

Abstract
The unfolded protein response (UPR), an endoplasmic reticulum (ER) stress-induced signaling cascade, is mediated by three major stress sensors IRE-1α, PERK, and ATF6α. Studies described the UPR as a critical network in selection, adaptation, and survival of cancer cells. While previous reviews focused mainly on solid cancer cells, in this review, we summarize the recent findings focusing on acute leukemias. We take into account the impact of the underlying genetic alterations of acute leukemia cells, the leukemia stem cell pool, and provide an outline on the current genetic, clinical, and therapeutic findings. Furthermore, we shed light on the important oncogene-specific regulation of individual UPR signaling branches and the therapeutic relevance of this information to answer the question if the UPR could be an attractive novel target in acute leukemias.
Keywords: Acute myeloid leukemia, Acute lymphoblastic leukemia, Leukemia stem cells, Unfolded protein response, XBP1, Small-molecule inhibitors
Introduction
Although the therapy of acute leukemias either originating from myeloid (acute myeloid leukemia (AML)) or lymphoid lineage (acute lymphoblastic leukemia (ALL)) has improved in recent decades, the heterogeneous genetic landscape of these diseases causes relapse in the majority of patients. The unfolded protein response (UPR) is a conserved adaptive signaling pathway aiming to restore protein homeostasis mainly in the ER. Recent studies suggest an important function in acute leukemias. In this review, we will summarize these results highlighting the druggability of the UPR and give an outlook of potential mechanisms.
The unfolded protein response
Cell survival is largely dependent on correct production, control, and folding of proteins. To maintain cellular protein homeostasis (proteostasis) and to be shielded against stress stimuli accumulating within the endoplasmic reticulum (ER) causing “ER stress,” cells are mainly dependent on the cytoprotective network of the UPR [1–3]. While under acute conditions, UPR activation acts pro-survival; continuous and chronic stress causes a shift of UPR signaling in acting pro-apoptotic [4, 5]. Thereby, understanding the complex role of the UPR is also understanding not only the specific pathways and molecules both in their time but also cell-specific context [3, 6].
The correct function of the UPR is mediated through three distinct signaling branches in a time- and stimuli-specific manner, namely inositol-requiring enzyme 1 alpha (IRE-1α), PKR-like ER kinase (PERK), and activating transcription factor 6 alpha (ATF6α) [7]. Under unstressed conditions, the stress sensors of PERK, IRE-1α, and ATF6α are maintained inactive through binding to the ER chaperone heat shock 70 kDa protein 5/78 kDa glucose-regulated protein (HSPA5/GRP78). Through various stimuli, such as accumulation of misfolded proteins within the ER, GRP78 binds with a higher affinity to their exposed hydrophobic domains, dissociating from UPR sensors, thereby priming IRE-1α and PERK for oligomerization and autotransphosphorylation [8] and revealing an ER export motif in ATF6α [7].
PERK-eIF2α
Upon activation, PERK phosphorylates eukaryotic translation initiation factor 2 subunit alpha translation initiation factor (eIF2α) at serine 51 (S51) [9], thereby attenuating global protein synthesis to reduce the number of proteins entering ER, imparting a pro-survival effect on the cells. This prevents assembly of the 80S ribosome translation initiation complex and allows selective expression of activating transcription factor 4 (ATF4). ATF4 then enters the nucleus activating ER stress-response genes involved in protein folding, antioxidant responses, autophagy, amino acid metabolism, and apoptosis promoting cell survival [10–13]. Moreover, PERK activation leads to nuclear factor (erythroid-derived 2)-like 2 (NRF2) phosphorylation and subsequent control of the antioxidant pathway [14].
ATF6α
The second branch is represented by the basic leucine zipper transcription factor ATF6α. ATF6α activation occurs in the Golgi complex following export from the ER upon ER stress [15]. ATF6α is then cleaved on both sides of the membrane by site-1 (S1P, also named membrane-bound transcription factor peptidase, site 1 MBTPS1) and site-2 proteases (S2P, also named membrane-bound transcription factor peptidase, site 2 MBTPS2) generating an active transcription factor through regulated intramembrane proteolysis [16]. Following its cleavage, the ATF6α cytosolic domain translocates to the nucleus and activates specific transcriptional programs that promote adaptation, including upregulation of various components of ER-associated degradation (ERAD) [17].
IRE-1α—XBP1/RIDD
The most evolutionary conserved arm of the UPR is mediated by IRE-1α, which is activated by autophosphorylation and oligomerization upon accumulation of misfolded proteins in the ER. IRE-1α contains an endoribonuclease (RNase) and a kinase domain. So far, the most described function of the RNase domain is to reduce ER load through unconventional splicing of the X-box binding protein 1 (XBP1) mRNA [18]. This unconventional splicing leads to removal of a 26-nucleotide intron. Recently, it was uncovered that a multimeric protein complex tRNA splicing ligase is responsible for ligation of IRE-1α-cleaved XBP1 mRNA 5′ and 3′ extremities of which the RNA 2′,3′-cyclic phosphate and 5′-OH ligase (RTCB, also named HSPC117/C22orf28) seems to be the most essential subunit [19–22] and demonstrated a physiological role in plasma-cell differentiation [19]. This causes a frameshift in the XBP1 reading frame, thus generating a transcriptionally active protein (XBP1s). XBP1s controls expression of genes involved in protein folding, secretion, ERAD, and lipid synthesis [23, 24]. IRE-1α RNase activity is also involved in RNA degradation (pathway known as regulated IRE-1α-dependent decay or RIDD) [25].
The complex cellular mechanisms which lead to activation of the UPR and in-depth genetic functions in solid cancer cells have already been extensively discussed in a number of excellent review articles [26, 27, 1, 6, 2, 3, 28, 29]. Thereby, we will focus here on the novel role of the UPR network in acute leukemias.
Role of the UPR in AML
AML originates from a myeloid-committed hematopoietic stem cell (HSC) and is characterized by acquisition of genetic and epigenetic changes [30–35]. These cause aberrant activation of signaling pathways [36–38] contributing to AML pathogenesis and progression [39]. Mechanistically, for instance, transcription factor CCAAT/enhancer binding protein alpha (C/EBPα) [40] is frequently deregulated by genomic mutations causing maturation defects [41, 42]. Others include internal tandem duplications in the FMS-like tyrosine kinase 3 gene (FLT3-ITD) which correlates with poor outcome [43] or the balanced translocation t(15;17) (namely PML-RARα) representing the main oncogenic driver of the acute promyelocytic leukemia subset [44].
With a median age of 66 years at diagnosis and an overall survival rate for older AML patients of less than 10 %, the treatment response has not changed dramatically within three decades [45–47]. In younger patients, remission rates are more promising, while also in this population, ultimately ~50 % of the patients relapse within the first 5 years [38, 39, 45–50].
