

Abstract
Mitral regurgitation, and other conditions marked by a pure isolated volume overload (VO) of the heart, result in a progressive form of eccentric left ventricular (LV) remodeling and dysfunction. As opposed to the more extensively studied pressure overload, there are no approved medical therapies because an understanding of the underlying pathological mechanisms at work in VO is lacking. Over the past twenty years, our laboratory has identified multiple key biological functions involved in the pathological remodeling in VO. Specifically, we have noted perturbed matrix homeostasis, detrimental adrenergic signaling, increased intracellular reactive oxygen species, and an intense inflammatory response that implicates mast cells and their product chymase, which appears to cause extensive remodeling both inside and outside the cardiomyocyte. How these multiple pathways intersect over the course of VO and their response to various single and combined interventions are now the subject of intense investigation.
Keywords: Mitral Regurgitation, Volume Overload, Extracellular Matrix, Myocardial Inflammation, Adrenergic Drive, Oxidative Stress, LV dysfunction, LV remodeling
Introduction
Isolated mitral regurgitation (MR) is defined as MR due to intrinsic valvular disease. It is different from ischemic MR or functional MR, in which the LV dilatation occurs due to ischemic heart disease or cardiomyopathy that can cause the MR despite normal mitral valve leaflets. Mitral regurgitation creates a unique hemodynamic stress by inducing a low pressure form of volume overload (VO) caused by ejection into the low pressure left atrium. Mechanisms of LV dysfunction in isolated MR are not well understood, nor are there any approved medical therapies for this condition. Vasodilator therapy in other forms of LV dysfunction reduces LV wall stress and improves LV function; however, studies in isolated MR show no beneficial effect on LV remodeling. This is also the case for standard medical therapy with renin-angiotensin system blockade.
Work in this laboratory was the first to show that, as opposed to other forms of heart failure in which renin-angiotensin system (RAS) blockade has been highly successful, chronic VO is associated with loss of interstitial collagen surrounding cardiomyocytes1 and a primary upregulation of the kallikrein kinin system.2 This upregulation of the kallikrein kinin system promotes collagen loss, as well as inflammatory cell infiltration, and neither are improved by ACE inhibitor therapy.1,3 Studies of prolonged inflammatory cytokine or inflammatory cell blockade in human heart failure with pure volume overload have not met success. Mechanistic studies of a pure VO are further fueled by the difficulty in determining optimal timing for surgical intervention in valvular regurgitation because of the well defined decrease in LV ejection fraction, and the not uncommon incidence of heart failure post-valve repair/replacement. These problems may be improved by an appropriate medical therapy, which has yet to be determined, for the extended period of observation of chronic MR prior to surgery.
Extracellular Matrix in LV Remodeling in VO
To better understand why RAS blockade fails to improve remodeling and function in both the dog with experimentally induced MR,1,4 and the rat with aorto-caval fistula (ACF)3,5, we performed gene arrays in both animal models.6–8 In the dog with four months of MR, there is a significant downregulation of multiple noncollagen microfibillar and glycoprotein genes essential to collagen assembly and total extracellular matrix (ECM) structure,6 which matches the loss of collagen weave that connects individual cardiomyocytes. We also found marked upregulation of pro-fibrotic growth factors. In particular, there is a down-regulation of transforming growth factor-beta (TGF-β) mRNA and protein activity, but upregulation of matrix metalloproteinase (MMP) genes and the antifibrotic kallikrein kinin system (Figure 1). In the rat with 15 weeks of ACF, we find patchy areas of LV endocardial perivascular fibrosis, despite the loss of collagen surrounding cardiomyocytes (Figure 2).7 As in the dog, there is no increase in hydroxyproline at any time point, despite the increase in multiple mRNAs for various collagens at two weeks, thereby reinforcing the failure to increase ECM production despite an increase in LV angiotensin II (Ang II) levels in both animal models at early and late time points.1,2,3,9 Taken together, these findings suggest a compartmentalization of collagen homeostasis in the volume overloaded heart, with collagen loss surrounding cardiomyocytes persisting throughout the course of volume overload, and a perivascular fibrosis in later stages largely in the LV endocardium, which is subjected to higher wall stress and lower myocardial blood flow due to the elevation in LV diastolic filling pressures.
Figure 1.

(A). Heatmap of the 659 genes altered greater than 1.5 fold (p<0.05) in the four week MR dog vs. baseline (1–4). Two-color gene array with dye swap was applied. Red = upregulation, black = no change, green = downregulation vs. baseline with scale of color corresponding to magnitude of fold-change. (B) Genes altered in MR related to ECM structure and (C) TGF-β pathway and ECM degradation. Previously reported in Zheng et al., Circulation 2009.
Figure 2. Expression of extracellular matrix protein in LV of sham and ACF rats.

LV interstitial collagen measured by (A) PASR staining and (B) hydroxyproline content in 2, 5 and 15 week age-matched sham and ACF rats. (C) Representative images of perivascular and endocardial collagen staining by PASR. Excessive collagen accumulation is observed in perivascular LV areas in ACF rats. (D) Representative gel-zymography image demonstrating MMP-2 activity in 2, 5 and 15 weeks sham and ACF rats. Results are expressed as fold-changes to corresponding control. (E) Representative western blot with tubulin loading controls and (F) immunohistological staining of periostin in age-matched sham and ACF rats. Endo: endocardium; V: vessel. Bars: 20um. *P<0.05, **P< 0.01 vs. sham. Previously reported in Chen et al., Am J Physiol, 2011.
