.02 (d, J = 15.9 Hz, 1H, =CH), 7.69 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 394.1246; found 394.1266. Anal. (C19H23NO6S) C, H, N.
(E)-2′,6′-Dimethoxy-4′-hydroxystyryl-4-methoxy-3-nitrobenzylsulfone (8s)
A mixture of 4-methoxy-3-nitrobenzylsulfonylacetic acid 6a (2.05 g, 5 mmol), 2,6-dimethoxy-4-hydroxybenzaldehyde (1.02 g, 5.5 mmol), benzoic acid (92 mg, 0.75 mmol), and piperidine (55 mg, 0.65 mmol) in toluene (50 mL) was refluxed for 2–4 h with continuous removal of water using a Dean-Stark water separator. Reaction completion was determined by TLC (9:1 chloroform/methanol on silica gel plate). The solvent was evaporated and to the residue water was added and extracted with ethylacetate. The organic phase was washed with saturated sodium bicarbonate solution, dilute hydrochloric acid, water and dried over anhydrous sodium sulfate. The organic phase filtered, evaporated the solvent under vacuo yielded a crude product 8s. The pure compound 8s was obtained following purification by silica gel flash column chromatography (chloroform). Yield, 48%; light yellow solid, mp 222–224 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.77 (s, 6H, 2 X OCH3), 3.91 (s, 3H, OCH3), 4.51 (s, 2H, CH2), 5.79 (s, 1H, OH), 6.11 (s, 2H, Ar-H), 6.99 (d, J = 15.6 Hz, 1H, =CH), 7.37 (d, J = 9.0 Hz, 1H, Ar-H), 7.48 (d, J = 15.6 Hz, 1H, CH=), 7.62 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.86 (d, J = 2.1 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 410.08; found 410.09. Anal. (C18H19NO8S) C, H, N.
(E)-2′,6′-Dimethoxy-4′-hydroxystyryl-4-methoxy-3-aminobenzylsulfone (8t)
The title compound was obtained by the reduction of (E)-2′,6′-dimethoxy-4′-hydroxystyryl-4-methoxy-3-nitrobenzylsulfone 8s following the procedure as described in compound 8b. Yield, 45%; light yellow solid, mp 132–134 °C. 1H NMR (CDCl3, 400 MHz): δ 3.67 (s, 6H, 2 X OCH3), 3.76 (s, 3H, OCH3), 4.07 (s, 2H, CH2), 5.75 (bs, 1H, OH), 5.95 (s, 2H, Ar-H), 6.65 – 6.69 (m, 3H, Ar-H), 6.92 (d, J = 15.6 Hz, 1H, =CH), 7.73 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 380.1090; found 380.0184. Anal. (C18H21NO6S) C, H, N.
(E)-2′,6′-Dimethoxy-4′-phenoxybutanoicacidstyryl-4-methoxy-3-aminobenzylsulfone (8u)
Step 1: (E)-2′,6′-dimethoxy-4′-phenoxybutanoic acidstyryl-4-methoxy-3-nitrobenzylsulfone
The condensation of 4-methoxy-3-nitrobenzylsulfonylacetic acid 6a with 4-(4-formyl-3,5-dimethoxyphenoxy) butyric acid following the procedure as described in method A resulted in the desired product (E)-2′,6′-dimethoxy-4′-phenoxybutanoic acidstyryl-4-methoxy-3-nitrobenzylsulfone. Yield, 56%; pale yellow solid, mp 198–200 °C. 1H NMR (CDCl3, 400 MHz): δ 2.11–2.18 (m, 2H, CH2), 2.61 (t, J = 7.1 Hz, 2H, CH2), 3.83 (s, 6H, 2 X OCH3), 3.97 (s, 3H, OCH3), 4.08 (t, J = 6.1 Hz, 2H, CH2), 4.23 (s, 2H, CH2), 6.09 (s, 2H, Ar-H), 7.02 (d, J = 15.6 Hz, 1H, =CH), 7.11 (d, J = 8.7 Hz, 1H, Ar-H), 7.63 (dd, J = 8.7, 2.3 Hz, 1H, Ar-H), 7.78 (d, J = 15.6 Hz, 1H, CH=), 7.84 (d, J = 2.2 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 496.1199; found 496.1984. Anal.Calcd. for C22H25NO10S: C, 53.33%; H, 5.09%; N, 2.83%. Found: C, 53.42%; H, 5.01%; N, 2.87%.
Step 2: (E)-2′,6′-Dimethoxy-4′-phenoxybutanoic acidstyryl-4-methoxy-3-aminobenzylsulfone (8u)
The title compound was obtained by the reduction of (E)-2′,6′-dimethoxy-4′-phenoxybutanoic acidstyryl-4-methoxy-3-nitrobenzylsulfone following the procedure as described in compound 8b. Yield, 48%; light yellow solid, mp 176–178 °C. 1H NMR (CDCl3, 400 MHz): δ 2.04–2.10 (m, 2H, CH2), 2.53 (t, J = 7.1 Hz, 2H, CH2), 3.75 (s, 6H, 2 X OCH3), 3.77 (s, 3H, OCH3), 4.00 (t, J = 6.0 Hz, 2H, CH2), 4.06 (s, 2H, CH2), 6.02 (s, 2H, Ar-H), 6.66–6.70 (m, 3H, Ar-H), 6.99 (d, J = 15.6 Hz, 1H, =CH), 7.77 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 466.1457; found 466.1443. Anal. (C22H27NO8S) C, H, N.
(E)-2′,4′,6′-Trimethoxystyryl-4-bromo-3-nitrobenzylsulfone (8v)
The title compound was obtained from 4-bromo-3-nitrobenzylsulfonylacetic acid 6c and 2, 4, 6-trimethoxybenzaldehyde following the procedure as described in method A. Yield, 56%; yellow solid, mp 186–188 °C. 1H NMR (CDCl3, 300 MHz): δ 3.84 (s, 6H, 2 X OCH3), 3.86 (s, 3H, OCH3), 4.73 (s, 2H, CH2), 6.10 (s, 2H, Ar-H), 7.03 (d, J = 15.9 Hz, 1H, =CH), 7.10 (d, J = 8.7 Hz, 1H, Ar-H), 7.63 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.80 (d, J = 15.6 Hz, 1H, CH=), 7.85 (d, J = 2.1 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 471.9987; found 471.9972. Anal. (C18H18BrNO7S) C, H, N.
(E)-2′,4′,6′-Trimethoxystyryl-4-bromo-3-aminobenzylsulfone (8w)
The title compound was obtained by the reduction of (E)-2′,4′,6′-trimethoxystyryl-4-bromo-3-nitrobenzylsulfone 8v following the procedure as described in compound 8b. Yield, 49%; white solid, mp 162–164 °C. 1H NMR (CDCl3, 300 MHz): δ 3.80 (s, 6H, 2 X OCH3), 3.84 (s, 3H, OCH3), 4.63 (s, 2H, CH2), 6.01 (s, 2H, Ar-H), 6.70 – 6.77 (m, 2H, Ar-H), 7.06 (d, J = 15.9 Hz, 1H, =CH), 7.27 (s, 1H, Ar-H), 7.85 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 442.0174; found 442.0189. Anal. (C18H20BrNO5S) C, H, N.
(E)-2′,4′,6′-Trimethoxystyryl-4-chloro-3-aminobenzylsulfone (8x)
Step 1: (E)-2′,4′,6′-Trimethoxystyryl-4-chloro-3-nitrobenzylsulfone
The condensation of 4-chloro-3-nitrobenzylsulfonylacetic acid 6d with 2,4,6-trimethoxybenzaldehyde following the procedure as described in method A resulted in the desired product (E)-2′,4′,6′-trimethoxystyryl-4-chloro-3-nitrobenzylsulfone. Yield, 51%; pale yellow solid, mp 173–174 °C. 1H NMR (CDCl3, 300 MHz): δ 3.76 (s, 6H, 2 X OCH3), 3.78 (s, 3H, OCH3), 4.06 (s, 2H, CH2), 6.02 (s, 2H, Ar-H), 6.59 (dd, J = 8.1, 2.1 Hz, 1H, Ar-H), 6.77 (d, J = 2.1 Hz, 1H, Ar-H), 7.00 (d, J = 15.6 Hz, 1H, =CH), 7.13 (d, J = 8.1 Hz, Ar-H), 7.75 (d, J = 15.9 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 428.0489; found 428.0476. Anal.Calcd. for C18H18ClNO7S: C, 50.53%; H, 4.24%; N, 3.27%. Found: C, 50.72%; H, 4.29%; N, 3.24%.
Step 2: (E)-2′,4′,6′-Trimethoxystyryl-4-chloro-3-aminobenzylsulfone (8x)
The title compound was obtained by the reduction of (E)-2′,4′,6′-trimethoxystyryl-4-chloro-3-nitrobenzylsulfone following the procedure as described in compound 8b. Yield, 50%; pale yellow solid, mp 138–140 °C. 1H NMR (CDCl3, 300 MHz): δ 3.76 (s, 6H, 2 X OCH3), 3.78 (s, 3H, OCH3), 4.00 (br s, 2H, NH2), 4.06 (s, 2H, CH2), 6.02 (s, 2H, Ar-H), 6.59 (dd, J = 8.1, 2.1 Hz, 1H, Ar-H), 6.77 (d, J = 2.1 Hz, 1H, Ar-H), 6.95 (d, J = 15.6 Hz, 1H, =CH), 7.13 (d, J = 8.1 Hz, Ar-H), 7.75 (d, J = 15.9 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 398.0842; found 398.0848. Anal. (C18H20ClNO5S) C, H, N.