UPR and C/EBPα in AML
Studies showed that the IRE-1α/XBP1s branch of the UPR was activated in 17.4 % of AML cases [51, 52]. Besides the expression of XBP1s (16 out of 92 patients), equally that of GRP78 and the ER protein quality control lectin calreticulin were increased [52]. Characteristically, these patients were poor risk (44 % vs 25 %), although this was not significant, potentially because of the low patient numbers. The important role of calreticulin was then shown by the same research group, providing evidence that it was able to bind and block the translation of C/EBPα [53]. Here, calreticulin did bind to the C/EBPα mRNA and form a stem-loop secondary structure preventing translation. Similarly, the UPR-related molecule disulfide isomerase protein (PDI), a thiol-disulfide oxidoreductase residing in the ER lumen, did equally bind to this stem-loop region of the C/EBPα mRNA [54]. Together, they formed a complex and regulated the translation of C/EBPα. Still, this observation remains surprising as both calreticulin and PDI are localized in the lumen of the ER and are not predicted to be present in the same compartment as C/EBPα mRNA.
UPR and PML-RARα in AML
In the majority of PML-RARα+ AML patients, retinoic acid receptor alpha (RARα) is fused to the promyelocytic leukemia (PML) open reading frame on chromosome 15 [55]. The resulting fusion molecule PML-RARα acts as transcriptional repressor in a dominant-negative manner by blocking retinoic acid-induced myeloid differentiation [56]. In PML-RARα+ AML, only one study described a potential therapeutic role for the UPR. Mechanistically, wild-type RARα forms heterodimers with the soluble nuclear receptor co-repressor 1 (N-CoR) family of co-repressors mediating transcriptional repression [57, 58] and releases the co-repressors in response to cognate agonists such as all-trans-retinoic acid (ATRA) leading to myeloid differentiation [56]. Additionally, the N-CoR protein is critical for transcriptional repression by the tumor suppressor Max dimerization protein 1 (MAD) [59, 60].
Here, a mechanism is through increased binding to N-CoR by PML-RARα [60]. This binding causes an abnormal protein conformation and insolubility of the N-CoR protein. The misfolded N-CoR protein is then recruited to the ER and targeted by the ERAD system. Thereby, the tumor-suppressive function of MAD is missing. This suggests that the UPR is a critical promoter of aberrant activation of PML-RARα through ubiquitination of N-CoR [61] (Fig. 1).
Fig. 1.

A schematic overview on how PML-RARα causes ER stress in AML is provided. Here, under physiological conditions, RARα can dissociate from coreceptors such as N-CoR. In PML-RARα+ AML, the binding is significantly increased. This leads to a conformational change of the N-CoR protein. The misfolded N-CoR protein is degraded by ERAD through activation of ATF6α. Through the lack of soluble and functional N-CoR, protein myeloid differentiation is prevented and activity of the tumor suppressor MAD is reduced
UPR and hypoxia in AML
Hypoxic environments such as the bone marrow, where the majority of leukemia cells reside [62], lead to activation of the hypoxia-inducible factor (HIF) pathway. HIF is a heterodimer consisting of an unstable alpha subunit (such as HIF-1α) and a stable beta subunit (HIF-1β) binding DNA through hypoxia-response elements. Under normoxic conditions, HIF members are hydroxylated and thereby inactivated by prolyl hydroxylase domain family members generating a binding site for the von Hippel-Lindau (pVHL) tumor suppressor. This leads to ubiquitination of HIF-α when oxygen is available. Under hypoxic conditions through the functional lack of pVHL, HIF-α accumulates and dimerizes with HIF-β family members activating several hundred genes [63].
PML-RARα has been shown to transcriptionally activate HIF-1α, while this was not through direct physical interaction. Furthermore, downregulation of HIF-1α by shRNA negatively affected self-renewal, migration, and neo-angiogenesis of PML-RARα+ AML cells. Additionally, inhibition of HIF-1α together with ATRA was synergistic [64]. The correlation between expression of HIF factors and the prognosis of AML patients is divergent. For instance, HIF-1α negatively impacted survival of normal karyotype (NK) AML patients, where low HIF-1α expression was associated with improved event-free survival (p = 0.04, hazard ratio = 0.22). Multivariate analysis showed that HIF-1α expression was an independent prognostic marker [65]. A very recent study showed an opposite result, where HIF-2α expression was not associated with poor prognosis. HIF-2α expression was even higher in good risk subgroups such as inv(16) (p = 0.0031) and t(15;17) (p = 0.04) [66, 67].
Hypoxic conditions are well-described stress inducers leading to activation of the UPR [68]. The link between hypoxia and UPR in AML was suggested by the description that HIF-2α protected AML cells against ER stress [69]. Genetic silencing of HIF-2α did not only affect survival of long-term repopulating HSCs, but also caused an increase of ER stress and production of reactive oxygen species (ROS). The increased production of ROS and UPR led to apoptosis in AML cells [69].
In PML-RARα+ AML cell lines, such as HL60, the link between oxidative stress and UPR was shown as activation of the NADPH oxidase by phorbol-12-myristate-13-acetate (PMA) led to increased ROS production and caused ER stress as shown by expression and phosphorylation of different UPR genes including GRP78, eIF2α, and XBP1s [70].
Role of the UPR in B-ALL
In contrast to AML, ALL originates from either the B-cell (B-ALL) or T-cell lineage (T-ALL). B-ALL is the most common type of childhood leukemia and is frequently found in adults. Here, other B-ALL specific genetic alterations such as fusion genes, e.g., translocation between breakpoint cluster region and tyrosine kinase abelson murine leukemia viral oncogene homolog 1 (BCR-ABL1) [71], activating point mutations such as NRASG12D, rearrangements in the mixed lineage leukemia gene (MLLr) [72], and aberrations in fundamental genes of B-cell development, e.g., paired box 5 (PAX5) and ikaros family zinc finger protein 1 (IKZF1) [73, 74], define B-ALL progression as well as subsequent relapse [75, 76, 74, 77].
Mutations in the RAS/RAF pathway have been identified in ~20–30 % of B-ALL patients correlating with poor prognosis [78]. Interestingly, the recent identification that RAS mutations are significantly increased in relapsed B-ALL patients further underlines their critical clinical importance [79]. Therapeutically, activating RAS mutations such as NRASG12D is very challenging as they confer resistance towards chemotherapy [80]. More importantly, a pivotal disadvantage of targeted tyrosine kinase inhibitor (TKI) therapy, such as imatinib is paradoxical activation (off-target) of the RAS pathway [81].