In addition to the lack of global extracellular matrix production in the pure VO, McDermot and coworkers report that chronic isolated MR in the dog does not trigger substantial increases in the rate of protein synthesis, but instead a decrease in the rate of protein degradation.10 This matches the modest (at most) increase in LV mass seen in this model and is consistent with studies from both the Carabello laboratory11 and our laboratory12,13, which demonstrate extensive myofibrillar degeneration/loss with the characteristic long and thin cardiomyocytes. In preliminary studies by the late Dr. George Cooper years ago, he found a greater than two-fold decrease in total tubulin in the MR dog heart (unpublished data). Careful inspection of Western blots in Figure 3 demonstrates that, despite equal protein loading in the 15 week ACF rats, there is a decrease in tubulin expression. Thus, in the pure VO, the loss of extracellular interstitial collagen is matched by an intracellular loss of cytoskeletal and myofibrillar proteins. Current experiments in our laboratory are studying the connection of increased cardiomyocyte oxidative stress and MMP activation as the cause of myofibrillar and cytoskeletal breakdown. However, the lack of adequate replacement of collagen and myofibrils is a question that requires further investigation.
Figure 3. Heatmap demonstrating upregulated inflammation-related genes identified by DAVID analysis and inflammatory cell infiltration and protein expression of inflammation related genes.

(A) Upregulated genes related to inflammation and immune response at 2, 5 and 15 week. (B) Number of neutrophils (MPO+) and (C) macrophages (CD68+) in ACF and age-matched shams at each time point. Representative western blots demonstrating expression of (D) NF-κB p65, (E) HMOX-1 (heme oxygenase 1) with densitometric analysis after normalization to tubulin. *P < 0.05, **P < 0.01 vs. sham. Previously reported in Chen et al., Am J Physiol, 2011.
In part related to this “failure of hypertrophy” is the loss of cellular surface proteins that connect the collagen weave to the cell surface, resulting in the dephosphorylation of focal adhesion kinase (FAK) in both the dog with isolated MR14 and rat with ACF.15 An intact focal adhesion complex is essential for cell growth, survival, and myofibril assembly. As opposed to the volume overloaded MR heart, increased FAK phosphorylation has been documented in animal models of pressure overload.16 Interestingly, mice with selective inactivation of cardiomyocyte FAK demonstrate increased myofibrillar degeneration, elongation and thinning of cardiomyocytes, and eccentric LV remodeling and heart failure after six months.17 The disruption of FAK and cellular integrins are also associated with disruption, disorganization, and decreased respiratory capacity of subsarcolemmal mitochondria early in the course of volume overload in the rat with ACF, and this is prevented by MMP inhibition in the acute 24 hour ACF.18
Inflammatory Cell Infiltration in Volume Overload
The pure stretch of VO also demonstrates a molecular and cellular inflammatory response that contributes to MMP activation and ECM degradation. This inflammatory feature is consistent with an early and persistent increase in mast cells and chymase activity in the MR dog and ACF rat.1,2,3,5,9 As opposed to the constant increase in mast cells, there is a cyclical pattern of polymorphonuclear and macrophage infiltration only seen in the very early (acute) and late (decompensated) stages of ACF in the rat (Figure 3).7 Mast cells contain a collection of cytokines and proteolytic enzymes, including tryptase and chymase, which are released upon stress and activate MMPs. Mast cell stabilizing drugs have been reported to improve LV remodeling in the rat.19 However, in the dog with chronic MR, long term treatment with a mast cell stabilizer had no effect on LV remodeling and worsened cardiomyocyte shortening and calcium transients;20 this was most likely due to the stabilizers’ calcium entry blocking effect, which is essential in preventing mast cell degranulation.
As with studies in human heart failure, prolonged inflammatory cytokine or inflammatory cell blockade in pure volume overload have not met with success. Neutrophil infiltration blockade attenuates the MMP activation, ECM degradation, and myocyte apoptosis induced by ACF at 24 hours, as well as the development of eccentric hypertrophy induced by ACF at 2 and 3 weeks. In spite of this acute protective effect, however, sustained neutrophil depletion for more than 4 weeks results in adverse cardiac remodeling with further increase in cardiac dilatation and macrophage infiltration.21 In a similar fashion, we have found that chronic TNF-α blockade failed to prevent eccentric LV remodeling or improve LV function after 2 or 4 weeks of ACF. It is of interest that the potent antioxidant protein, HMOX-1, was upregulated 5-fold at 2 weeks ACF and was significantly decreased by TNF-α blockade (Figure 4). Further, the increase in HMOX-1 is directly linked to TNF-α and not the hemodynamic stress of VO, because TNF-α neutralization decreased HMOX-1 transcript and protein expression but did not affect LV end-diastolic dimension (LVEDD) or pressure (LVEDP). Thus, TNF-α blockade actually abrogates the potentially beneficial effects of HMOX-1 and other cell-protective molecules in the course of volume overload. Further, it is now established that TNF receptor 1 exacerbates, and TNF receptor 2 ameliorates, LV remodeling, hypertrophy, NF-κB, inflammation, and apoptosis in heart failure, suggesting that future drug strategies should specifically target TNF receptor 1 effects.22 Finally, our gene array results from the volume overload of ACF in the rat demonstrate that the 24 hour,8 2 week, and 15 week time points7 are marked by global inflammatory gene expression, while the 5 week interval was relatively quiescent for inflammatory gene expression. Thus, the expression of inflammatory cytokines in the MR heart may be dependent upon the clinical stage of LV remodeling and/or heart failure symptoms.