(E)-2′,4′,6′-Trimethoxystyryl-4-chloro-2-aminobenzylsulfone (8y)
Step 1: (E)-2′,4′,6′-Trimethoxystyryl-4-chloro-2-nitrobenzylsulfone
The condensation of 4-chloro-2-nitrobenzylsulfonylacetic acid 6e with 2,4,6-trimethoxybenzaldehyde following the procedure as described in method A resulted in the desired product (E)-2′,4′,6′-trimethoxystyryl-4-chloro-2-nitrobenzylsulfone. Yield, 54%; pale yellow solid, mp 206–208 °C. 1H NMR (CDCl3, 300 MHz): δ 3.79 (s, 6H, 2 X OCH3), 3.84 (s, 3H, OCH3), 4.27 (s, 2H, CH2), 6.10 (s, 2H, Ar-H), 6.59 (dd, J = 8.1, 2.1 Hz, 1H, Ar-H), 6.84 (d, J = 2.1 Hz, 1H, Ar-H), 7.10 (d, J = 15.6 Hz, 1H, =CH), 7.21 (d, J = 8.1 Hz, Ar-H), 7.82 (d, J = 15.9 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 428.0489; found 428.0469. Anal.Calcd. for C18H18ClNO7S: C, 50.53%; H, 4.24%; N, 3.27%. Found: C, 50.45%; H, 4.20%; N, 3.22%.
Step 2: (E)-2′,4′,6′-Trimethoxystyryl-4-chloro-2-aminobenzylsulfone (8y)
The title compound was obtained by the reduction of (E)-2′,4′,6′-trimethoxystyryl-4-chloro-2-nitrobenzylsulfone following the procedure as described in compound 8b. Yield, 48%; yellow solid, mp 184–186 °C. 1H NMR (CDCl3, 300 MHz): δ 3.86 (s, 6H, 2 X OCH3), 3.88 (s, 3H, OCH3), 4.27 (s, 2H, CH2), 4.50 (br s, 2H, NH2),6.12 (s, 2H, Ar-H), 6.74 (dd, J = 8.1, 2.1 Hz, 1H, Ar-H), 6.79 (d, J = 2.1 Hz, 1H, Ar-H), 6.99 (d, J = 8.1 Hz, Ar-H), 7.11 (d, J = 15.9 Hz, 1H, =CH), 7.92 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 398.0842; found 398.0836. Anal. (C18H20ClNO5S) C, H, N.
(E)-2′,4′,6′-Trimethoxystyryl-4-fluoro-3-nitrobenzylsulfone (8z)
The title compound was obtained from 4-fluoro-3-nitrobenzylsulfonylacetic acid 6f and 2,4,6-trimethoxybenzaldehyde following the procedure as described in method A. Yield, 53%; light yellow solid, mp 161–163 °C. 1H NMR (CDCl3, 300 MHz): δ 3.85 (s, 6H, 2 X OCH3), 3.87 (s, 3H, OCH3), 4.28 (s, 2H, CH2), 6.10 (s, 2H, Ar-H), 7.02 (d, J = 15.6 Hz, 1H, =CH), 7.31 (dd, J = 8.7, 1.8 Hz, 1H, Ar-H), 7.69–7.74 (m, 1H, Ar-H), 7.75 (d, J = 15.3 Hz, 1H, CH=), 8.07 (dd, J = 6.9, 2.1 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 412.0791; found 412.0774. Anal. (C18H18FNO7S) C, H, N.
(E)-2′,4′,6′-Trimethoxystyryl-4-fluoro-3-aminobenzylsulfone (8aa)
The title compound was obtained by the reduction of (E)-2′,4′,6′-trimethoxystyryl-4-fluoro-3-nitrobenzylsulfone 8z following the procedure as described in compound 8b. Yield, 46%; pale yellow solid, mp 129–131 °C. 1H NMR (CDCl3, 300 MHz): δ 3.76 (s, 6H, 2 X OCH3), 3.78 (s, 3H, OCH3), 4.09 (s, 2H, CH2), 6.02 (s, 2H, Ar-H), 6.57 – 6.62 (m, 1H, Ar-H), 6.80 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 6.85 (dd, J = 8.4, 2.7 Hz, 1H, Ar-H), 6.96 (d, J = 15.9 Hz, 1H, =CH), 7.27 (s, 1H, Ar-H), 7.76 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 382.1087; found 382.1105. Anal. (C18H20FNO5S) C, H, N.
(E)-2′,4′,6′-Trifluorostyryl-4-methoxy-3-aminobenzylsulfone (8ab)
Step 1: (E)-2′,4′,6′-Trifluorostyryl-4-methoxy-3-nitrobenzylsulfone
The condensation of 4-methoxy-3-nitrobenzylsulfonylacetic acid 6a with 2,4,6-trifluorobenzaldehyde following the procedure as described in method A resulted in the desired product (E)-2′,4′,6′-trifluorostyryl-4-methoxy-3-nitrobenzylsulfone. Yield, 52%; yellow solid, mp 141–143 °C. 1H NMR (CDCl3, 300 MHz): δ 3.89 (s, 3H, OCH3), 4.28 (s, 2H, CH2), 6.20 (s, 2H, Ar-H), 6.80–6.90 (m, 2H, Ar-H), 6.97–7.05 (m, 1H, Ar-H), 7.10 (d, J = 15.9 Hz, 1H, =CH), 7.62 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 388.0388; found 388.0379. Anal.Calcd. for C16H12F3NO5S: C, 49.61%; H, 3.12%; N, 3.62%. Found: C, 49.75%; H, 3.17%; N, 3.68%.
Step 2: (E)-2′,4′,6′-Trifluorostyryl-4-methoxy-3-aminobenzylsulfone (8ab)
The title compound was obtained by the reduction of (E)-2′,4′,6′-trifluorostyryl-4-methoxy-3-nitrobenzylsulfone following the procedure as described in compound 8b. Yield, 48%; light yellow solid, mp 110–112 °C. 1H NMR (CDCl3, 300 MHz): δ 3.86 (s, 3H, OCH3), 4.18 (s, 2H, CH2), 6.67–6.83 (m, 5H, Ar-H), 7.01 (d, J = 16.2 Hz, 1H, =CH), 7.50 (d, J = 15.9 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 358.0646; found 358.0637. Anal. (C16H14F3NO3S) C, H, N.
(E)-2′,4′,5′-Trifluorostyryl-4-methoxy-3-aminobenzylsulfone (8ac)
Step 1: (E)-2′,4′,5′-Trifluorostyryl-4-methoxy-3-nitrobenzylsulfone
The condensation of 4-methoxy-3-nitrobenzylsulfonylacetic acid 6a with 2,4,5-trifluorobenzaldehyde following the procedure as described in method A resulted in the desired product (E)-2′,4′,5′-trifluorostyryl-4-methoxy-3-nitrobenzylsulfone. Yield, 51%; pale yellow solid, mp 161–163 °C. 1H NMR (CDCl3, 300 MHz): δ 3.91 (s, 3H, OCH3), 4.19 (s, 2H, CH2), 6.67 (dd, J = 8.1, 2.1 Hz, 1H, Ar-H), 6.74–6.77 (m, 2H, Ar-H), 6.80 (d, J = 15.6 Hz, 1H, =CH), 6.97–7.05 (m, 1H, Ar-H), 7.17–7.24 (m, 1H, Ar-H), 7.43 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 388.0388; found 388.0394. Anal.Calcd. for C16H12F3NO5S: C, 49.61%; H, 3.12%; N, 3.62%. Found: C, 49.50%; H, 3.16%; N, 3.69%.
Step 2: (E)-2′,4′,5′-Trifluorostyryl-4-methoxy-3-aminobenzylsulfone (8ac)
The title compound was obtained by the reduction of (E)-2′,4′,5′-trifluorostyryl-4-methoxy-3-nitrobenzylsulfone following the procedure as described in compound 8b. Yield, 46%; white solid, mp 132–134 °C. 1H NMR (CDCl3, 300 MHz): δ 3.91 (s, 3H, OCH3), 4.19 (s, 2H, CH2), 6.67 (dd, J = 8.1, 2.1 Hz, 1H, Ar-H), 6.74–6.77 (m, 2H, Ar-H), 6.80 (d, J = 15.6 Hz, 1H, =CH), 6.97–7.05 (m, 1H, Ar-H), 7.17–7.24 (m, 1H, Ar-H), 7.43 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 358.0646; found 358.0627. Anal. (C16H14F3NO3S) C, H, N.
Preparation of 3-Nitro-4-methoxybenzyl mercaptan (9). (Scheme 4)
Step1: 3-Nitro-4-methoxybenzylisothiouronium salt (12)
A solution of 3-nitro-4-methoxybenzyl bromide 2a (10.0 g, 41 mmol) and thiourea (10.0 g, 131 mmol) in 50 mL water was heated under reflux for 2 h. The reaction mixture was cooled, stirred at room temperature for 2 h and filtered the solid. The resulted dried product used in next step without further purification. The yield of this reaction was 90%, giving a white solid with a melting point 174–176 °C. 1H NMR (CDCl3, 300 MHz): δ 3.91 (s, 3H, OCH3), 4.51 (s, 2H, CH2), 7.39 (d, J = 8.7 Hz, 1H, Ar-H), 7.72 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.98 (d, J = 2.1 Hz, 1H, Ar-H), 9.12 (br s, 4H, 2 X NH2). HRMS: m/z calcd [M + H] 323.9939; found 323.9929. Anal. (C9H14 BrN3O3S) C, H, N.
Step 2: 3-Nitro-4-methoxybenzyl merccaptan (9)
The isothiouronium salt 12 was decomposed by boiling several times with concentrated ammonia and hexane (100 mL, 15:85). Concentration of the combined hexane extracts provided crude 9. The pure compound 9 was obtained on silica gel flash column chromatography (hexane/ethyl acetate, 4:1). The yield of this reaction was 55%, giving a yellow solid with a melting point 48–49 °C. 1H NMR (CDCl3, 300 MHz): δ 1.74 (t, J = 7.5 Hz, SH), 3.67 (d, J = 7.5 Hz, 2H, CH2), 3.89 (s, 3H, OCH3), 6.98 (d, J = 8.4 Hz, 1H, Ar-H), 7.45 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 7.76 (d, J = 2.4 Hz, 1H, Ar-H). HRMS: m/z calcd [M - H] 198.0303; found 198.0320. Anal. (C8H9NO3S) C, H, N.