UPR and BCR-ABL1 in B-ALL
The Philadelphia chromosome (Ph) is the result of a reciprocal translocation of BCR on chromosome 22 (region q11) and ABL1 on chromosome 9 (region q34) resulting in the constitutive active tyrosine kinase BCR-ABL1 [82]. The BCR-ABL1 fusion gene defines a high-risk subset (Ph+ ALL) correlating with very poor survival rates [83].
Initial studies showed that both XBP1s and GRP78 were higher expressed in Ph+ leukemia cell lines, while detailed functional studies were missing [84]. Additionally, withdrawal of imatinib in Ph+ leukemia caused hy peractivation of the BCR-ABL1 kinase leading to metabolic reprogramming accommodated with an increase of ATP production through glycolysis. This led to increased ER swelling and stress and subsequent activation of XBP1s and CCAAT/enhancer-binding protein homologous protein (CHOP) [85], a known inducer of UPR-related apoptosis [86].
We showed that for instance, GRP78, IRE-1α, and XBP1 were upregulated in the high-risk Ph+ and MLLr+ B-ALL subsets [87]. Additionally, their promoters were also hypomethylated in B-ALL cases. Furthermore, genetic deletion of Grp78 or Xbp1 caused apoptosis in mouse models of BCR-ABL1 und NRAS-mutated B-ALL. Mechanistically, expression of XBP1s was linked to BCR-ABL1 kinase activity. Both genetic and pharmacological inhibition of BCR-ABL1 downstream signaling such as signal transducer and activator of transcription 5 (STAT5), mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK), or protein kinase B alpha (AKT) led to reduction of XBP1s expression. BTB and CNC homology 1, basic leucine zipper transcription factor 2 (BACH2), a tumor suppressor in B-ALL [88], was also able to negatively regulate expression of XBP1s [87].
In the Ph+ ALL subsets, TKIs such as imatinib or dasatinib directly targeting the BCR-ABL fusion oncogene are essential for a successful therapy [89, 73, 83, 90]. In Ph+ ALL primary cases, imatinib treatment downregulated XBP1s expression, while not entirely abolishing it [87]. This suggests that a potential dual strategy targeting both the IRE-1α/XBP1 axis with a TKI could be beneficial.
Clinically, high mRNA expression of XBP1 correlated with poor prognosis [87], both in univariate and multivariate analysis in B-ALL, even in the Ph+ ALL subset.
UPR and stem cells
In the physiological stem cell hierarchy, the UPR has been shown to regulate the self-renewal capacities of HSCs. Through single cell analysis, it was possible to study exact expression levels in individual progenitor populations [91]. Gene expression analysis revealed that in HSCs, PERK was predominately activated compared to downstream progenitor populations (Fig. 2) [92].
Fig. 2.

The expression profile of different UPR genes is shown by microarray analysis in HSC and different progenitor populations (multipotent progenitors (MPP), common myeloid progenitors (CMP), multipotent lymphoid progenitors (MLP), megakaryocytic-erythroid progenitors (MEP) and granulocyte-monocyte progenitors (GMP). The hierarchical tree is based on van Galen et al.
Further experiments showed that HSCs were sensitive to induced ER stress, for instance through ER stressing agents such as tunicamycin. HSCs were equally more sensitive towards ER-induced apoptosis, when compared to progenitor populations, which was mainly mediated by activation of the PERK-eIF2α-ATF4-CHOP pathway. A pharmacological PERK inhibitor (GSK2606414) caused apoptosis in the HSC population, while progenitor populations were resistant, even at high concentrations [92].
While these studies were conducted in physiological HSC populations, they raise the question, if similarly individual UPR genes play an important role in leukemia stem/initiating cells (LSCs/LICs). LICs share common characteristics of normal HSCs such as self-renewal, pluripotency, and quiescence [93] and are considered a fundamental source of relapse [94–96]. Considering the probability of trans-differentiation of cancerous stem cells into non-stem cell state and vice versa, they are usually not affected by conventional and even targeted therapies [94]. Nevertheless, thus far, an extensive study addressing whether leukemia stem cells are dependent on either global activation of the UPR or individual branches of the UPR is still missing. This could have direct therapeutic implications, since small-molecule inhibitors which target both PERK and IRE-1α signaling are currently available [87, 97–102].
The UPR as a druggable target
Concept of proteotoxicity as a therapeutic approach
The concept of proteotoxicity was mainly studied in secretory cells such as plasma cells, which as antibody-producing cells are strongly dependent on a well-developed secretory system and are pruned to potential protein overload. Here, proteasomal degradation represents the main pathway for ERAD [103]. Thereby, most studies focused on a better understanding, how plasma-cell derived malignancies, namely multiple myeloma, could be targeted through the proteasome. This led to development of the proteasome inhibitor bortezomib which blocks the 20S proteasome with great clinical success [104]. Despite great initial clinical success, extensive use caused multiple mechanisms of resistance [105] including point mutations, such as a point mutation (G322A) in the β-subunit of the binding pocket of bortezomib [106]. Another mechanism of resistance is activation of alternative degradation pathways including the aggresome [107], upregulation of heat shock proteins, or dedifferentiation of multiple myeloma cells to become less dependent on an effective ER stress control [108]. These developments have to be considered, despite the fact that disrupting proteostasis is a potential novel approach in acute leukemias (Fig. 3).
Fig. 3.
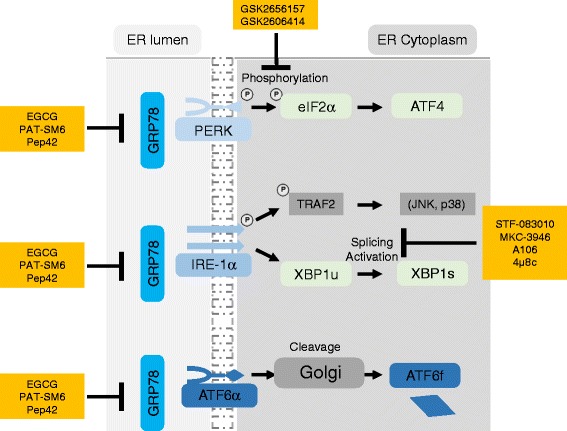
A schematic overview of the UPR network and the currently available inhibitors targeting the individual signaling molecules and subsequent pathways
Targeting PERK
As the UPR has already been suggested as a potential oncogenic network, the therapeutic efforts to date have focused mainly on the PERK/eIF2α and IRE-1α/XBP1 signaling axes. In the case of PERK, selective ATP-competitive PERK kinase inhibitors such as GSK2606414 or GSK2656157 [97, 98] were anti-proliferative in multiple cancer models in vivo including multiple myeloma. So far, pharmacological inhibition of PERK kinase activity has not yet been shown to be effective in leukemia cells. Still, a potential hint in this direction is that loss of PERK kinase activity using dominant-negative mutants already showed to negatively affect leukemia survival, albeit in chronic myeloid leukemia, but not acute leukemia cells [109].