Figure 4. Western blot of HMOX-1 in 2 week ACF rats.
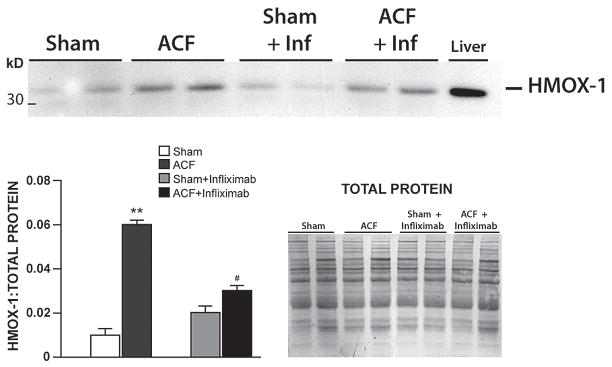
HMOX-1 in sham, ACF, sham + infliximab (Inf) and ACF + Inf with total protein demonstrated by commassie stain. **P < 0.01 vs. sham, # P < 0.01 vs. ACF. Unpublished data.
At this time it is not clear how the extracellular matrix loss in the pure volume overload can be therapeutically targeted, especially in view of the results in other models which demonstrated that prolonged MMP inhibition results in excessive collagen deposition (fibrosis), and that blockade of the bradykinin type 2 (BK2) receptor results in a marked increase in blood pressure.23 It is of interest that kallikrein, interstitial fluid bradykinin, and ACE2 activity are increased in rats with the VO of ACF at 4 and 15 weeks.2 These in vivo anti-fibrotic responses in the rat with ACF were also recapitulated in isolated adult rat fibroblasts and cardiomyocytes after cyclical cell stretch, demonstrating increases in Kallikrein mRNA and bradykinin and MMP-2 activation.2 Interestingly, blockade of kallikrein with the nonspecific protease inhibitor aprotinin prevented the decrease in collagen with chronic ACF and improved systolic function while having no effect on LV dilatation.2
Chymase has offered an interesting target for ECM degradation because mast cell infiltration and chymase activity are increased at all stages of VO in the rat and dog.1–3,5–9,23 In addition to an Ang II-forming capacity from Ang I that is 20-fold higher than ACE,24–26 chymase directly activates MMPs,27–30 degrades fibronectin,31 and activates kallikrein.32 The addition of chymase to isolated smooth muscle cells33 or neonatal cardiomyocytes34 results in cell death due to chymase degradation of cell adhesion proteins and disruption of the focal adhesion complex. A newly described propeptide cleaved from angiotensinogen, Ang-(1–12), may be an alternate substrate for the formation of biologically active angiotensins.35,36 Although the enzymatic mechanism responsible for cardiac Ang-(1–12) formation from angiotensinogen remains an open question, a recent study suggested a chymase-mediated mechanism in the ischemia/reperfusion rat heart.37 In collaboration with Ferrario and coworkers we have reported that this chymase-mediated Ang II formation from Ang-(1–12) represents the predominant mechanism of Ang II formation in the left atria of humans undergoing corrective surgery for chronic atrial fibrillation38, and also in the human LV.39
It is important to note that the many unique features of isolated MR in the dog—MMP activation, increased bradykinin, ECM loss, disruption of the focal adhesion complex, and cardiomyocyte myofibrillar loss—are all either directly or indirectly beneficially affected by chronic inhibition of chymase, which leads to an improvement in LV systolic function and cardiomyocyte shortening.13
Increased Adrenergic Drive in VO
Another key factor in the pure VO is the role of increased adrenergic drive, which is central to both early and late stage volume overload of MR in both animal models40–42 and humans.43 Prolonged excessive adrenergic stimulation has a cytotoxic effect on cardiomyocytes,44 in particular breakdown and loss of myofibrillar proteins in the cardiomyocyte of the dog and human with isolated MR.11–13 Using a cardiac microdialysis technique in open-chest, anesthetized dogs in the early two and four week phase of the volume overload of isolated MR, we have shown that maximal electrical stimulation of the stellate ganglion activates intrathoracic adrenergic neurons to release norepinephrine (NE) and epinephrine (EP) into the cardiac interstitial fluid (ISF), and that β1 receptor blockade (β1-RB) mitigates the excessive sympathetic neuronal EP and NE release.45,46 Neural ganglia cells in the heart express plentiful β receptors, and therefore a paracrine/autocrine feed-forward loop can be created where EP acts on the neuronal β2 receptors and stimulates further release of NE and EP.40,41 This positive feedback can lead to abrupt and substantial increases in catecholamine exposure of cardiomyocytes, fibroblasts, and other cardiac cells. After synaptic release, NE and EP undergo metabolism and can be cleared by neurons (uptake 1); NE and EP can also be taken up by non-neuronal cells (uptake 2).47 The remaining NE and EP escape these processes and enter the general circulation, and this is known as “spillover”. Importantly, our studies in the dog indicate that measuring trans-cardiac plasma NE and EP concentrations substantially underestimates sympathetic efferent neuronal NE and EP release into the cardiac ISF space.48 Thus, in addition to the direct effects on cardiomyocyte toxicity, β1-RB provides the additional benefit of reducing further catecholamine release form neural ganglia cells into the ISF during volume overload.