General Procedure for the Preparation of (E)-Styryl benzyl sulfone (8). Method B (Scheme 3). Preparation of 4′-Methoxyphenacyl 3-nitro-4-methoxybenzyl sulfide (11)
To a cooled solution of sodium hydroxide (100 mmol) in absolute methanol (50 mL), taken in a 250 mL round-bottomed flask, 3-nitro-4-methoxybenzyl mercaptan 9 (100 mmol) was added slowly and the reaction mixture was stirred for 5 minutes. An appropriate 4-methoxyphenacyl bromide 10 (100 mmol) was added in portions to the contents of the flask and stirred for 2 h. After completion of the reaction (TLC, monitoring, hexane/ethylacetate, 8:2 on silica gel plate), the contents of the flask were poured into crushed ice and the compound formed was washed with ice-cold water and dried to get 4′-methoxyphenacyl-3-nitro-4-methoxybenzyl sulfide 11. The yield of this reaction was 95%, giving a yellow solid with a melting point 87–89 °C. 1H NMR (CDCl3, 300 MHz): δ 3.55 (s, 2H, CH2), 3.67 (s, 2H, CH2), 3.81(s, 3H, OCH3), 3.88 (s, 3H, OCH3), 6.87 (d, J = 9.0 Hz, 2H, Ar-H), 6.96 (d, J = 8.4 Hz, 2H, Ar-H), 7.49 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 7.79 (d, J = 2.4 Hz, 1H, Ar-H), 7.85 (d, J = 8.7 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 348.0827; found 348. 0811. Anal. (C17H17NO5S) C, H, N.
Preparation of 4′-Methoxyphenacyl-3-nitro-4-methoxybenzyl sulfone (13)
The crude 4′-methoxyphenacyl-3-nitro-4-methoxybenzyl sulfide 11 (50 mmol) in glacial acetic acid (100 mL) was taken in a 250 mL round-bottomed flask and 30% hydrogen peroxide (60 mL) was added in portions at frequent intervals. Then the reaction mixture was kept at room temperature for 24 h. The solid, if any formed was separated by filtration and the filtrate was poured onto crushed ice. The compound separated was filtered, washed with water, dried and added to the first crop, if any. The total product on recrystallization from methanol afforded pure 4′-Methoxyphenacyl-3-nitro-4-methoxybenzyl sulfone (13). Yield, 76%; white solid, mp 172–174 °C. 1H NMR (CDCl3, 300 MHz): δ 3.83 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 4.31 (s, 2H, CH2), 4.45 (s, 2H, CH2), 6.87 (d, J = 9.0 Hz, 2H, Ar-H), 7.06 (d, J = 8.7 Hz, 2H, Ar-H), 7.67 (dd, J = 8.7, 2.1 Hz, 1H, Ar-H), 7.90 (d, J = 9.0 Hz, 1H, Ar-H), 7.98 (d, J = 2.1 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 380.0726; found 380.0709. Anal. (C17H17NO7S) C, H, N.
Preparation of 2-(3-Nitro-4-methoxybenzylsulfonyl)-1-(4′-methoxyphenyl)ethanol (14)
To anhydrous tetrahydrofuran (THF) solution (20 mL) of 4-Methoxyphenacyl benzyl sulfone 13 (10 mmol) maintained at 0 °C, was added NaBH4 (10 mmol) slowly under N2 atmosphere. The reaction mixture was maintained at 0 °C for 1 h. After completion of the reaction monitored by TLC (hexane/ethylacetate, 8:2 on silica gel plate), the contents were poured on to crushed ice. The solid that separated out was filtered, washed with water and dried under vacuum to yield 14. Yield, 56%; white solid, mp 152–154 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.43 (dd, J = 9.9, 4.6 Hz, 2H, CH-CH2), 3.73 (s, 3H, OCH3), 3.94 (s, 3H, OCH3), 4.58 (dd, J = 15.7, 13.6 Hz, 2H, SO2CH2), 5.05 (m, 1H, CHOH), 6.02 (d, J = 4.2 Hz, 1H, CH), 6.90 (d, J = 8.6 Hz, 2H, Ar-H), 7.32 (d, J = 8.6 Hz, 2H, Ar-H), 7.42 (d, J = 9.0 Hz, 1H, Ar-H), 7.69 (dd, J = 8,7, 1.8 Hz, 1H, Ar-H), 7.93 (d, J = 1.8 Hz, 1H, Ar-H). HRMS: m/z calcd [M+H] 382.0882; found 382.0916. Anal. (C17H19NO7S) C, H, N.
Preparation of (E)-4′-Methoxystyryl-3-nitro-4-methoxybenzyl sulfone (8c)
p-Toluenesulfonic acid (1 mmol) was added in one portion to a mixture of 2-(3-Nitro-4-methoxybenzylsulfonyl)-1-(4′-methoxyphenyl)ethanol 14 (5 mmol) in anhydrous toluene (25 mL) at room temperature and under N2 atmosphere. The temperature was raised to 120 °C, and the mixture was refluxed for 2–4 h using Dean-Stark water separator. After completion of the reaction monitored by TLC (chloroform, on silica gel plate), the reaction mixture was concentrated under reduced pressure and then quenched by the addition of water (25 mL). The aqueous layer was neutralized with a saturated aqueous solution of sodium hydrogen carbonate and extracted with dichloromethane (3 × 25mL). The combined organic extracts were washed with brine (2 × 25 mL) dried over Na2SO4, filtered and the solvent was evaporated under reduced pressure to afford crude product, which on recrystallization in 2-propanol afforded the desired product 8c. Yield: 65%; white solid, 184–186 °C. Analytical data is same as 8c obtained by method A.
General Procedure for the Preparation of 2,4,6-trimethoxyphenyl acetylene (15, Scheme 6)
Scheme 6.
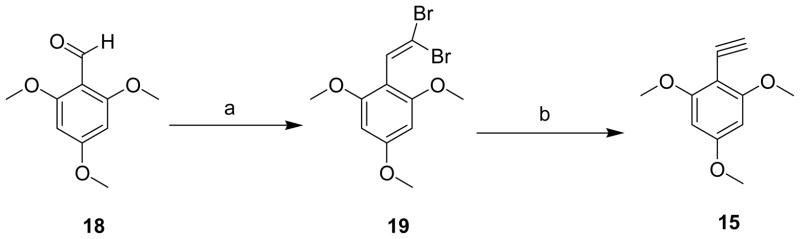
Synthesis of 2,4,6-trimethoxyphenyl acetylenea
a Reagents and conditions: (a)Triphenylphosphine, tetrabromomethane, CH2Cl2, 5 °C, 30 min; (b) n-BuLi, dry THF, −78 °C, 15 min.
Step 1: 2-(2′,2′-dibromovinyl)-1,3,5-trimethoxybenzene (19)
To a solution of 2,4,6-trimethoxybenzaldehyde 18 (5.0 g, 25.5 mmol), and triphenyl phosphine (13.37 g, 51.0 mmol) in anhydrous dichloromethane (60 mL) was added a solution of tetrabromomethane (9.8 g, 30.0 mmol) in dichloromethane (10 mL), keeping the temperature kept below 5 °C. The reaction mixture was stirred for additional 30 min. After completion of the reaction monitored by TLC (hexane/ethylacetate, 9:1 on silica gel plate), the reaction mixture was filtered and concentrated in vacuo. The residue was purified by flash column chromatography (hexane/ethylacetate, 9:1), resulted pure 19. The yield of this reaction was 72%, giving a white solid with a melting point 128–130 °C. 1H NMR (CDCl3, 300 MHz): δ 3.82 (s, 6H, 2 X OCH3), 3.83 (s, 3H, OCH3), 6.12 (s, 2H, Ar-H), 7.19 (s, 1H, CH=). HRMS: m/z calcd [M +H] 352.9133; found 352.9112. Anal. (C11H12Br2O3) C, H.
Step 2: 2,4,6-Trimetoxyphenyl acetylene (15)
A solution of 2-(2′,2′-dibromovinyl)-1,3,5-trimethoxybenzene 19 (7.42 g, 21.3 mmol) in dry tetrahydrofuran (135 mL) was cooled to −78 °C, and slowly added n-butyllithium (18.0 mL, 45.0 mmol). The mixture was stirred for further 15 min at −78 °C. After completion of the reaction monitored by TLC (hexane/ethylacetate, 9:1 on silica gel plate), water added (60 mL) and extracted with ethyl acetate (2 × 100 mL). The organic phase was washed with water, dried over anhydrous sodium sulphate, filtered and concentrated in vacuo and the residue was purified by flash column chromatography (hexane/ethyl acetate, 9:1) resulted pure 15. The yield of this reaction was 67%, giving a white solid with a melting point 119–122 °C. 1H NMR (CDCl3, 300 MHz): δ 3.43 (s, 1H, CH), 3.77 (s, 3H, OCH3), 3.82 (s, 6H, 2 X OCH3), 6.04 (s, 2H, Ar-H). HRMS: m/z calcd [M +H] 193.0786; found 193.0764. Anal. (C11H12O3) C, H.
Synthesis of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfide (17) and (Z)-2′,4′,6′-trimethoxystyry-4-methoxy-3-nitrolbenzyl sulfide (16). (Scheme 5)
A hexane solution of triethyl borane (10.5 mL, 10.1 mmol) was added to a solution of 2,4,6-trimethoxyphenyl acetylene 15 (5.0 g, 10.1 mmol) and 4-methoxy-3-nitrobenzyl thiol 9a (2.49g, 12.12 mmol) in benzene (100 mL) at 25 °C under nitrogen atmosphere. The resulting mixture was stirred for 2 h at 25 °C. After completion of the reaction monitored by TLC (hexane/ethylacetate, 9:1 on silica gel plate), the reaction mixture was quenched with 1 molar ammonium chloride solution and extracted with ethyl acetate. The organic phase was washed with water, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo and the residue was purified by column chromatography (hexane/ethyl acetate, 9:1 and with gradual increasing in polarity) resulted as a stereo isomeric mixture (E/Z = 60/40).