Targeting GRP78
Targeting GRP78 has been achieved by epigallocatechin gallate (EGCG), a green tea extract [110] which binds to the ATP-binding site of GRP78. This binding modulates its ATPase activity leading to conformational conversion into the inactive oligometric form. In B-ALL cells, treatment with EGCG caused apoptosis [111]. Additionally, targeting GRP78 also sensitized B-ALL cells towards vincristine, a chemotherapeutic drug commonly used in the therapy of B-ALL patients.
In a second approach, Pep42, a cyclic 13-mer peptide [112], was used to target both cell surface and intracellular GRP78 after receptor-mediated endocytosis and was equally able to induce apoptosis in B-ALL cell lines [111]. So far, two clinical studies are studying GRP78 as a therapeutic target. EGCG is currently being evaluated in Alzheimer’s disease (clinical trial ID: NCT00951834), while a novel monoclonal IgM antibody (PAT-SM6) against GRP78 [113, 114] is being evaluated in multiple myeloma with moderate clinical response (clinical trial ID: NCT01727778) [115]. Single treatment with PAT-SM6 led to a stable disease in four out of 12 patients (Table 1).
Table 1.
Overview of clinical trials investigating the therapeutic usefulness of the UPR
| Compound | Molecular target | Disease | Stage of clinical development | References |
|---|---|---|---|---|
| Epigallocatechin gallate (EGCG) | GRP78 | Alzheimer’s disease | Clinical, phase 2 and 3 | NCT00951834 |
| PAT-SM6 | GRP78 | Multiple myeloma | Clinical, phase 1 | NCT01727778, Rasche et al. [115] |
Targeting IRE-1α/XBP1s signaling
Most currently available small-molecule inhibitors block the RNase activity either through direct inhibition of the RNase [101, 102] or through modulation of the kinase domain [99]. The currently available preclinical small-molecule inhibitors (MKC-3946, STF-083010, A-I06 and 4μ8c) aim to prevent XBP1 mRNA splicing and production of the transcriptionally active XBP1s protein [87, 97–102, 116]. In the concept of using proteotoxicity as a therapeutic approach, most initial studies focused on multiple myeloma with only partially convincing results. For instance, treatment with MKC-3946 led to apoptosis in multiple myeloma cells [101]; nevertheless, this is in contrast to other studies which suggest that multiple myeloma cells can survive without functional XBP1s [108].
In B-ALL, we tested the efficacy of IRE-1α inhibition and were able to show that treatment with different preclinical IRE-1α inhibitors caused apoptosis in primary B-ALL cases in a dose-dependent manner [87]. XBP1s splicing was significantly reduced in primary B-ALL cases, and this was accommodated with ER stress as shown by increased accumulation of a specific ER-tracker [117]. Pharmacological inhibition of IRE-1α was able to cause not only apoptosis, but also cell cycle arrest in B-ALL xenografts and prolonged survival of B-ALL-bearing mice in vivo [87].
Nevertheless, small-molecule inhibitors for IRE-1α seem to vary substantially in their efficacy and specificity [99, 116, 100, 101]. The identification of a new class of hydroxy-aryl-aldehydes of IRE-1α inhibitors [118] might help to improve our understanding of small-molecule inhibitor design [119], allowing improved inhibition of IRE-1α RNase activity.
Additionally, the identification of the RIDD mechan ism, through which IRE-1α can splice mRNAs containing a specific consensus sequence which is recognized by the RNase domain [120], suggests that genetic and pharmacological results may vary, depending for instance on the type of cancer, or lead to activation of other secondary pathways.For instance, pharmacological inhibition of the RNase domain of IRE-1α predominantly blocked splicing of XBP1, while RIDD remained intact [121]. This suggests that splicing of RIDD targets and XBP1 differs substantially, and both should be considered as distinct pharmacological targets.
A final aspect is the acquisition of secondary mutations or resistance, a major therapeutic challenge for instance in TKI-treatment. Until now, no mutations for XBP1 have been identified yet in B-ALL patients, suggesting that this pathway is active. Still, the identification of two loss-of-function mutations of XBP1 in multiple myeloma [108] suggests that the use of IRE-1α inhibitors could lead to acquisition of mutations in acute leukemias [122, 108] (Table 2).
Table 2.
Overview of preclinical studies studying the therapeutic usefulness of the UPR
| Compound | Molecular target | Disease | Stage of clinical development | References |
|---|---|---|---|---|
| STF-083010 | IRE-1 | Multiple myeloma | Preclinical | Papandreou et al. [99] |
| ALL | Kharabi Masouleh et al. [87] | |||
| CLL | Kriss et al. [116] | |||
| A106 | IRE-1 | CLL | Preclinical | Kriss et al. [116] |
| ALL | Kharabi Masouleh et al. [87] | |||
| MKC-3946 | IRE-1 | Multiple myeloma | Preclinical | Volkmann et al. [100] |
| Mimura et al. [101] | ||||
| 4μ8c | IRE-1 | Multiple myeloma | Preclinical | Cross et al. [102] |
| GSK2606414 | PERK | Multiple myeloma | Preclinical | Axten et al. [98] |
| GSK2656157 | Pancreatic cancer | |||
| Epigallocatechin gallate (EGCG) | GRP78 | ALL | Preclinical | Uckun et al. [111] |
| PAT-SM6 | GRP78 | Melanoma | Preclinical | Rosenez et al. [113] |
| Multiple myeloma | Rasche et al. [114] | |||
| Pep42 | GRP78 | ALL | Preclinical | Uckun et al. [111] |
Concluding remarks
Taken together, the role of the UPR in acute leukemia subsets has only recently begun to be elucidated, while further studies are required to answer several questions and comprehend the global impact of UPR signals in such disease. Several studies indicate that different UPR genes play an important role in both AML and B-ALL. Still, both diseases are quite heterogeneous, and it has to be elucidated how different oncogenes cause activatio of UPR. For instance, BCR-ABL1 kinase signaling significantly differs to that of MLLr. Still, in both B-ALL subsets, XBP1s expression was highly upregulated [87]. Additionally, the impact of the UPR on LSCs is unknown. The study by van Galen et al. suggests that different progenitor populations show distinct expression and dependencies on the UPR [92]. Translating these findings to LSCs suggests that LSCs might not be dependent equally on all UPR pathway, rather only on individual pathways, while such a study has not yet been conducted.
Finally, the finding that expression of XBP1s is significantly reduced but not abolished upon treatment with different pharmacological inhibitors (MEK, AKT, or BCR-ABL) suggests that potential combinational therapies with IRE-1α inhibitors might be useful [87].