The combined loss of interstitial collagen with cardiomyocyte myofibrillar proteins and breakdown of the focal adhesion complex are improved by β1-RB.14,15 In the dog with isolated MR, β1-RB also results in significant improvements in cardiomyocyte function, calcium homeostasis, and LV function, but no improvement in extracellular matrix loss, LV dilatation, or cardiomyocyte elongation.11,49 In patients with severe MR and normal LV ejection fraction (EF), β-RB is associated with a survival benefit in patients, regardless of the presence or absence of coronary artery disease.50 We have completed a phase IIb clinical trial of 38 asymptomatic subjects with moderate to severe isolated MR, who were randomized to either Placebo or β1-RB (Toprol-XL; average dose of drug: 75 mg q day) for two years.51 Magnetic resonance imaging with 3-dimensional analysis was performed at baseline and at approximate 6-month intervals. Rate of progression analysis was performed for primary end-point variables. Significant treatment effects were found on LVEF (p=0.0060) and LV early diastolic filling rate (p=0.0011), such that in untreated patients these systolic and diastolic parameters decreased over time on an intention to treat analysis. However, β1-AR blockade, as in the dog with isolated MR, does not attenuate the LV dilatation. Thus, it would appear that chronic β1-RB protects the cardiomyocyte from excessive adrenergic drive; but the question remains, is valve surgery the only treatment that can arrest the continued LV dilatation.
Increased Cardiomyocyte Oxidative Stress in VO
A major step forward in identifying a potentially important target in an isolated volume overload was recently accomplished by examination of LV biopsies taken from patients with isolated MR. These patients had decreased LV systolic function 6 months post-mitral valve (MV) repair, despite LV ejection fractions (EF) > 60% prior to surgery.12 These LVs had significant cardiomyocyte myofibrillar degeneration along with increased protein nitration and lipofuscin accumulation, consistent with an increased formation of reactive oxygen and nitrogen species, and activity of the protein xanthine oxidase (XO). In addition to being a major source of reactive oxygen species, XO is linked to bioenergetic dysfunction since its substrates derive from ATP catabolism. Correspondingly, there was also evidence of aggregates of small mitochondria in cardiomyocytes, which is generally considered a response to a cellular bioenergetic deficit.
To determine whether XO activation is linked to the hemodynamic stress of VO, we induced a 24 hour ACF in adult rats and isolated cardiomyocytes from sham and ACF left ventricles. Isolated cardiomyocytes from ACF LVs demonstrated increased XO activity, hydrogen peroxide and superoxide formation, increased MMP activity, and disruption of the subsarcolemmal mitochondrial network (Figure 5).18,51 The global MMP-inhibitor PD166793 preserved interstitial collagen, integrin-α5 and the SSM structural arrangement, and prevented the decrease in state 3 mitochondrial respiration. These studies established a link between cardiomyocyte oxidative stress, MMP activation, and a disruption of the cytoskeletal network, which is a new emerging field for non-collagen targets of MMPs in cardiomyocyte ischemia/reperfusion injury.52
Figure 5. Oxidative stress in cardiomyocytes 24hr ACF.

(A) Fluorescence microscopy of cardiomyocytes from sham and 24hr ACF rats probed with H2DCFDA and DHE dyes for H2O2 and O2·− formation (respectively). (B) Quantitation of fluorescence intensity in cardiomyocytes from sham (white) and ACF (grey) rats. *p<0.05 vs. sham n=3 rats/group. (C) XO activity in sham and 24 hr ACF rat cardiomyocytes. (D) Overlay of collagen I and III, and Integrin α-5 with cytochrome oxidase 4 CcOX4 a marker of mitochondrial density and DAPI at higher power demonstrates a prominent loss of CcOX4 and Integrin α-5 in the subsarcolemmal area of cardiomyocytes in the ACF LV, which is restored by global MMP inhibitor PD 166793. Previously reported in Ulasova et al., J Mol Cell Cardiol, 2011.
To gain further insight into the role of oxidative stress in MR, we subsequently showed that cyclical stretch of adult rat cardiomyocytes caused increased oxidative stress, mitochondrial swelling, and cytoskeletal and myofibrillar disruption—all of which were prevented by XO inhibitor or the mitochondrial targeted antioxidant drug MitoQ (Figure 6).51 More importantly, MitoQ prevented XO activation and myofibrillar degeneration, suggesting that the mitochondria themselves represent a more direct target in preventing oxidative stress, XO activation, and cellular remodeling in cardiomyocyte stretch. As mentioned previously, increased adrenergic drive is central to early and late stage MR. In the dog and human with isolated MR, β1-RB results in improvement in cardiomyocyte function, calcium homeostasis, and LV function, but no improvement in LV dilatation and cardiomyocyte elongation. However, β1-RB in the dog with isolated MR has no effect on a two-fold increase in LV XO activity (unpublished data), suggesting that additional protection against cardiomyocyte mitochondrial reactive oxygen species (ROS) production may be synergistic with β1-RB in the treatment of isolated MR. These studies have led us to hypothesize that the pure VO of isolated MR causes continual cellular damage emanating from pure stretch-mediated mitochondrial ROS production. This, when combined with continual and mast cell infiltration and degranulation, sets up a self-perpetuating process of cytoskeletal and myofibrillar breakdown without replacement, as described by McDermott and colleagues. Taken together, this suggests that a combined therapeutic approach of β1-RB and direct targeting of cardiomyocyte oxidative stress may be required to attenuate the adverse LV remodeling in VO.