(E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfide (17)
The yield of this reaction was 60%, giving a white solid with a melting point 121–123 °C. 1H NMR (CDCl3, 300 MHz): δ 3.80 (s, 6H, 2 X OCH3), 3.81 (s, 3H, OCH3), 3.93 (s, 2H, CH2S), 3.96 (s, 3H, OCH3), 6.10 (s, 2H, Ar-H), 6.83 (d, J = 15.6 Hz, 1H, CH=), 7.00 (d, J = 15.6 Hz, 1H, =CH), 7.05 (d, J = 8.7 Hz, 1H, Ar-H), 7.59 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.80 (d, J = 2.4 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 392.1090; found 392.1084. Anal. (C19H21NO6S) C, H, N.
(Z)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfide (16)
The yield of this reaction was 40%, giving a white solid with a melting point 102–104 °C. 1H NMR (CDCl3, 300 MHz): δ 3.75 (s, 12H, 4 X OCH3), 3.86 (s, 2H, CH2S), 6.06 (s, 2H, Ar-H), 6.14 (d, J = 10.2 Hz, 1H, CH=), 6.49 (d, J = 10.5 Hz, 1H, =CH), 6.94 (d, J = 8.4 Hz, 1H, Ar-H), 7.43 (dd, J = 8.7, 2.1 Hz, 1H, Ar-H), 7.75 (d, J = 1.8 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 392.1090; found 392.1084. Anal. (C19H21NO6S) C, H, N.
Synthesis of (E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-aminobenzyl sulfide (24)
The title compound was obtained by the reduction of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-nitrobenzylsulfide 17 following the procedure as described in compound 8b. Yield, 43%; pale yellow solid, mp 106–108 °C. 1H NMR (CDCl3, 300 MHz): δ 3.73 (s, 6H, 2 X OCH3), 3.76 (s, 6H, 2 X OCH3), 3.80 (s, 2H, CH2S), 6.03 (s, 2H, Ar-H), 6.63–6.71 (m, 2H, Ar-H), 6.74 (d, J = 15.6 Hz, 1H, CH=), 7.01 (d, J = 15.9 Hz, 1H, =CH), 7.30 (s, 1H, Ar-H). HRMS: m/z calcd [M + H] 362.1348; found 362.1339. Anal. (C19H23NO4S) C, H, N.
Synthesis of (E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfoxide (20)
A aqueous 30% hydrogen peroxide (0.56 mL, 5.2 mmol) was added to a stirred solution of (E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfide 17 (1.0 g, 2.6 mmol) in 1,1,1,3,3,3-hexafluoro-2-propanol (10 mL) at 25 °C. The reaction was monitored by TLC. After the complete disappearance of the sulfide (2 h), the excess hydrogen peroxide was quenched with saturated sodium sulfite (Na2SO3) solution (5.0 mL) and the fluorous organic phase containing the sulfoxide was separated. After removal of the solvent under vacuo, sulfoxide 20 was obtained as a semisolid. Flash column chromatography (chloroform) gave pure 20. The yield of this reaction was 91%, giving a white solid with a melting point 148–150 °C. 1H NMR (CDCl3, 300 MHz): δ 3.75 (s, 6H, 2 X OCH3), 3.77 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 3.96–4.00 (d, J = Hz, 2H, CH2S), 6.01 (s, 2H, Ar-H), 6.96 (d, J = 15.9 Hz, 1H, CH=), 7.02 (d, J = 8.7 Hz, 1H, Ar-H), 7.36 (d, J = 15.6 Hz, 1H,=CH), 7.46 (dd, J = 8.7, 2.1 Hz, 1H, Ar-H), 7.71 (d, J = 2.1 Hz, 1H, Ar-H). HRMS: m/z calcd [M + H] 408.1039; found 408.1022. Anal. (C19H21NO7S) C, H, N.
Synthesis of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzyl sulfoxide (21)
The title compound was obtained by the reduction of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-nitrobenzylsulfoxide 20 following the procedure as described in compound 8b. Yield, 46%; pale yellow solid, mp 131–133 °C. 1H NMR (CDCl3, 300 MHz): δ 3.77 (br s, 12H, 4 X OCH3), 3.81 (br s, 2H, NH2), 3.92 (d, J = 12.6 Hz, 2H, CH2S), 6.04 (s, 2H, Ar-H), 6.60–6.69 (m, 3H, Ar-H), 7.08 (d, J = 15.6 Hz, 1H, CH=), 7.43 (d, J = 15.6 Hz, 1H, =CH). HRMS: m/z calcd [M + H] 378.1297; found 378.1280. Anal. (C19H23NO5S) C, H, N.
Synthesis of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfone (22 & 8l)
The title compound was obtained by the oxidation of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfoxide 20 according to the procedure reported in the literature.7 The yield of this reaction was 62%; yellow solid, mp 184–186 °C. The analytical data are in accord with 8l.
Synthesis of (E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-aminobenzyl sulfone (23 & 8p)
Method A
The title compound was obtained by the reduction of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-nitrobenzylsulfone 22 following the procedure as described in compound 8b. The yield of this reaction was 52%; light yellow solid, mp 146–148 °C. The analytical data are in accord with 8p.
Method B
The title compound was obtained by the oxidation of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzyl sulfide 24. To a solution of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzyl sulfide 24 (903 mg, 2.5 mmol) in anhydrous dichloromethane (20 mL) at 0 °C was added m-chloroperoxybenzoic acid (1.29 g, 7.5 mmol) slowly under nitrogen atmosphere and the resulting mixture was stirred at 0 °C to room temperature for 3 h. After completion of the reaction (TLC monitoring, chloroform on silica gel plate), 10% sodium hydrogen carbonate solution (25 mL) was added slowly and stirred for 10 min and separated the aqueous layer. The organic layer washed with water (2 X 25 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography (chloroform) to obtain the title compound 24. The yield of this reaction was 64%; light yellow solid, mp 146–148 °C. The analytical data are in accord with 8p.
Synthesis of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzyl sulfoxide (21, Scheme 7)
Step 1: 4-Methoxy-3-nitrobenzyl sulfinylacetic acid (25)
A vigorously stirred solution of sodium hydroxide (0.58 g, 14.5 mmol) and deionized water (30 mL) was treated with 4-methoxy-3-nitrobenzylthioacetic acid 5a (3,30 g, 12.1 mmol). The resulting suspension was stirred at ambient temperature for 10 min. To this was added sodium bicarbonate (8.00 g, 95 mmol) and acetone (10 mL) and cooled to 0 °C. The oxone® solution (4.85 g in 20 mL of 4 × 10−4 M EDTA) was added over 10 min keeping the reaction below 5 °C. The suspension was stirred for 5 min and immediately quenched at 2 °C with sodium bisulfate (3 g in 6 mL of deionized water) and stirred for 15 min. Ethyl acetate (75 mL) was added and the solution was acidified with 6N (aq) HCl (18 mL). The aqueous phase was isolated, treated with sodium chloride (15.0 g), and re-extracted with ethyl acetate (75 mL). The organic layers were combined and washed with deionized water (50 mL), washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and concentrated under vacuo. The crystals formed were dried under vacuum to afford pure 25. The yield of this reaction was 89%, giving a white solid with a melting point 122–124 °C. 1H NMR (CDCl3, 300 MHz): δ 3.42 (s, 2H, -SCH2), 3.90 (s, 3H, OCH3), 4.02 (s, 2H, CH2S), 7.42 (d, J = 8.4 Hz, 1H, Ar-H), 7.69 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.92 (d, J = 2.1 Hz, 1H, Ar-H), 12.90 (br s, 1H, COOH). HRMS: m/z calcd [M - H] 272.0307; found 272.0300. Anal.Calcd. for C10H11NO6S: C, 43.95%; H, 4.06%; N, 5.13%. Found: C, 44.06%; H, 4.09%; N, 5.17%.
Step 2: (E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfoxide (20)
The title compound was obtained from 4-methoxy-3-nitrobenzylsulfinylacetic acid 25 and 2,4,6-trimethoxybenzaldehyde following the procedure as described in method A and scheme 1. The yield of this reaction was 48%. The analytical data are in accord with 20 as reported in scheme 5.
Step 3: (E)-2′, 4′, 6′-Trimethoxystyryl-4-methoxy-3-aminobenzyl sulfoxide (21)
The title compound was obtained by the reduction of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-nitro-benzylsulfoxide 20 following the procedure as described in compound 8b. The yield of this reaction was 42%. The analytical data are in accord with 21 as reported in scheme 5.
General Procedure for the Preparation of Amino esters (26, Scheme 8)
Sodium acetate (32.8 g, 400 mmol) was dissolved in ethanol (200 mL). Methyl 2-bromoacetate (61.1 g, 400 mmol) was added to the above solution and refluxed for 10 min. To the cooled reaction mixture compound (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone 8p or (E)-3′,4′,5′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone 8q (39.35 g, 100 mmol) was added and then refluxed for 48 h. After completion of the reaction monitored by TLC (chloroform/methanol, 9:1 on silica gel plate), the reaction mixture was concentrated under vacuum and poured into ice water. The solid formed was filtered, washed with water and dried under vacuum. The crude product on purification from ethanol resulted analytical pure product 26. The following amino esters were prepared using the above procedure.