Acknowledgements
The research in our group is supported by generous funding from the Ernst Jung Foundation, German Cancer Aid, RWTH START, and RWTH START UP to BKM; by Breast Cancer Campaign, Irish Cancer Soc, and Belgian grant (IAP) to AS.
Footnotes
Competing interests
THB receives research funding from Celgene. All other authors have declared no competing interests.
Authors’ contributions
BKM drafted the manuscript. The other authors critically read and edited the paper. All authors read and approved the final manuscript.
References
- 1.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 2.Clarke HJ, Chambers JE, Liniker E, Marciniak SJ. Endoplasmic reticulum stress in malignancy. Cancer Cell. 2014;25(5):563–73. doi: 10.1016/j.ccr.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 3.Chambers JE, Marciniak SJ. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 2. Protein misfolding and ER stress. Am J Physiol Cell Physiol. 2014;307(8):C657–70. doi: 10.1152/ajpcell.00183.2014. [DOI] [PubMed] [Google Scholar]
- 4.Gorman AM, Healy SJ, Jager R, Samali A. Stress management at the ER: regulators of ER stress-induced apoptosis. Pharmacol Ther. 2012;134(3):306–16. doi: 10.1016/j.pharmthera.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 5.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833(12):3460–70. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chevet E, Hetz C, Samali A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015;5(6):586–97. doi: 10.1158/2159-8290.CD-14-1490. [DOI] [PubMed] [Google Scholar]
- 7.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2(6):326–32. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 8.Shamu CE, Walter P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996;15(12):3028–39. [PMC free article] [PubMed] [Google Scholar]
- 9.Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7(6):1165–76. doi: 10.1016/S1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- 10.Rzymski T, Milani M, Singleton DC, Harris AL. Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle. 2009;8(23):3838–47. doi: 10.4161/cc.8.23.10086. [DOI] [PubMed] [Google Scholar]
- 11.Ye J, Koumenis C. ATF4, an ER stress and hypoxia-inducible transcription factor and its potential role in hypoxia tolerance and tumorigenesis. Curr Mol Med. 2009;9(4):411–6. doi: 10.2174/156652409788167096. [DOI] [PubMed] [Google Scholar]
- 12.Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol. 2008;40(1):14–21. doi: 10.1016/j.biocel.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 13.Wek RC, Anthony TG. EXtENDINg beta cell survival by UPRegulating ATF4 translation. Cell Metab. 2006;4(5):333–4. doi: 10.1016/j.cmet.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 14.Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem. 2004;279(19):20108–17. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- 15.Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3(1):99–111. doi: 10.1016/S1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 16.Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6(6):1355–64. doi: 10.1016/S1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 17.Brodsky JL, Skach WR. Protein folding and quality control in the endoplasmic reticulum: recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol. 2011;23(4):464–75. doi: 10.1016/j.ceb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uemura A, Oku M, Mori K, Yoshida H. Unconventional splicing of XBP1 mRNA occurs in the cytoplasm during the mammalian unfolded protein response. J Cell Sci. 2009;122(Pt 16):2877–86. doi: 10.1242/jcs.040584. [DOI] [PubMed] [Google Scholar]
- 19.Jurkin J, Henkel T, Nielsen AF, Minnich M, Popow J, Kaufmann T, et al. The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J. 2014;33(24):2922–36. doi: 10.15252/embj.201490332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu Y, Liang FX, Wang X. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol Cell. 2014;55(5):758–70. doi: 10.1016/j.molcel.2014.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kosmaczewski SG, Edwards TJ, Han SM, Eckwahl MJ, Meyer BI, Peach S, et al. The RtcB RNA ligase is an essential component of the metazoan unfolded protein response. EMBO Rep. 2014;15(12):1278–85. doi: 10.15252/embr.201439531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ray A, Zhang S, Rentas C, Caldwell KA, Caldwell GA. RTCB-1 mediates neuroprotection via XBP-1 mRNA splicing in the unfolded protein response pathway. J Neurosci. 2014;34(48):16076–85. doi: 10.1523/JNEUROSCI.1945-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, et al. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell. 2007;27(1):53–66. doi: 10.1016/j.molcel.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 24.Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, et al. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002;16(4):452–66. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maurel M, Chevet E, Tavernier J, Gerlo S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem Sci. 2014;39(5):245–54. doi: 10.1016/j.tibs.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 26.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14(9):581–97. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 27.Yadav RK, Chae SW, Kim HR, Chae HJ. Endoplasmic reticulum stress and cancer. J Cancer Prevention. 2014;19(2):75–88. doi: 10.15430/JCP.2014.19.2.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hetz C, Chevet E. Theme series—UPR in cancer. Semin Cancer Biol. 2015. doi:10.1016/j.semcancer.2015.04.008. [DOI] [PubMed]
- 29. Dejeans N, Barroso K, Fernandez-Zapico ME, Samali A, Chevet E. Novel roles of the unfolded protein response in the control of tumor development and aggressiveness. Semin Cancer Biol. 2015. doi:10.1016/j.semcancer.2015.04.007. [DOI] [PubMed]
- 30.Abdel-Wahab O, Levine RL. Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood. 2013;121(18):3563–72. doi: 10.1182/blood-2013-01-451781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conway O’Brien E, Prideaux S, Chevassut T. The epigenetic landscape of acute myeloid leukemia. Adv Hematol. 2014;2014:103175. doi: 10.1155/2014/103175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eriksson A, Lennartsson A, Lehmann S. Epigenetic aberrations in acute myeloid leukemia: early key events during leukemogenesis. Exp Hematol. 2015. doi:10.1016/j.exphem.2015.05.009. [DOI] [PubMed]