Figure 6. Cyclical stretch causes myofibrillar structural damage and mitochondrial swelling in rat cardiomyocytes.

Upper panels: Transmission electron microscopic TEM) images of unstretched cardiomyocyte control, control + allopurinol (allo, 250 μM), control + Mito-Q (50 nM); 4,500x and 17,000x. Lower panels: TEM images of cardiomyocytes after 3 hour stretch treated with 250 μM allo or 50 nM Mito-Q. Stretch causes breakdown of myofilaments and Z-line (black arrows) and mitochondrial swelling (white arrows) in stretched cells vs. controls. Z-line structural integrity and mitochondrial morphology are preserved by MitoQ. Previously reported in Gladden et al., Free Rad Biol Med, 2011.
Summary and Future Perspectives
The marked increase in oxidative stress in our in vitro studies of stretched cardiomyocytes—that is prevented by MitoQ—has now directed our focus to prevention of ROS release form the numerous mitochondria in the cell (which occupy 40% of the cardiomyocyte interior volume). We are now testing the hypothesis that inhibition of mitochondrial ROS production will prevent: 1) MMP activation and digestion of cytoskeletal components, 2) the impressive loss of myofibrils, and 3) the disruption of interfibrillar mitochondrial structure and their connection to contractile units of the sarcomere. Figure 7 demonstrates the underlying myocardial events that are operating throughout the course of volume overload. Future directions will study the how the increased adrenergic drive, protease (chymase) and MMP activation, and mitochondrial-derived ROS production are interrelated and whether these are independent or interrelated processes in the pathophysiology of the unique hemodynamic stress of a pure stretch on the myocardium without a systolic pressure overload.
Figure 7. Mechanisms in the LV remodeling in a pure volume overload.

VO has a number of simultaneous events with acute induction including increased adrenergic drive and cardiomyocyte oxidative stress. In addition, there is an acute influx of macrophages and neutrophils that abate while mast cell infiltration and degranulation continue throughout the course of VO. The release of chymase into the cardiac interstitium is associated with an increase in interstitial Ang II but also activation of MMPs and degradation of fibronectin, which combined with increased oxidative stress, leads to cardiomyocyte elongation and adverse LV eccentric remodeling. The failure to produce collagen despite an increase in Ang II may be counterbalanced by an increase in Kallikrein/bradykinin and ACE2, which also result in the antifibrotic effects of Ang-(1–7). For all of these reasons, ACE inhibition increases BK2–dependent chymase release from mast cells (60), which may further counteract a direct effect of ACE inhibition on Ang II formation by activating the chymase pathway of Ang II formation via Ang I and Ang-(1–12).
References
- 1.Dell’Italia LJ, Balcells E, Meng QC, Su X, Schultz D, Bishop SP, Machida N, Straeter-Knowlen I, Hankes GH, Dillon R, Cartee RE, Oparil S. Volume overload cardiac hypertrophy is unaffected by ACE inhibitor treatment in the dog. Am J Physiol. 1997;273:H961–H970. doi: 10.1152/ajpheart.1997.273.2.H961. [DOI] [PubMed] [Google Scholar]
- 2.Wei CC, Chen Y, Powell LC, Zheng J, Shi K, Bradley WE, Powell PC, Ahmad S, Ferrario CM, Dell’Italia LJ. Cardiac kallikrein-kinin system is upregulated in chronic volume overload and mediates an inflammatory induced collagen loss. PLoS ONE. 2012;7(6):e40110. doi: 10.1371/journal.pone.0040110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei CC, Lucchesi PA, Tallaj J, Bradley WE, Powell P, Dell’Italia LJ. Cardiac interstitial bradykinin and mast cells modulate the pattern of left ventricular remodeling in volume overload in the rat. Am J Physiol. 2003;54:H784–H792. doi: 10.1152/ajpheart.00793.2001. [DOI] [PubMed] [Google Scholar]
- 4.Perry GJ, Wei CC, Su X, Bishop SP, Hankes GH, Dillon GH, Mukherjee R, Spinale FG, Dell’Italia LJ. Afterload reduction and blockade of the tissue renin angiotensin system does not improve left ventricular and cardiomyocyte remodeling in chronic mitral regurgitation. J Am Coll Cardiol. 2002;39:1374–1379. doi: 10.1016/s0735-1097(02)01763-1. [DOI] [PubMed] [Google Scholar]
- 5.Ryan TD, Rothstein E, Aban I, Tallaj J, Husain A, Lucchesi PA, Dell’Italia LJ. Left ventricular eccentric remodeling is mediated by bradykinin and precedes myocyte elongation in rats with volume overload. J Am Coll Cardiol. 2007;49:811–821. doi: 10.1016/j.jacc.2006.06.083. [DOI] [PubMed] [Google Scholar]