(E)-Methyl 2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)-acetate (26a)
Sodium acetate (3.28 g, 39 mmol) was dissolved in methanol (20 mL). Methyl 2-bromoacetate (6.11 g, 40 mmol) was added to the above solution and refluxed for 10 min. To the cooled reaction mixture compound (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone 8p (3.95 g, 10 mmol) was added and then reflux for 4–6 h. The reaction mixture was concentrated under vacuum and poured into ice water. The formed precipitate was filtered, washed with water and dried under vacuum. The crude product on recrystallization from ethanol resulted pure product 26a. Yield, 70%; white solid, mp 150–152 °C. 1H NMR (CDCl3, 300 MHz): δ 3.69 (s, 3H, OCH3), 3.75 (s, 6H, 2 X OCH3), 3.78 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 3.81 (d, J = 5.4 Hz, 2H, CH2), 4.09 (s, 2H, CH2), 4.74 (t, J = 5.4 Hz, 1H, NH), 6.01(s, 2H, Ar-H), 6.41 (d, J = 1.8 Hz, 1H, Ar-H), 6.61–6.68 (m, 2H, Ar-H), 6.97 (d, J = 15.6 Hz, 1H, =CH), 7.73 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 466.1457; found 466.1446. Anal. (C22H27NO8S) C, H, N.
(E)-Methyl 2-(2-methoxy-5-((3′,4′,5′-trimethoxystyrylsulfonyl)methyl)phenylamino)-acetate (26b)
The title compound was obtained by the alkylation of (E)-3′,4′,5′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone 8q with methyl 2-bromoacetate following the procedure as described in compound 26. Yield, 63%; light brown solid, mp 82–84 °C. 1H NMR (CDCl3, 400 MHz): δ 3.67 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 3.81 (s, 6H, 2 X OCH3), 3.82 (s, 3H, OCH3), 3.86 (d, J = 5.4 Hz, 2H, CH2), 4.14 (s, 2H, CH2), 4.53 (t, J = 5.4 Hz, 1H, NH), 6.52 (d, J = 15.5 Hz, 1H, =CH), 6.57 (s, 2H, Ar-H), 6.62 (dd, J = 8.2, 2.1 Hz, 1H, Ar-H), 6.67 (d, J = 8.2 Hz, 1H, Ar-H), 6.70 (d, J = 2.1 Hz, 1H, Ar-H), 7.24 (d, J = 15.4 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 466.1457; found 466.1465. Anal. (C22H27NO8S) C, H, N.
(E)-Ethyl 3-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)propanoate (26c)
The title compound was obtained by the alkylation of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone 8p with ethyl 3-bromopropionate following the procedure as described in compound 26. Yield, 59%; light green solid, mp 72–74 °C. 1H NMR (CDCl3, 300 MHz): δ 1.26 (t, J = 7.2 Hz, 3H, CH3), 2.58 (t, J = 6.3 Hz, 2H, CH2), 3.41 (t, J = 6.6 Hz, 2H, CH2), 3.82 (s, 9H, 3 X OCH3), 3.84 (s, 3H, OCH3), 4.04–4.10 (m, 2H, OCH2), 4.17 (s, 2H, CH2), 5.06 (t, J = 5.4 Hz, 1H, NH), 6.09 (s, 2H, Ar-H), 6.61–6.64 (m, 1H, Ar-H), 6.66–6.71 (m, 2H, Ar-H), 7.06 (d, J = 15.6 Hz, 1H, =CH), 7.84 (d, J = 15.9 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 480.1614; found 480.1647. Anal. (C23H29NO8S) C, H, N.
(E)-Methyl 2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)-propanoate (26d)
The title compound was obtained by the alkylation of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone 8p with methyl 2-bromopropionate following the procedure as described in compound 26. Yield, 60%; white solid, mp 178–180 °C. 1H NMR (CDCl3, 400 MHz): δ 1.24 (d, J = 6.9Hz, 3H, CH3), 3.50 (s, 3H, OCH3), 3.61 (s, 6H, 2 X OCH3), 3.63 (s, 3H, OCH3), 3.64 (s, 3H, OCH3), 3.90–3,92 (m, 1H, CH), 3.94 (s, 2H, CH2), 4.52 (br s, 1H, NH), 5.88 (s, 2H, Ar-H), 6.32 (d, J = 1.7 Hz, 1H, Ar-H), 6.47 (dd, J = 8.1, 1.7 Hz, 1H, Ar-H), 6.52 (d, J = 8.1 Hz, 1H, Ar-H), 6.81 (d, J = 15.6 Hz, 1H, =CH), 7.59 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 480.1579; found 480.1568. Anal. (C23H29NO8S) C, H, N.
(E)-Methyl 2″,2″-difluoro-2(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)-phenylamino)acetate (26e)
The title compound was obtained by the alkylation of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone 8p with methyl 2-chloro-2,2-difluoroacetate following the procedure as described in compound 26. Yield, 57%; white solid, mp186–188 °C. 1H NMR (CDCl3, 300 MHz): δ 3.74 (s, 3H, OCH3), 3.83 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 4.22 (s, 2H, CH2), 6.09 (s, 2H, Ar-H), 6.86 (d, J = 8.4 Hz, 1H, Ar-H), 7.09 (d, J = 15.6 Hz, 1H, =CH), 7.11 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 7.19 (s, 1H, Ar-H), 7.84 (d, J = 15.6 Hz, 1H, CH=), 8.08 (brs, 1H, NH). HRMS: m/z calcd [M + H] 502.1269; found 502.1284. Anal. (C22H25F2NO8S) C, H, N.
(E)-Methyl 3″,3″,3″-trifluoro-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)-methyl)phenylamino)propanoate (26f)
The title compound was obtained by the alkylation of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone 8p with methyl 2-bromo-3,3,3-trifluoropropanoate following the procedure as described in compound 26. Yield, 56%; white solid, mp 192–194 °C. 1H NMR (CDCl3, 300 MHz): δ 3.49 (s, 3H, OCH3), 3.51 (s, 6H, 2 X OCH3), 3.55 (s, 3H, OCH3), 3.57 (s, 3H, OCH3), 3.95–4.16 (m, 1H, CH), 4.22 (s, 2H, CH2), 4.85 (t, J = 9.0 Hz, 1H, NH), 5.92 (s, 2H, Ar-H), 6.23 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 6.40 (d, J = 8.1 Hz, 1H, Ar-H), 6.53 (d, J = 2.1 Hz, 1H, Ar-H), 6.80 (d, J = 15.6 Hz, 1H, =CH), 7.55 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 534.1345; found 534.1339. Anal. (C23H26F3NO8S) C, H, N.
(E)-Methyl 2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)-2-methylpropanoate (26g)
The title compound was obtained by the alkylation of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone 8p with methyl 2-bromo2-methylpropionate following the procedure as described in compound 26. Yield, 60%; white solid, mp 170–172 °C. 1H NMR (CDCl3, 500 MHz): δ 1.52 (s, 6H, 2 X CH3), 3.68 (s, 3H, OCH3), 3.81 (s, 6H, 2 X OCH3), 3.82 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 4.11 (s, 2H, CH2), 6.06 (s, 2H, Ar-H), 6.41 (s, 1H, Ar-H), 6.69–6.71 (m, 2H, Ar-H), 7.02 (d, J = 15.5 Hz, 1H, =CH), 7.80 (d, J = 15.5 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 494.1770; found 494.1761. Anal. (C24H31NO8S) C, H, N.
(E)-Methyl 2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)-2-phenylacetate (26h)
The title compound was obtained by the alkylation of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone 8p with methyl α-bromophenylacetate following the procedure as described in compound 26. Yield, 63%; white solid, mp 94–96 °C. 1H NMR (CDCl3, 300 MHz): δ 3.71 (s, 3H, OCH3), 3.83 (s, 6H, 2 X OCH3), 3.86 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 4.06 (s, 2H, CH2), 4.99 (d, J = 6.0 Hz, 1H, CH), 5.43(d, J = 6.3 Hz, 1H, NH), 6.10 (s, 2H, Ar-H), 6.36 (d, J = 1.8 Hz, 1H, Ar-H), 6.67 (dd, J = 8.1, 1.8 Hz, 1H, Ar-H), 6.73 (d, J = 8.1 Hz, 1H, Ar-H), 6.99 (d, J = 15.6 Hz, 1H, =CH), 7.29–7.35 (m, 3H, Ar-H), 7.42 (dd, J = 8.1, 1.8 Hz, 2H, Ar-H), 7.77 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 542.1770; found 542.1778. Anal. (C28H31NO8S) C, H, N.
(E)-Methyl 2-(4″-fluorophenyl)-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)-methyl)phenylamino)acetate (26i)
The title compound was obtained by the alkylation of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone 8p with methyl 2-bromo-2-(4-fluorophenyl) acetate following the procedure as described in compound 26. Yield, 60%; white solid, mp 152–154 °C. 1H NMR (CDCl3, 500 MHz): δ 3.72 (s, 3H, OCH3), 3.82 (s, 6H, 2 X OCH3), 3.86 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 4.05 (s, 2H, CH2), 4.98 (d, J = 2.5 Hz, 1H, CH), 6.10 (s, 2H, Ar-H), 6.33 (d, J = 2.0 Hz, 1H, Ar-H), 6.69 (dd, J = 8.5, 2.0 Hz, 1H, Ar-H), 6.73 (d, J = 8.5 Hz, 1H, Ar-H), 6.96–6.99 (m, 2H, Ar-H), 7.00 (d, J = 15.5Hz, 1H, =CH), 7.37–7.40 (m, 2H, Ar-H), 7.76 (d, J = 16.0 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 560.1676; found 560.1656. Anal. (C28H30FNO8S) C, H, N.
(E)-Methyl 2-(4″-chlorophenyl)-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)-methyl)phenylamino)acetate (26j)
The title compound was obtained by the alkylation of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone 8p with methyl 2-bromo-2-(4-chlorophenyl)acetate following the procedure as described in compound 26. Yield, 64%; white solid, mp 154–156 °C. 1H NMR (CDCl3, 300 MHz): δ 3.72 (s, 3H, OCH3), 3.83 (s, 6H, 2 X OCH3), 3.86 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 4.05 (s, 2H, CH2), 4.96 (s, 1H, CH), 6.11 (s, 2H, Ar-H), 6.31 (s, 1H, Ar-H), 6.67–6.75 (m, 2H, Ar-H), 6.99 (d, J = 15.6 Hz, 1H, =CH), 7.25–7.28 (m, 2H, Ar-H), 7.54 (d, J = 8.4 Hz, 2H, Ar-H), 7.76 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 576.1381; found 576.1367. Anal. (C28H30ClNO8S) C, H, N.