- 33.Lamba G, Zaidi SK, Luebbers K, Verschraegen C, Stein GS, Rosmarin A. Epigenetic landscape of acute myelogenous leukemia—moving toward personalized medicine. J Cell Biochem. 2014;115(10):1669–72. doi: 10.1002/jcb.24853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mazzarella L, Riva L, Luzi L, Ronchini C, Pelicci PG. The genomic and epigenomic landscapes of AML. Semin Hematol. 2014;51(4):259–72. doi: 10.1053/j.seminhematol.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 35.Gutierrez SE, Romero-Oliva FA. Epigenetic changes: a common theme in acute myelogenous leukemogenesis. J Hematol Oncol. 2013;6:57. doi: 10.1186/1756-8722-6-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hatlen MA, Wang L, Nimer SD. AML1-ETO driven acute leukemia: insights into pathogenesis and potential therapeutic approaches. Front Med. 2012;6(3):248–62. doi: 10.1007/s11684-012-0206-6. [DOI] [PubMed] [Google Scholar]
- 37.Lo-Coco F, Hasan SK. Understanding the molecular pathogenesis of acute promyelocytic leukemia. Best Pract Res Clin Haematol. 2014;27(1):3–9. doi: 10.1016/j.beha.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 38.Sakamoto KM, Grant S, Saleiro D, Crispino JD, Hijiya N, Giles F, et al. Targeting novel signaling pathways for resistant acute myeloid leukemia. Mol Genet Metab. 2015;114(3):397–402. doi: 10.1016/j.ymgme.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roboz GJ. Current treatment of acute myeloid leukemia. Curr Opin Oncol. 2012;24(6):711–9. doi: 10.1097/CCO.0b013e328358f62d. [DOI] [PubMed] [Google Scholar]
- 40.Friedman AD, Keefer JR, Kummalue T, Liu H, Wang QF, Cleaves R. Regulation of granulocyte and monocyte differentiation by CCAAT/enhancer binding protein alpha. Blood Cells Mol Dis. 2003;31(3):338–41. doi: 10.1016/S1079-9796(03)00135-9. [DOI] [PubMed] [Google Scholar]
- 41.Nerlov C. C/EBPalpha mutations in acute myeloid leukaemias. Nat Rev Cancer. 2004;4(5):394–400. doi: 10.1038/nrc1363. [DOI] [PubMed] [Google Scholar]
- 42.Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27(3):263–70. doi: 10.1038/85820. [DOI] [PubMed] [Google Scholar]
- 43.Levis M. FLT3 mutations in acute myeloid leukemia: what is the best approach in 2013? Hematology Am Soc Hematol Educ Program. 2013;2013:220–6. doi: 10.1182/asheducation-2013.1.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Braekeleer E, Douet-Guilbert N, De Braekeleer M. RARA fusion genes in acute promyelocytic leukemia: a review. Expert Rev Hematol. 2014;7(3):347–57. doi: 10.1586/17474086.2014.903794. [DOI] [PubMed] [Google Scholar]
- 45.Burnett AK, Hills RK, Milligan DW, Goldstone AH, Prentice AG, McMullin MF, et al. Attempts to optimize induction and consolidation treatment in acute myeloid leukemia: results of the MRC AML12 trial. J Clin Oncol. 2010;28(4):586–95. doi: 10.1200/JCO.2009.22.9088. [DOI] [PubMed] [Google Scholar]
- 46.DiNardo CD, Cortes JE. New treatment for acute myelogenous leukemia. Expert Opin Pharmacother. 2015;16(1):95–106. doi: 10.1517/14656566.2015.981527. [DOI] [PubMed] [Google Scholar]
- 47.Montalban-Bravo G, Garcia-Manero G. Novel drugs for older patients with acute myeloid leukemia. Leukemia. 2015;29(4):760–9. doi: 10.1038/leu.2014.244. [DOI] [PubMed] [Google Scholar]
- 48.Erba HP. Finding the optimal combination therapy for the treatment of newly diagnosed AML in older patients unfit for intensive therapy. Leuk Res. 2015;39(2):183–91. doi: 10.1016/j.leukres.2014.11.027. [DOI] [PubMed] [Google Scholar]
- 49.Fernandez HF, Sun Z, Yao X, Litzow MR, Luger SM, Paietta EM, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med. 2009;361(13):1249–59. doi: 10.1056/NEJMoa0904544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walter RB, Estey EH. Management of older or unfit patients with acute myeloid leukemia. Leukemia. 2015;29(4):770–5. doi: 10.1038/leu.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schardt JA, Mueller BU, Pabst T. Activation of the unfolded protein response in human acute myeloid leukemia. Methods Enzymol. 2011;489:227–43. doi: 10.1016/B978-0-12-385116-1.00013-3. [DOI] [PubMed] [Google Scholar]
- 52.Schardt JA, Weber D, Eyholzer M, Mueller BU, Pabst T. Activation of the unfolded protein response is associated with favorable prognosis in acute myeloid leukemia. Clin Cancer Res. 2009;15(11):3834–41. doi: 10.1158/1078-0432.CCR-08-2870. [DOI] [PubMed] [Google Scholar]
- 53.Schardt JA, Eyholzer M, Timchenko NA, Mueller BU, Pabst T. Unfolded protein response suppresses CEBPA by induction of calreticulin in acute myeloid leukaemia. J Cell Mol Med. 2010;14(6B):1509–19. doi: 10.1111/j.1582-4934.2009.00870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haefliger S, Klebig C, Schaubitzer K, Schardt J, Timchenko N, Mueller BU, et al. Protein disulfide isomerase blocks CEBPA translation and is up-regulated during the unfolded protein response in AML. Blood. 2011;117(22):5931–40. doi: 10.1182/blood-2010-08-304485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Collins SJ. Retinoic acid receptors, hematopoiesis and leukemogenesis. Curr Opin Hematol. 2008;15(4):346–51. doi: 10.1097/MOH.0b013e3283007edf. [DOI] [PubMed] [Google Scholar]
- 56.Grignani F, Fagioli M, Alcalay M, Longo L, Pandolfi PP, Donti E, et al. Acute promyelocytic leukemia: from genetics to treatment. Blood. 1994;83(1):10–25. [PubMed] [Google Scholar]
- 57.Lonard DM, Lanz RB, O’Malley BW. Nuclear receptor coregulators and human disease. Endocr Rev. 2007;28(5):575–87. doi: 10.1210/er.2007-0012. [DOI] [PubMed] [Google Scholar]
- 58.Loinder K, Soderstrom M. Functional analyses of an LXXLL motif in nuclear receptor corepressor (N-CoR) J Steroid Biochem Mol Biol. 2004;91(4–5):191–6. doi: 10.1016/j.jsbmb.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 59.Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty CD, Torchia J, et al. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387(6628):43–8. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- 60.Khan MM, Nomura T, Kim H, Kaul SC, Wadhwa R, Shinagawa T, et al. Role of PML and PML-RARalpha in Mad-mediated transcriptional repression. Mol Cell. 2001;7(6):1233–43. doi: 10.1016/S1097-2765(01)00257-X. [DOI] [PubMed] [Google Scholar]
- 61.Khan MM, Nomura T, Chiba T, Tanaka K, Yoshida H, Mori K, et al. The fusion oncoprotein PML-RARalpha induces endoplasmic reticulum (ER)-associated degradation of N-CoR and ER stress. J Biol Chem. 2004;279(12):11814–24. doi: 10.1074/jbc.M312121200. [DOI] [PubMed] [Google Scholar]