- 6.Zheng J, Chen Y, MD, Pat B, Dell’Italia LA, Tillson M, Dillon AR, Powell P, Shi K, Shah N, Denney T, Husain A, Dell’Italia LJ. Microarray identifies extensive downregulation of noncollagen extracellular matrix and pro-fibrotic growth factor genes in chronic isolated MR in the dog. Circulation. 2009;119:2086–2095. doi: 10.1161/CIRCULATIONAHA.108.826230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y, Pat B, Gladden JD, Zheng J, Powell P, Wei CC, Cui X, Darley-Usmar V, Husain A, Dell’Italia LJ. Dynamic molecular and histopathological changes in inflammation and extracellular matrix turnover in the transition to heart failure in isolated volume overload. Am J Physiol. 2011;300:H2251–H2260. doi: 10.1152/ajpheart.01104.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y-W, Pat B, Zheng J, Cain L, Powell P, Sabri K, Husain A, Dell’Italia LJ. Tumor Necrosis Factor-α Produced in Cardiomyocytes Mediates a Predominant Myocardial Inflammatory Response to Stretch in Early Volume Overload. J of Mol Cell Cardiol. 2010;49:70–78. doi: 10.1016/j.yjmcc.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stewart JA, Jr, Wei CC, Brower GL, Rynders PE, Hankes GH, Dillon AR, Lucchesi PA, Janicki JS, Dell’Italia LJ. Cardiac mast cell- and chymase-mediated matrix metalloproteinase activity and left ventricular remodeling in mitral regurgitation in the dog. J Mol Cell Cardiol. 2003;35(3):311–319. doi: 10.1016/s0022-2828(03)00013-0. [DOI] [PubMed] [Google Scholar]
- 10.Matsuo T, Carabello BA, Nagamoto Y, Koide M, Hamawake M, Zile MR, McDermott PJ. Mechanisms of cardiac hypertrophy in canine volume overload. Am J Physiol. 1998;275:H65–H74. doi: 10.1152/ajpheart.1998.275.1.h65. [DOI] [PubMed] [Google Scholar]
- 11.Tsutsui H, Spinale FG, Nagatsu M, Schmid PG, Ishihara K, DeFreyte G, Cooper G, Carabello BA. Effects of chronic beta-adrenergic blockade on the left ventricular and cardiocyte abnormalities of chronic canine mitral regurgitation. J Clin Invest. 1994;93:2639–2648. doi: 10.1172/JCI117277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmed M, Gladden JD, Litovsky S, McGiffin D, Gupta H, Lloyd S, Denney T, Dell’Italia LJ. Myofibrillar degeneration, oxidative stress and post surgical systolic dysfunction in patients with isolated mitral regurgitation and pre surgical left ventricular ejection fraction > 60% J Am Coll Cardiol. 2009;55:671–679. doi: 10.1016/j.jacc.2009.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pat B, Killingsworth C, Shi K, Zheng J, Powell P, Chen Y, Walcott G, Ahmed M, Desai R, Gladden JD, Wei CC, Hase N, Kobayashi T, Granzier H, Denney T, Tilson M, Dillon AR, Husain A, Dell’Italia LJ. Chymase inhibition prevents fibronectin and myofibrillar loss and improves cardiomyocyte function and LV torsion angle in dogs with isolated mitral regurgitation. Circulation. 2010;122:1488–1595. doi: 10.1161/CIRCULATIONAHA.109.921619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabri A, Rafiq K, Kolpakov MA, Dillon AR, Dell’Italia LJ. Impaired focal adhesion signaling in the course of volume overload due to mitral regurgitation in the dog. Effect of Beta-1 Adrenergic Receptor Blockade. Circulation Res. 2008;102:1127–1136. doi: 10.1161/CIRCRESAHA.107.163642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seqqat R, Guo X, Rafiq K, Kolpakov MA, Guo J, Koch WJ, Houser SR, Dell’Italia LJ, Sabri A. Beta1-adrenergic receptors promote focal adhesion signaling downregulation and myocyte apoptosis in acute volume overload. J Mol Cell Cardiol. 2012;53(2):240–249. doi: 10.1016/j.yjmcc.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franchini KG, Torsoni AS, Soares PH, Saad MJ. Early Activation of the multicomponent signaling complex associated with focal adhesion kinase induced by pressure overload in the rat heart. Circ Res. 2000;87:558–565. doi: 10.1161/01.res.87.7.558. [DOI] [PubMed] [Google Scholar]
- 17.Peng X, Kraus MS, Wei H, Shen TL, Pariaut R, Alcaraz A, Ji G, Cheng L, Yang Q, Kotlikoff MI, Chen J, Chien K, Gu H, Guan JL. Inactivation of focal adhesion kinase in cardiomyocytes promotes eccentric cardiac hypertrophy and fibrosis in mice. J Clin Invest. 2006;116:217–227. doi: 10.1172/JCI24497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ulasova E, Gladden JD, Zheng J, Chen Y, Pat B, Powell P, Zhmijewski J, Ballinger S, Darley-Usmar V, Dell’Italia LJ. Extracellular matrix loss in acute volume overload causes structural alterations and dysfunction in cardiomyocyte subsarcolemmal mitochondria. J Mol Cell Cardiol. 2010;50:147–156. doi: 10.1016/j.yjmcc.2010.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brower GL, Janicki JS. Pharmacologic inhibition of mast cell degranulation prevents left ventricular remodeling induced by chronic volume overload in rats. J Card Fail. 2005;11(7):548–556. doi: 10.1016/j.cardfail.