(E)-Methyl 2-(4″-bromophenyl)-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)-methyl)phenylamino)acetate (26k)
The title compound was obtained by the alkylation of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone 8p with methyl 2-bromo-2-(4-bromophenyl)acetate following the procedure as described in compound 26. Yield, 62%; white solid, mp 150–152 °C. 1H NMR (CDCl3, 400 MHz): δ 3.48 (s, 3H, OCH3), 3.59 (s, 6H, 2 X OCH3), 3.62 (s, 3H, OCH3), 3.65 (s, 3H, OCH3), 3.81 (s, 2H, CH2), 4.71(s, 1H, CH), 5.87 (s, 2H, Ar-H), 6.07 (d, J = 1.8 Hz, 1H, Ar-H), 6.44 (dd, J = 8.1, 1.8 Hz, 1H, Ar-H), 6.50 (d, J = 8.1 Hz, 1H, Ar-H), 6.75 (d, J = 15.6 Hz, 1H, =CH), 7.05–7.08 (m, 2H, Ar-H), 7.16–7.19 (m, 2H, Ar-H), 7.53 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 620.0876; found 620.0859. Anal. (C28H30BrNO8S) C, H, N.
(E)-Methyl 2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)-2-(4″-methoxyphenyl)acetate (26l)
The title compound was obtained by the alkylation of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone 8p with methyl-2-bromo-2-(4-methoxyphenyl)acetate following the procedure as described in compound 26. Yield, 61%; white solid, mp 182–184 °C. 1H NMR (CDCl3, 500 MHz): δ 3.70 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 3.83 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 4.06 (s, 2H, CH2), 4.91(s, 1H, CH), 6.10 (s, 2H, Ar-H), 6.37 (d, J = 1.5 Hz, 1H, Ar-H), 6.68 (dd, J = 8.1, 1.5 Hz, 1H, Ar-H), 6.73 (d, J = 8.1 Hz, 1H, Ar-H), 6.84 (d, J = 9.0 Hz, 2H, Ar-H), 7.00 (d, J = 15.5 Hz, 1H, =CH), 7.33 (d, J = 8.0 Hz, 2H, Ar-H), 7.77 (d, J = 16.0 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 572.1876; found 572.1896. Anal. (C29H33NO9S) C, H, N.
General Procedure for the Preparation of Amino acids (27, Scheme 8)
To a solution of amine ester 26 (46.5g, 100 mmol) in ethanol (200 mL), 20% aqueous sodium hydroxide solution (200 mL) was added. The reaction mixture was refluxed for 2.5 h. After completion of the reaction (TLC, monitoring, chloroform/methanol, 9:1 on silica gel plate), the solvent removed under vacuum and the remainder was acidified by acetic acid to pH 4. The solid that formed was filtered and dried to get the crude amino acid 27 which on crystallization from acetone (2 X 25 mL) resulted in analytically pure amino acid 27 as white crystals.
(E)-2-(2-Methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)acetic acid (27a)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)acetate 26a following the procedure as described in compound 27. Yield, 50%; pale yellow solid, mp 172–174 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.78 (s, 2H, NH-CH2), 3.86 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 3.92 (s, 6H, 2X OCH3), 4.33 (s, 2H, CH2), 6.35 (s, 2H, Ar-H), 6.48 (d, J = 1.8 Hz, 1H, Ar-H), 6.65 (dd, J = 8.1, 1.8 Hz, 1H, Ar-H), 6.86 (d, J = 8.1 Hz, 1H, Ar-H), 7.17 (d, J = 15.9 Hz, 1H, =CH), 7.62 (d, J = 15.6 Hz, 1H, CH=). 13 C NMR (DMSO-d6, 75 MHz): δ 172.1, 163.5, 160.8, 146.3, 136.9, 132.7, 123.6, 121.3, 119.2, 111.9, 109.3, 102.5, 90.6, 60.3, 55.9, 55.5, 55.4, 44.4, 30.6. HRMS: m/z calcd [M - H] 450.1301; found 450.1311. Anal. (C21H25NO8S) C, H, N.
(E)-2-(2-Methoxy-5-((3′,4′,5′-trimethoxystyrylsulfonyl)methyl)phenylamino)acetic acid (27b)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(2-methoxy-5-((3′,4′,5′-trimethoxystyrylsulfonyl)methyl)phenylamino)acetate 26b following the procedure as described in compound 27. Yield, 55%; light yellow solid, mp 112–114 °C. 1H NMR (CDCl3, 400 MHz): δ 3.78 (s, 3H, OCH3), 3.79 (s, 6H, 2 X OCH3), 3.81 (s, 3H, OCH3), 3.87 (s, 2H, CH2), 4.15 (s, 2H, CH2), 6.44 (d, J = 1.8 Hz, 1H, Ar-H), 6.55 (d, J = 15.4 Hz, 1H, =CH), 6.59 (s, 2H, Ar-H), 6.62–6.66 (m, 1H, Ar-H), 6.69 (d, J = 8.2 Hz, 1H, Ar-H), 7.26 (d, J = 15.5 Hz, 1H, CH=). HRMS: m/z calcd [M - H] 450.1301; found 450.1314. Anal. (C21H25NO8S) C, H, N.
(E)-3-(2-Methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)propanoic acid (27c)
The title compound was obtained by the hydrolysis of (E)-ethyl 3-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)propanoate 26c following the procedure as described in compound 27. Yield, 51%; white solid, mp 130–132 °C. 1H NMR (CDCl3, 300 MHz): δ 2.56 (t, J = 6.3 Hz, 2H, CH2), 3.34 (t, J = 6.3 Hz, 2H, CH2), 3.75 (s, 6H, 2 X OCH3), 3.76 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 4.11 (s, 2H, CH2), 6.01 (s, 2H, Ar-H), 6.57 (s, 1H, Ar-H), 6.64–6.71 (m, 2H, Ar-H), 6.99 (d, J = 15.9 Hz, 1H, =CH), 7.76 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M - H] 464.1457; found 464.1471. Anal. (C22H27NO8S) C, H, N.
(E)-2-(2-Methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)propanoic acid (27d)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)propanoate 26d following the procedure as described in compound 27. Yield, 57%; light yellow solid, mp 116–118 °C. 1H NMR (DMSOd6, 400 MHz): δ 1.07 (d, J = 6.6 Hz, 3H, CH3), 3.57 (s, 3H, OCH3), 3.60 (s, 3H, OCH3), 3.64 (s, 6H, 2 X OCH3), 3.79–3.83 (m, 1H, CH), 4.06 (s, 2H, CH2), 6.08 (s, 2H, Ar-H), 6.26 (s, 1H, Ar-H), 6.35(d, J = 7.5 Hz, 1H, Ar-H), 6.58 (d, J = 8.1 Hz, 1H, Ar-H), 6.90 (d, J = 15.6 Hz, 1H, =CH), 7.35 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M - H] 464.1457; found 464.1446. Anal. (C22H27NO8S) C, H, N.
(E)-2″,2″-Difluoro-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)-phenylamino)acetic acid (27e)
The title compound was obtained by the hydrolysis of (E)-methyl 2″,2″-difluoro-2(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)-acetate 26e following the procedure as described in compound 27. Yield, 59%; white solid, mp 196–198 °C. 1H NMR (CDCl3, 300 MHz): δ 3.85 (s, 6H, 2 X OCH3), 3.87 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 4.12 (s, 2H, CH2), 6.12 (s, 2H, Ar-H), 6.58 (d, J = 8.1 Hz, 1H, Ar-H), 6.89 (d, J = 8.4 Hz, 1H, Ar-H), 7.06 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 7.29 (d, J = 15.6 Hz, 1H, =CH), 7.84 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M - H] 486.1112; found 486.1101. Anal. (C21H23F2NO8S) C, H, N.
(E)-3″,3″,3″-Trifluoro-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)-phenylamino)propanoic acid (27f)
The title compound was obtained by the hydrolysis of (E)-methyl 3″,3″,3″-trifluoro-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino) propanoate 26f following the procedure as described in compound 27. Yield, 53%; white solid, mp 180–182 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.78 (s, 3H, OCH3), 3.83 (s, 6H, 2 X OCH3), 3.87 (s, 3H, OCH3), 4.05 (d, J = 6.3 Hz, CH), 4.12 (s, 2H, CH2), 6.28 (s, 2H, Ar-H), 6.61 (d, J = 8.4 Hz, 1H, Ar-H), 6.76 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 7.01–7.09 (m, 1H, Ar-H), 7.10 (d, J = 15.6 Hz, 1H, =CH), 7.57 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M - H] 518.1175; found 518.1186. Anal. (C22H24F3NO8S) C, H, N.
(E)-2-(2-Methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)-2-methylpropanoic acid (27g)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)-2-methylpropanoate 26g following the procedure as described in compound 27. Yield, 61%; white solid, mp 142–144 °C. 1H NMR (CDCl3, 500 MHz): δ 1.48 (s, 6H, 2 X CH3), 3.83 (s, 6H, 2 X OCH3), 3.84 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 4.14 (s, 2H, CH2), 6.07 (s, 2H, Ar-H), 6.52 (s, 1H, Ar-H), 6.79 (d, J = 8.0 Hz, 1H, Ar-H), 6.85 (d, J = 8.1 Hz, 1H, Ar-H), 7.05 (d, J = 16.0 Hz, 1H, =CH), 7.78 (d, J = 15.5 Hz, 1H, CH=). HRMS: m/z calcd [M - H] 478.1614; found 478.1605. Anal. (C23H29NO8S) C, H, N.