- 62.Greim H, Kaden DA, Larson RA, Palermo CM, Rice JM, Ross D, et al. The bone marrow niche, stem cells, and leukemia: impact of drugs, chemicals, and the environment. Ann N Y Acad Sci. 2014;1310:7–31. doi: 10.1111/nyas.12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 64.Coltella N, Percio S, Valsecchi R, Cuttano R, Guarnerio J, Ponzoni M, et al. HIF factors cooperate with PML-RARalpha to promote acute promyelocytic leukemia progression and relapse. EMBO Mol Med. 2014;6(5):640–50. doi: 10.1002/emmm.201303065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Deeb G, Vaughan MM, McInnis I, Ford LA, Sait SN, Starostik P, et al. Hypoxia-inducible factor-1alpha protein expression is associated with poor survival in normal karyotype adult acute myeloid leukemia. Leuk Res. 2011;35(5):579–84. doi: 10.1016/j.leukres.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 66.Visani G, Bernasconi P, Boni M, Castoldi GL, Ciolli S, Clavio M, et al. The prognostic value of cytogenetics is reinforced by the kind of induction/consolidation therapy in influencing the outcome of acute myeloid leukemia—analysis of 848 patients. Leukemia. 2001;15(6):903–9. doi: 10.1038/sj.leu.2402142. [DOI] [PubMed] [Google Scholar]
- 67.Delaunay J, Vey N, Leblanc T, Fenaux P, Rigal-Huguet F, Witz F, et al. Prognosis of inv(16)/t(16;16) acute myeloid leukemia (AML): a survey of 110 cases from the French AML Intergroup. Blood. 2003;102(2):462–9. doi: 10.1182/blood-2002-11-3527. [DOI] [PubMed] [Google Scholar]
- 68.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8(11):851–64. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- 69.Rouault-Pierre K, Lopez-Onieva L, Foster K, Anjos-Afonso F, Lamrissi-Garcia I, Serrano-Sanchez M, et al. HIF-2alpha protects human hematopoietic stem/progenitors and acute myeloid leukemic cells from apoptosis induced by endoplasmic reticulum stress. Cell Stem Cell. 2013;13(5):549–63. doi: 10.1016/j.stem.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 70.Kuwabara WM, Zhang L, Schuiki I, Curi R, Volchuk A, Alba-Loureiro TC. NADPH oxidase-dependent production of reactive oxygen species induces endoplasmatic reticulum stress in neutrophil-like HL60 cells. PLoS One. 2015;10(2):e0116410. doi: 10.1371/journal.pone.0116410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ilaria RL, Jr. Pathobiology of lymphoid and myeloid blast crisis and management issues. Hematology Am Soc Hematol Educ Program. 2005:188–94. doi:10.1182/asheducation-2005.1.188. [DOI] [PubMed]
- 72.de Boer J, Walf-Vorderwulbecke V, Williams O. In focus: MLL-rearranged leukemia. Leukemia. 2013;27(6):1224–8. doi: 10.1038/leu.2013.78. [DOI] [PubMed] [Google Scholar]
- 73.Bernt KM, Hunger SP. Current concepts in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia. Front Oncol. 2014;4:54. doi: 10.3389/fonc.2014.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood. 2015;125(26):3977–87. doi: 10.1182/blood-2015-02-580043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Woo JS, Alberti MO, Tirado CA. Childhood B-acute lymphoblastic leukemia: a genetic update. Exp Hematol Oncol. 2014;3:16. doi: 10.1186/2162-3619-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45(3):242–52. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005–15. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McCubrey JA, Steelman LS, Kempf CR, Chappell WH, Abrams SL, Stivala F, et al. Therapeutic resistance resulting from mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR signaling pathways. J Cell Physiol. 2011;226(11):2762–81. doi: 10.1002/jcp.22647. [DOI] [PubMed] [Google Scholar]
- 79.Irving J, Matheson E, Minto L, Blair H, Case M, Halsey C, et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood. 2014;124(23):3420–30. doi: 10.1182/blood-2014-04-531871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Knight T, Irving JA. Ras/Raf/MEK/ERK pathway activation in childhood acute lymphoblastic leukemia and its therapeutic targeting. Front Oncol. 2014;4:160. doi: 10.3389/fonc.2014.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Packer LM, Rana S, Hayward R, O’Hare T, Eide CA, Rebocho A, et al. Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug-resistant chronic myeloid leukemia. Cancer Cell. 2011;20(6):715–27. doi: 10.1016/j.ccr.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Klein F, Feldhahn N, Harder L, Wang H, Wartenberg M, Hofmann WK, et al. The BCR-ABL1 kinase bypasses selection for the expression of a pre-B cell receptor in pre-B acute lymphoblastic leukemia cells. J Exp Med. 2004;199(5):673–85. doi: 10.1084/jem.20031637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maino E, Sancetta R, Viero P, Imbergamo S, Scattolin AM, Vespignani M, et al. Current and future management of Ph/BCR-ABL positive ALL. Expert Rev Anticancer Ther. 2014;14(6):723–40. doi: 10.1586/14737140.2014.895669. [DOI] [PubMed] [Google Scholar]
- 84.Tanimura A, Yujiri T, Tanaka Y, Hatanaka M, Mitani N, Nakamura Y, et al. The anti-apoptotic role of the unfolded protein response in Bcr-Abl-positive leukemia cells. Leuk Res. 2009;33(7):924–8. doi: 10.1016/j.leukres.2009.01.027. [DOI] [PubMed] [Google Scholar]
- 85.Dengler MA, Staiger AM, Gutekunst M, Hofmann U, Doszczak M, Scheurich P, et al. Oncogenic stress induced by acute hyper-activation of Bcr-Abl leads to cell death upon induction of excessive aerobic glycolysis. PLoS One. 2011;6(9):e25139. doi: 10.1371/journal.pone.0025139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li Y, Guo Y, Tang J, Jiang J, Chen Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim Biophys Sin. 2014;46(8):629–40. doi: 10.1093/abbs/gmu048. [DOI] [PubMed] [Google Scholar]
- 87.Kharabi Masouleh B, Geng H, Hurtz C, Chan LN, Logan AC, Chang MS, et al. Mechanistic rationale for targeting the unfolded protein response in pre-B acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2014;111(21):E2219–28. doi: 10.1073/pnas.1400958111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Swaminathan S, Huang C, Geng H, Chen Z, Harvey R, Kang H, et al. BACH2 mediates negative selection and p53-dependent tumor suppression at the pre-B cell receptor checkpoint. Nat Med. 2013;19(8):1014–22. doi: 10.1038/nm.3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Miller GD, Bruno BJ, Lim CS. Resistant mutations in CML and Ph(+)ALL—role of ponatinib. Biologics. 2014;8:243–54. doi: 10.2147/BTT.S50734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schultz KR, Bowman WP, Aledo A, Slayton WB, Sather H, Devidas M, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children’s oncology group study. J Clin Oncol. 2009;27(31):5175–81. doi: 10.1200/JCO.2008.21.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Laurenti E, Doulatov S, Zandi S, Plumb I, Chen J, April C, et al. The transcriptional architecture of early human hematopoiesis identifies multilevel control of lymphoid commitment. Nat Immunol. 2013;14(7):756–63. doi: 10.1038/ni.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.van Galen P, Kreso A, Mbong N, Kent DG, Fitzmaurice T, Chambers JE, et al. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature. 2014;510(7504):268–72. doi: 10.1038/nature13228. [DOI] [PubMed] [Google Scholar]