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Pat B, Killingsworth C, Denney T, Tillson M, Dillon SR, Husain A, Dell’Italia LJ. Mast cell stabilizer worsens left ventricular function and cardiomyocyte function and calcium homeostasis in dogs with isolated mitral regurgitation. J Card Fail. 2010;16:769–777. doi: 10.1016/j.cardfail.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kolpakov MA, Seqqat R, Rafiq K, Xi K, Margulies KB, Libonati J, Powell P, Houser S, Dell’Italia LJ, Sabri A. Pleitropic effects of neutrophils on myocyte apoptosis and left ventricular remodeling during early volume overload. J Mol Cell Cardiol. 2009;47:634–645. doi: 10.1016/j.yjmcc.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamid T, Gu Y, Ortines RV, Bhattacharya C, Wang G, Xuan YT, Prabhu SD. Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: role of nuclear factor-kappaB and inflammatory activation. Circulation. 2009;119(10):1386–2013. doi: 10.1161/CIRCULATIONAHA.108.802918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brower GL, Chancey AL, Thanigaraj S, Matsubara BB, Janicki JS. Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity. Am J Physiol Heart Circ Physiol. 2002;283(2):H518–525. doi: 10.1152/ajpheart.00218.2000. [DOI] [PubMed] [Google Scholar]
- 24.Dell’Italia LJ, Husain A. Dissecting the role of chymase in angiotensin II formation and heart and blood vessel diseases. Curr Opin Cardiol. 2002;17(4):374–9. doi: 10.1097/00001573-200207000-00009. [DOI] [PubMed] [Google Scholar]
- 25.Urata H, Healy B, Stewart RW, Bumpus FM, Husain A. Angiotensin II-forming pathways in normal and failing human hearts. Circ Res. 1990;66:883–890. doi: 10.1161/01.res.66.4.883. [DOI] [PubMed] [Google Scholar]
- 26.Urata H, Kinoshita A, Misono KS, Bumpus FM, Husain A. Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J Biol Chem. 1990;265(36):22348–22357. [PubMed] [Google Scholar]
- 27.Urata H. Chymase and matrix metalloproteinase. Hypertens Res. 2007;30(1):3–4. doi: 10.1291/hypres.30.3. [DOI] [PubMed] [Google Scholar]
- 28.Fang KC, Raymond WW, Blount JL, Caughey GH. Dog mast cell alpha-chymase activates progelatinase B by cleaving Phe88-Gln89 and Phe91-Glu92 bonds of catalytic domain. J Biol Chem. 1997;272:25628–25635. doi: 10.1074/jbc.272.41.25628. [DOI] [PubMed] [Google Scholar]
- 29.Fang KC, Raymond WW, Lazarus SC, Caughey GH. Dog mastocytoma cells secrete a 92-kD gelatinase activated extracellularly by mast cell chymase. J Clin Invest. 1996;97:1589–1596. doi: 10.1172/JCI118583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saarinen J, Kalkkinen N, Welgus HG, Kovanen PT. Activation of human interstitial procollagenase through direct cleavage of the Leu83-Thr84 bond by mast cell chymase. J Biol Chem. 1994;269:18134–18140. [PubMed] [Google Scholar]
- 31.Vartio T, Seppa H, Vaheri A. Susceptibility of soluble and matrix fibronectins to degradation by tissue proteinases, mast cell chymase and cathepsin G. J Biol Chem. 1981;256:471–477. [PubMed] [Google Scholar]
- 32.Forteza R, Lauredo I, Abraham WM, Conner GE. Bronchial tissue kallikrein activity is regulated by hyaluronic acid binding. Am J Respir Cell Mol Biol. 1999;21:666–674. doi: 10.1165/ajrcmb.21.6.3651. [DOI] [PubMed] [Google Scholar]
- 33.Hara M, Matsumori A, Ono K, Kido H, Hwang MW, Miyamoto T, Iwasaki A, Okada M, Nakatani K, Sasayama S. Mast cells cause apoptosis of cardiomyocytes and proliferation of other intramyocardial cells in vitro. Circulation. 1999;100:1443–1449. doi: 10.1161/01.cir.100.13.1443. [DOI] [PubMed] [Google Scholar]
- 34.Leskinen MJ, Heikkila HM, Speer MY, Hakala JK, Laine M, Kouvanen PT, Lindstedt KA. Mast cell chymase induces smooth muscle cell apoptosis by disrupting NF-kappaB-mediated survival signaling. Exp Cell Res. 2006;312:1289–1298. doi: 10.1016/j.yexcr.2005.12.033. [DOI] [PubMed] [Google Scholar]
- 35.Nagata S, Kato J, Sasaki K, Minamino N, Eto T, Kitamura K. Isolation and identification of proangiotensin-12, a possible component of the renin-angiotensin system. Biochem Biophys Res Commun. 2006;350:1026–1031. doi: 10.1016/j.bbrc.2006.09.146. [DOI] [PubMed] [Google Scholar]
- 36.Jessup JA, Trask AJ, Chappell MC, Nagata S, Kato J, Kitamura K, Ferrario CM. Localization of the novel angiotensin peptide, angiotensin-(1–12), in heart and kidney of hypertensive and normotensive rats. Am J Physiol. 2008;294:H2614–2618. doi: 10.1152/ajpheart.91521.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prosser HC, Forster ME, Richards AM, Pemberton CJ. Cardiac chymase converts rat proAngiotensin-12 (PA12) to angiotensin II: effects of PA12 upon cardiac haemodynamics. Cardiovasc Res. 2009;82:40–50. doi: 10.1093/cvr/cvp003. [DOI] [PubMed] [Google Scholar]