(E)-2-(2-Methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)-2-phenylacetic acid (27h)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)-2-phenylacetate 26h following the procedure as described in compound 27. Yield, 60%; white solid, mp 124–126 °C. 1H NMR (CDCl3, 400 MHz): δ 3.75 (s, 6H, 2 X OCH3), 3.77 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 4.00(s, 2H, CH2), 4.86 (s, 1H, CH), 6.03 (s, 2H, Ar-H), 6.32 (d, J = 1.4 Hz, 1H, Ar-H), 6.66–6.69 (m, 2H, Ar-H), 6.94 (d, J = 15.6 Hz, 1H, =CH), 7.22–7.26 (m, 3H, Ar-H), 7.32–7.34 (m, 2H, Ar-H), 7.69 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M - H] 526.1614; found 526.1604. Anal. (C27H29NO8S) C, H, N.
(E)-2-(4″-Fluorophenyl)-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)-phenylamino)acetic acid (27i)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(4″-fluorophenyl)-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino) acetate 26i following the procedure as described in compound 27. Yield, 60%; light yellow solid, mp 82–84 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.79 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 4.14 (s, 2H, CH2), 4.42(s, 1H, CH), 6.29 (s, 2H, Ar-H), 6.33 (d, J = 2.0 Hz, 1H, Ar-H), 6.69 (dd, J = 8.5, 2.0 Hz, 1H, Ar-H), 6.73 (d, J = 8.5 Hz, 1H, Ar-H), 6.96–6.99 (m, 2H, Ar-H), 7.00 (d, J = 15.5 Hz, 1H, =CH), 7.37–7.40 (m, 2H, Ar-H), 7.76 (d, J = 16.0 Hz, 1H, CH=). HRMS: m/z calcd [M - H] 544.1520; found 544.1434. Anal. (C27H28FNO8S) C, H, N.
(E)-2-(4″-Chlorophenyl)-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)-phenylamino)acetic acid (27j)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(4″-chlorophenyl)-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino) acetate 26j following the procedure as described in compound 27. Yield, 60%; white solid, mp 172–174 °C. 1H NMR (CDCl3, 400 MHz): δ 3.55 (s, 6H, 2 X OCH3), 3.57 (s, 3H, OCH3), 3.60 (s, 3H, OCH3), 3.80 (s, 2H, CH2), 4.69(s, 1H, CH), 5.83 (s, 2H, Ar-H), 6.06 (d, J =1.8 Hz, 1H, Ar-H), 6.44 (dd, J = 8.1, 1.8 Hz, 1H, Ar-H), 6.47 (d, J = 8.1 Hz, 1H, Ar-H), 6.73 (d, J = 15.6 Hz, 1H, =CH), 7.00 (d, J = 7.8 Hz, 2H, Ar-H), 7.08 (d, J = 8.5 Hz, 2H, Ar-H), 7.49 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M - H] 560.1224; found 560.1212. Anal. (C27H28ClNO8S) C, H, N.
(E)-2-(4″-Bromophenyl)-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)-phenylamino)acetic acid (27k)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(4″-bromophenyl)-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino) acetate 26k following the procedure as described in compound 27. Yield, 60%; white solid, mp 178–179 °C. 1H NMR (DMSO-d6, 400 MHz): δ 3.93 (s, 6H, 2 X OCH3), 3.94 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 4.25 (s, 2H, CH2), 4.96(s, 1H, CH), 6.39 (s, 2H, Ar-H), 6.41 (d, J = 1.8 Hz, 1H, Ar-H), 6.69 (dd, J = 8.1, 1.8 Hz, 1H, Ar-H), 6.91 (d, J = 8.1 Hz, 1H, Ar-H), 7.14 (d, J = 15.6 Hz, 1H, =CH), 7.39 (d, J = 8.4 Hz, 2H, Ar-H), 7.57 (d, J = 8.4 Hz, 2H, Ar-H),7.61 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M - H] 604.0719; found 604.0731. Anal. (C27H28BrNO8S) C, H, N.
(E)-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)-2-(4″-methoxyphenyl)acetic acid (27l)
The title compound was obtained by the hydrolysis of (E)-(4″-Methoxyphenyl)-{2-methoxy-5-[2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonylmethyl]-phenylamino}acetic acid methyl ester 26l following the procedure as described in compound 27. Yield, 60%; white solid, mp 174–175 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.72 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 3.86 (s, 6H, 2 X OCH3), 3.87 (s, 3H, OCH3), 4.16 (s, 2H, CH2), 4.80(s, 1H, CH), 6.31 (s, 2H, Ar-H), 6.37 (d, J = 1.5 Hz, 1H, Ar-H), 6.62 (dd, J = 8.1, 1.5 Hz, 1H, Ar-H), 6.71 (d, J = 8.1 Hz, 1H, Ar-H), 6.82 (d, J = 9.0 Hz, 2H, Ar-H), 7.08 (d, J = 15.6 Hz, 1H, =CH), 7.27 (d, J = 8.7 Hz, 2H, Ar-H), 7.54 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M - H] 556.1720; found 556.1731. Anal. (C28H31NO9S) C, H, N.
Sodium (E)-2-(2-Methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)-acetate (ESTYBON ®) (ON 01910.Na) (28)
The title compound was obtained by the direct hydrolysis of (E)-methyl-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenyl amino)acetate 26a. To a solution of sodium hydroxide (3.95 g, 99 mmol) in water (11.5 mL) at 20 °C was added ethanol (40 mL), (E)-methyl 2-(2-methoxy-5-((2′,4′,6′-trimethoxystyryl sulfonyl)methyl)phenylamino)acetate 26a and dichloromethane (200 mL). The resulting mixture was stirred at room temperature for 3–4 h. After completion of the reaction monitored by TLC (chloroform/methanol, 9:1 on silica gel plate), charcoal was added and stirred for 30 min. The reaction mixture was filtered through celite and washed with ethanol (2 X 20 mL). The combined filterate was distilled at 50 °C until most of the solvent removed. Methyl Ethyl Ketone (70 mL) was added to the residue and distill of the methyl ethyl ketone at 50 °C. To the residue water (10 mL) was added and the resulting mixture was heated to 70 °C and maintained for 30 min. The reaction mixture cooled to room temperature and stirred for 2 h at room temperature. The solid formed filtered, washed with methyl ethyl ketone (2 X 20 mL) and dried to get the title compound 28. Yield, 80%; white solid, mp 174–178 °C. 1H NMR (D2O, 300 MHz): δ 3.46 (s, 2H, NH-CH2), 3.63 (s, 6H, 2 X OCH3), 3.72 (s, 3H, OCH3), 3.75(s, 3H, OCH3), 4.23 (s, 2H, CH2), 5.89 (s, 2H, Ar-H), 6.44 (d, J = 1.5 Hz, 1H, Ar-H), 6.59 (dd, J = 8.4, 1.5 Hz, 1H, Ar-H), 6.72 (d, J = 8.4 Hz, 1H, Ar-H), 6.91 (d, J = 15.6 Hz, 1H, =CH), 7.39 (d, J = 15.6 Hz, 1H, CH=). HRMS: m/z calcd [M + H] 510.1365; found 510.1378. Anal. (C21H24NNaO8S. 2H20) C, H, N.
Biology. Tissue Culture and Reagents
Paclitaxel was purchased from Sigma. Cell lines were purchased from ATCC. Cell lines were routinely grown in DMEM or RPM1 (CellGro) supplemented with 10% fetal bovine serum (Cell Generation Co) and 1 unit/mL penicillin-streptomycin (Gibco).
Cytotoxicity Assay
We have tested a number of tumor cell lines using a dose response end point assay system. The cells were grown in either DMEM or RPMI supplemented with 10% fetal bovine serum and 1unit/mL Penicillin-Streptomycin solution. The tumor cells were plated into 6 well dishes at a cell density of 1.0 × 105 cells/mL/well and compounds were added 24 h later at various concentrations. Cell counts were determined from duplicate wells after 96 h of treatment. The total number of viable cells was determined by trypan blue exclusion.
Flow Cytometry
Human prostate tumor cells, DU145 cells, and normal diploid human lung fibroblasts, HFL-1 cells, were grown in DMEM (Cellgro) supplemented with 10% fetal bovine serum and 1 unit/mL penicillin-streptomycin. The cells were plated onto 100 mm2 dishes at a cell density of 1.0 × 106 cells/dish, and 24 h later, they were treated with 2.5 μM of the compound. The cells were harvested 24 h after treatment. The cells were removed from the plate by trypsin digestion and combined with the non-attached cells found in the medium. The cell pellets were washed in phosphate buffered saline (PBS), and fixed in ice cold 70% ethanol for at least 24 h. The fixed cells were then washed with room temperature PBS and stained with propidium iodide (50 mg/mL) and RNase A (0.5 mg) for 30 min at 37 °C. The stained cells were then analyzed on a Becton-Dickinson (BD) (FACScan) flow cytometer and the data analyzed by cell cycle analysis software (Modfit, BD).
PARP Western
DU145 and HFL-1 cells were plated at a density of 3.0 × 106 cells per 150 mm2 plate and treated 24 h later with either DMSO or 28. The cells were collected 48 h treatment and cell pellets were frozen. The frozen cell pellets were lysed in 1% NP40/PBS lysis buffer containing protease inhibitors. Equal amounts of total cellular protein was then resolved on a 10%-SDS-polyacrylamide gel. The gels were transferred onto nitrocellulose paper (S/S), hybridized with anti-PARP antibodies (BD) and developed using ECL (Perkin-Elmer, MA) solution.
Cellular Viability and Caspase 3/7 Activity
Exponentially growing A549 cells were seeded in a white walled 96-well plate at a density of 3,600 cells/well in 100 μl of DMEM containing 10% FBS and 1% Pen/Strep. Cells were then allowed to adhere overnight at 37 °C in an incubator. The next day, cells were treated with varying concentrations of 28 or DMSO and then returned to the incubator. 24 h later, plates were removed from the incubator and 20 μL of CellTiter-Blue® Reagent (Promega Cat # G8080) was added individually to each well following manufacturer’s instructions. Plates were slowly shaken for 0.5 min and then returned to the incubator. After 3 h, fluorescence was read using a Glomax 96-well plate reader. Next, 120 μL of Caspase Glo® 3/7 Reagent (Promega Cat # G8090) was added to each well per manufacturer’s instructions. Plates were slowly shaken for 0.5 min and allowed to develop at room temperature for 2 h. At the end of this period, luminescence was read using a Glomax 96-well plate reader.