- 93.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 94.Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15(9):494–501. doi: 10.1016/j.tcb.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 95.Bhatia R, Holtz M, Niu N, Gray R, Snyder DS, Sawyers CL, et al. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood. 2003;101(12):4701–7. doi: 10.1182/blood-2002-09-2780. [DOI] [PubMed] [Google Scholar]
- 96.Holtz M, Forman SJ, Bhatia R. Growth factor stimulation reduces residual quiescent chronic myelogenous leukemia progenitors remaining after imatinib treatment. Cancer Res. 2007;67(3):1113–20. doi: 10.1158/0008-5472.CAN-06-2014. [DOI] [PubMed] [Google Scholar]
- 97.Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013;73(6):1993–2002. doi: 10.1158/0008-5472.CAN-12-3109. [DOI] [PubMed] [Google Scholar]
- 98.Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) J Med Chem. 2012;55(16):7193–207. doi: 10.1021/jm300713s. [DOI] [PubMed] [Google Scholar]
- 99.Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117(4):1311–4. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Volkmann K, Lucas JL, Vuga D, Wang X, Brumm D, Stiles C, et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J Biol Chem. 2011;286(14):12743–55. doi: 10.1074/jbc.M110.199737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mimura N, Fulciniti M, Gorgun G, Tai YT, Cirstea D, Santo L, et al. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood. 2012;119(24):5772–81. doi: 10.1182/blood-2011-07-366633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A. 2012;109(15):E869–78. doi: 10.1073/pnas.1115623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tsai YC, Weissman AM. The unfolded protein response, degradation from endoplasmic reticulum and cancer. Genes Cancer. 2010;1(7):764–78. doi: 10.1177/1947601910383011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Moreau P, Attal M, Facon T. Frontline therapy of multiple myeloma. Blood. 2015;125(20):3076–84. doi: 10.1182/blood-2014-09-568915. [DOI] [PubMed] [Google Scholar]
- 105.Niewerth D, Jansen G, Assaraf YG, Zweegman S, Kaspers GJ, Cloos J. Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist Updat. 2015;18:18–35. doi: 10.1016/j.drup.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 106.Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008;112(6):2489–99. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- 107.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143(7):1883–98. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Leung-Hagesteijn C, Erdmann N, Cheung G, Keats JJ, Stewart AK, Reece DE, et al. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell. 2013;24(3):289–304. doi: 10.1016/j.ccr.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kusio-Kobialka M. Pro-survival signaling pathways activated by BCR-ABL oncogene in chronic myelogenous leukemia cells: the role of the perk-EIF2alpha signal path and acetylation of p53. Postepy Biochem. 2014;60(4):401–6. [PubMed] [Google Scholar]
- 110.Ermakova SP, Kang BS, Choi BY, Choi HS, Schuster TF, Ma WY, et al. (-)-Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res. 2006;66(18):9260–9. doi: 10.1158/0008-5472.CAN-06-1586. [DOI] [PubMed] [Google Scholar]
- 111.Uckun FM, Qazi S, Ozer Z, Garner AL, Pitt J, Ma H, et al. Inducing apoptosis in chemotherapy-resistant B-lineage acute lymphoblastic leukaemia cells by targeting HSPA5, a master regulator of the anti-apoptotic unfolded protein response signalling network. Br J Haematol. 2011;153(6):741–52. doi: 10.1111/j.1365-2141.2011.08671.x. [DOI] [PubMed] [Google Scholar]
- 112.Liu Y, Steiniger SC, Kim Y, Kaufmann GF, Felding-Habermann B, Janda KD. Mechanistic studies of a peptidic GRP78 ligand for cancer cell-specific drug delivery. Mol Pharm. 2007;4(3):435–47. doi: 10.1021/mp060122j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rosenes Z, Mulhern TD, Hatters DM, Ilag LL, Power BE, Hosking C, et al. The anti-cancer IgM monoclonal antibody PAT-SM6 binds with high avidity to the unfolded protein response regulator GRP78. PLoS One. 2012;7(9):e44927. doi: 10.1371/journal.pone.0044927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rasche L, Duell J, Morgner C, Chatterjee M, Hensel F, Rosenwald A, et al. The natural human IgM antibody PAT-SM6 induces apoptosis in primary human multiple myeloma cells by targeting heat shock protein GRP78. PLoS One. 2013;8(5):e63414. doi: 10.1371/journal.pone.0063414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rasche L, Duell J, Castro IC, Dubljevic V, Chatterjee M, Knop S, et al. GRP78-directed immunotherapy in relapsed or refractory multiple myeloma—results from a phase 1 trial with the monoclonal immunoglobulin M antibody PAT-SM6. Haematologica. 2015;100(3):377–84. doi: 10.3324/haematol.2014.117945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kriss CL, Pinilla-Ibarz JA, Mailloux AW, Powers JJ, Tang CH, Kang CW, et al. Overexpression of TCL1 activates the endoplasmic reticulum stress response: a novel mechanism of leukemic progression in mice. Blood. 2012;120(5):1027–38. doi: 10.1182/blood-2011-11-394346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Carlini L, Manley S. Live intracellular super-resolution imaging using site-specific stains. ACS Chem Biol. 2013;8(12):2643–8. doi: 10.1021/cb400467x. [DOI] [PubMed] [Google Scholar]
- 118.Sanches M, Duffy NM, Talukdar M, Thevakumaran N, Chiovitti D, Canny MD, et al. Structure and mechanism of action of the hydroxy-aryl-aldehyde class of IRE1 endoribonuclease inhibitors. Nat Commun. 2014;5:4202. doi: 10.1038/ncomms5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Smith AD, Roda D, Yap TA. Strategies for modern biomarker and drug development in oncology. J Hematol Oncol. 2014;7(1):70. doi: 10.1186/s13045-014-0070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186(3):323–31. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tam AB, Koong AC, Niwa M. Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep. 2014;9(3):850–8. doi: 10.1016/j.celrep.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang FM, Galson DL, Roodman GD, Ouyang H. Resveratrol triggers the pro-apoptotic endoplasmic reticulum stress response and represses pro-survival XBP1 signaling in human multiple myeloma cells. Exp Hematol. 2011;39(10):999–1006. doi: 10.1016/j.exphem.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]