- 38.Ahmad S, Simmons T, Varagic J, Moniwa N, Chappell MC, Ferrario CM. Chymase-dependent generation of angiotensin II from angiotensin-(1–12) in human atrial tissue. PLoS ONE. 2011;6:e28501. doi: 10.1371/journal.pone.0028501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahmad S, Varagic J, Moniwa N, Wei CC, Dell’Italia LJ, Ferrario CM. Chymase-mediated angiotensin II generation from angiotensin-(1–12) in left ventricular tissue of normal human subjects. J Am Soc Hypertens. 2013;7(2):128–136. doi: 10.1016/j.jash.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tallaj J, Wei CC, Hankes GH, Holland M, Rynders P, Dillon AR, Ardell JL, Armour JA, Lucchesi PA, Dell’Italia LJ. β1-adrenergic receptor blockade attenuates angiotensin II-mediated catecholamine release into the cardiac interstitium in mitral regurgitation. Circulation. 2003;108:225–230. doi: 10.1161/01.CIR.0000079226.48637.5A. [DOI] [PubMed] [Google Scholar]
- 41.Hankes GH, Ardell JL, Tallaj J, Wei C-C, Aban I, Holland M, Rynders P, Dillon SR, Cardenale R, Hoover DA, Armour JA, Husain A, Dell’Italia LJ. β1-adrenoreceptor blockade mitigates norepinephrine release into the cardiac interstitium in mitral regurgitation in the dog. Am J Physiol. 2006;291:H147–H151. doi: 10.1152/ajpheart.00951.2005. [DOI] [PubMed] [Google Scholar]
- 42.Nagatsu M, Zile MR, Tsutsui H, Schmid PG, DeFreyte G, Cooper G, 4th, Carabello BA. Native β-adrenergic support for left ventricular dysfunction in experimental mitral regurgitation normalizes indexes of pump and contractile function. Circulation. 1994;89:818–826. doi: 10.1161/01.cir.89.2.818. [DOI] [PubMed] [Google Scholar]
- 43.Mehta RH, Supiano MA, Oral H, Grossman PM, Montgomery DS, Smith MJ, Starling MR. Compared with control subjects, the systemic sympathetic nervous system is activated in patients with mitral regurgitation. Am Heart J. 2003;145:1078–1085. doi: 10.1016/S0002-8703(03)00111-X. [DOI] [PubMed] [Google Scholar]
- 44.Mann DL, Kent RL, Parsons B, Cooper G. Adrenergic effects on the biology of the adult mammalian cardiocyte. Circulation. 1992;85:790–804. doi: 10.1161/01.cir.85.2.790. [DOI] [PubMed] [Google Scholar]
- 45.Armour JA. Intrinsic cardiac neurons involved in cardiac regulation possess alpha 1-, alpha 2, beta 1- and beta 2-adrenoreceptors. Can J Cardiol. 1997;13:277–284. [PubMed] [Google Scholar]
- 46.Newton GE, Azevedo ER, Parker JD. Inotropic and sympathetic responses to the intracoronary infusion of a β2-receptor agonist: A human in vivo study. Circulation. 1999;99:2402–2407. doi: 10.1161/01.cir.99.18.2402. [DOI] [PubMed] [Google Scholar]
- 47.Esler M, Jennings G, Lambert G, Meredith I, Horne M, Eisenhofer G. Overflow of catecholamine neurotransmitters to the circulation: source, fate and function. Physiol Rev. 1990;70(4):963–985. doi: 10.1152/physrev.1990.70.4.963. [DOI] [PubMed] [Google Scholar]
- 48.Farrell DM, Wei CC, Tallaj J, Ardell JL, Hageman GR, Armour JA, Hageman GR, Bradley WE, Dell’Italia LJ. Angiotensin II modulates catecholamine release into the interstitial fluid of the canine myocardium in-vivo. Am J Physiol. 2001;281:H813–H822. doi: 10.1152/ajpheart.2001.281.2.H813. [DOI] [PubMed] [Google Scholar]
- 49.Pat B, Killingsworth C, Zheng J, Denney T, Powell P, Tillson M, Dillon SR, Dell’Italia LJ. Dissociation between cardiomyocyte function and remodeling with beta-adrenergic receptor blockade in isolated canine mitral regurgitation. Am J Physiol. 2008;295(6):H2321–2337. doi: 10.1152/ajpheart.00746.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Varadarajan P, Joshi N, Appel D, Duvvuri L, Pai RG. Effect of beta-blocker therapy on survival in patients with severe mitral regurgitation and normal left ventricular ejection fraction. Am J Cardiol. 2008;102:611–615. doi: 10.1016/j.amjcard.2008.04.029. [DOI] [PubMed] [Google Scholar]
- 51.Ahmed MI, Aban I, Lloyd SG, Gupta H, Howard G, Inusah S, Peri K, Robinson J, Smith P, McGiffin D, Denney TS, Dell’Italia LJ. A randomized controlled Phase IIb trial of Beta-1 receptor blockade in isolated degenerative mitral regurgitation. J Am Coll Cardiol. 2012;60:833–838. doi: 10.1016/j.jacc.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gladden JD, Ulasova E, Chen Y, Ahed M, Zellickson B, Baman M, Ballinger S, Darley-Usmar V, Dell’Italia LJ. Novel insights into interactions between mitochondria and xanthine oxidase in cardiac volume overload. Free Rad Biol Med. 2011;51:1975–1984. doi: 10.1016/j.freeradbiomed.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kandasamy AD, Chow AK, Ali MA, Schulz R. Matrix metalloproteinase-2 and myocardial oxidative stress injury: beyond the matrix. Cardiovasc Res. 2010;85(3):413–423. doi: 10.1093/cvr/cvp268. [DOI] [PubMed] [Google Scholar]