Nude mouse assay
Female athymic (NCR-nu/nu, Taconic) nude mice were injected with 0.5–1.0 x107 BT20 cells subcutaneously in the hind leg using a 1 mL tuberculin syringe equipped with a 271/2 gauge needle. Approximately 14 days later, mice were paired (N=9) and injected with 200 mg/Kg 28 or Phosphate buffered saline as the vehicle control. The intravenous injections were performed in the mouse tail vein using a 1 mL tuberculin syringe equipped with a 30 gauge needle. The animals were injected following a Q2D X 20 schedule. Tumor measurements (two dimensions) were done three times per week using traceable digital vernier calipers (Fisher). Tumor volume was calculated using the following equation: V= (Lx(S2)p/6, where L is the longer and S is the shorter of the two dimensions. Body weight was determined during each measurement. The animals were observed for signs of toxicity. The time of tumor volume doubling was calculated and the T-C value (difference in the average times post treatment for tumors of the treated groups to attain a doubling in volume compared to the average of the control group) was determined. We did not observe body weight loss of more than 10% in any group nor were there any animal deaths. All studies were performed under the guidelines of Temple University IACUC.
Supplementary Material
Acknowledgments
This work was supported by grants from NIH (CA 109820) and Onconova Therapeutics Inc.
Abbreviations
- MDS
myelodysplstic syndromes
- AML
acute myeloid leukemia
- PI3K
Phosphoinositide 3-kinase
- BIM
Bcl-2-interacting modulator of cell death
- FDA
food and drug Administration
- DNA
deoxyribonucleic acid
- CDKs
cyclin dependent kinases
- JNK
Janus kinase
- p-mTOR
mammalian target of rapamycin
Footnotes
Supporting Information Available: Elemental analysis data. This material is available free of charge via the Internet at http://pubs.acs.org Conflict of Interest Statement: Dr. E.P. Reddy is a stockholder, Board member, grant recipient and paid consultant of Onconova Therapeutics Inc. Dr. M.V.R. Reddy is a stock holder and paid consultant of Onconova Inc. Dr. S. Cosenza is a paid consultant of Onconova Therapeutics Inc.
References
- 1.Reddy MVR, Mallireddigari MR, Cosenza SC, Pallela VR, Iqbal NM, Robell KA, Kang AD, Reddy EP. Design, Synthesis, and Biological Evaluation of (E)-Styrylbenzylsulfones as Novel Anticancer Agents. J Med Chem. 2008;51:86–100. doi: 10.1021/jm701077b. [DOI] [PubMed] [Google Scholar]
- 2.Gumireddy K, Reddy MVR, Cosenza SC, Boominathan R, Baker SJ, Papathi N, Jiang J, Holland J, ReddY EP. ON01910, A non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer cell. 2005;7:275–286. doi: 10.1016/j.ccr.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Scott BL, Deeg HJ. Myelodysplastic Syndromes. Annu Rev Med. 2010;61:345–358. doi: 10.1146/annurev.med.051308.132852. [DOI] [PubMed] [Google Scholar]
- 4.Disperati P, Ichim CV, Tkachuk D, Chun K, Schuh AC, Wells RA. Progression of myelodysplasia to acute lymphoblastic leukaemia: Implications for disease biology. Leuk Res. 2006;30:233–239. doi: 10.1016/j.leukres.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 5.Mufti GJ, Bennett JM, Goasguen J, Bain BJ, Irith B, Brunning R, Cazzola M, Fenaux P, Germing U, Hellstrom-Lindberg E, Jinnai I, Manabe A, Matsuda A, Niemeyer CM, Sanz G, Tomonaga M, Vallespi T, Yoshimi A. Diagnosis and classification of myleodysplastic syndrome: international Working Group on Morphology of myelodysplastic syndrome (IWGM-MDS) consensus proposals for the definition and enumeration of myeloblasts and ring sideroblasts. Haematologica. 2008;93:1712–1717. doi: 10.3324/haematol.13405. [DOI] [PubMed] [Google Scholar]
- 6.Prasad A, Park IW, Allen H, Zhang X, Reddy MVR, Boominathan R, Reddy EP, Groopman JE. Styryl sulfonyl compounds inhibit translation of cyclin D1 in mantle cell lymphoma cells. Oncogene. 2007;26:5635–5642. doi: 10.1038/onc.2008.502. [DOI] [PubMed] [Google Scholar]
- 7.(a) Reddy MVR, Reddy S. Synthesis of α, β-Unsaturated Suflones. Acta Chim Hung. 1984;115:269–271. [Google Scholar]; (b) Reddy DB, Reddy NS, Reddy MVR, Balasubramanyam S. Preparation of styryl benzyl sulfones and 1, 2-bis-(styrylsulfonyl-methyl)-4,5-dimethylbenzenes. Org Prep Proc Int. 1988;20:205–212. [Google Scholar]
- 8.Reddy AK, Lohray BB, Bhushan V, Reddy AS, Mamidi NVSR, Reddy PP, Saibaba V, Reddy NJ, Suryaprakash A, Misra P, Vikramadithan RK, Rajagopalan R. Novel Antidiabetic and Hypolipidemic Agents.5. Hydroxyl versus Benzyloxy Containing Chroman Derivatives. J Med Chem. 1999;42:3265–3278. doi: 10.1021/jm9805541. [DOI] [PubMed] [Google Scholar]
- 9.Pinney KG, Mejia MP, Villalobos VM, Rosenquist BE, Pettit GR, Verdier-Pinard P, Hamel E. Synthesis and Biological Evaluation of Aryl Azide Derivatives of Combretastatin A-4 as Molecular Probes for Tubulin. Bioorg Med Chem. 2000;8:2417–2425. doi: 10.1016/s0968-0896(00)00176-0. [DOI] [PubMed] [Google Scholar]
- 10.Beugelmans R, Roussi G, Zamora EG, Carbonnelle A-C. Synthetic Studies Towards Western and Eastern Macropolypeptide Subunits of Kistamycin. Tetrahedron. 1999;55:5089–5112. [Google Scholar]
- 11.Kelly JL, Linn JA, Selway JWT. Synthesis and Antirhinovirus Activity of 6-(Dimethylamino)-2-(trifluoromethyl)-9-(substituted benzyl)-9H-purines. J Med Chem. 1989;32:1757–1763. doi: 10.1021/jm00128a016. [DOI] [PubMed] [Google Scholar]
- 12.(a) Reddy MVR, Reddy S, Reddy DB. Facile method for the synthesis of 2-(arylsulfonyl)-1-phenyl-3-aryl-2-propen-1-ones. Sulfur Lett. 1987;7:43–48. [Google Scholar]; (b) Russell Llyod B, Anthony D, Chantal Renee F, Richard Francis L. Novel Knoevenagel condensation of a β-keto sulfone and a β-carboalkoxy sulfone. Sulfur Lett. 1999;23:11–31. [Google Scholar]; (c) Reddy MM, Venkat RP, Reddy EP, Reddy MVR. Sequential Reduction and Dehydration of Phenacyl-(E)-Styryl Sulfones to Unsymmetrical (E, E)-Bis(styryl) Sulfones. Synthesis. 2005:3639–3643. [Google Scholar]; (d) Touati A, Cazaux L. Synthesis of sulfonamides, sulfonates and thiosulfonates which are inhibitors of coniferyl alcohol dehydrogenase. J Soc Alger Chim. 1996;6:39–52. [Google Scholar]
- 13.Gee SJ, Miyamoto T, Goodrow MH, Buster D, Hammock BD. Development of an Enzyme-Linked Immunosorbent Assay for the Analysis of the Thiocarbamate Herbicide Molinate. J Agric Food Chem. 1988;36:863–870. [Google Scholar]
- 14.Ichinose Y, Wakamatsu K, Nozaki K, Birbaum J-L, Oshima K, Utimoto K. Et3B Induced Radical Addition of Thiols to Acetylenes. Chem Lett. 1987:1647–1650. [Google Scholar]
- 15.Ravikumar KS, Zhang YM, Begue J-P, Bonnet-Delpon D. Role of Hexafluoro-2-propanol in Selective Oxidation of Sulfide to Sulfoxide: Efficient Preparation of Glycosyl Sulfoxides. Eur J Org Chem. 1998:2937–2940. [Google Scholar]
- 16.Nielsen SF, Kharazmi A, Christensen SB. Modification of the α, β-Double Bond In Chalcones only Marginally Affect the Antiprotozoal Activities. Bioorg Med Chem. 1998;6:937–945. doi: 10.1016/s0968-0896(98)00051-0. [DOI] [PubMed] [Google Scholar]
- 17.Webb KS. A Mild, Inexpensive and Practical Oxidation of Sulfides. Tet Lett. 1994;35:3457–3460. [Google Scholar]
- 18.Grever MR, Schepartz SA, Chabner BA. The National Cancer Institute:Cancer Drug Discovery and Development Program. Seminars in Oncology. 1992;19:622–663. [PubMed] [Google Scholar]
- 19.Harker WG, Sikic BI. Multidrug (pleiotropic) resistance in doxorubicin selected variants of the human sarcoma cell line MES-SA. Cancer Res. 1985;45:4091–4096. [PubMed] [Google Scholar]
- 20.Fujimori A, Arker WG, Kohlhagen G, Hoki Y, Pommier Y. Mutation at the catalytic site of topoisomerase 1 in CEM/C2, a human leukemia cell line resistant to camptothecin. Cancer Res. 1995;55:1339–1346. [PubMed] [Google Scholar]
- 21.Soldani C, Scovassi AI. Poly (ADP-ribose) polymerase-1 clevage during apoptosis: an update. Apoptosis. 2002;7:321–328. doi: 10.1023/a:1016119328968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.