

Abstract
The hypothalamus is the principal regulator of body weight and energy balance. It modulates both energy intake and energy expenditure by sensing the energy status of the body through neural inputs from the periphery as well as direct humoral inputs. Leptin, an adipokine, is one of the humoral factors responsible for alerting the hypothalamus that enough energy is stored in the periphery. Plasma leptin levels are positively linked to adiposity; leptin suppress energy intake and stimulates energy expenditure. However, prolonged increases in plasma leptin levels due to obesity cause leptin resistance, affecting both leptin access to hypothalamic neurons and leptin signal transduction within hypothalamic neurons. Decreased sensing of peripheral energy status through leptin may lead to a positive energy balance and gradual gains in weight and adiposity, further worsening leptin resistance. Leptin resistance, increased adiposity, and weight gain are all associated with aging in both humans and animals. Central insulin resistance is associated with similar observations. Therefore, improving the action of humoral factors in the hypothalamus may prevent gradual weight gain, especially during middle age. SIRT1 is a NAD+-dependent protein deacetylase with numerous substrates, including histones, transcription factors, co-factors, and various enzymes. SIRT1 improves both leptin sensitivity and insulin sensitivity by decreasing the levels of several molecules that impair leptin and insulin signal transduction. SIRT1 and NAD+ levels decrease with age in the hypothalamus; increased hypothalamic SIRT1 levels prevent age-associated weight gain and improve leptin sensitivity in mice. Therefore, preventing the age-dependent loss of SIRT1 function in the hypothalamus could improve the action of humoral factors in the hypothalamus as well as central regulation of energy balance.
Keywords: aging, energy homeostasis, energy sensing, inflammation, insulin resistance, leptin resistance, sirtuin, ubiquitin-proteasome system
Introduction
In this review, I will first highlight the impact of obesity prevalence and the importance of age-associated weight gain in this context. Next, I will provide an overview of the central mechanisms for regulating body weight, including a discussion of central leptin/insulin resistance during aging and obesity. Finally, I will explore how hypothalamic SIRT1 may be involved in age-associated weight gain and diet-induced obesity by regulating leptin and insulin sensitivity in the central nervous system. I propose that hypothalamic SIRT1 dysfunction could be a cause of weight gain associated with aging and diet.
Underestimated Prevalence of Obesity-Associated Health Issues
Obesity is a worldwide health concern. Based on the World Health Organization’s definition of obesity [body mass index (BMI) >30 kg/m2], more than 600 million adults were obese and more than 1.9 billion adults were overweight (BMI 25–30 kg/m2) in 2014 (1). In other words, ~13% of the world’s adult population (11% of men and 15% of women) was obese in 2014. These numbers are alarming, but they underestimate the actual impact on health care caused by obesity throughout the world.
Ethnic groups exhibit differing susceptibilities to obesity-associated diseases, such as diabetes. In particular, Asians are more vulnerable to the detrimental effects of obesity than Caucasians. The prevalence of diabetes among Asian descendants living in the United States and Canada with BMI of 25 kg/m2 is equal to that of Caucasians with BMI of 30 kg/m2 (2, 3). This phenomenon is partly due to different pancreatic beta-cell reserve capacity (4), but Asians also have more visceral adipose mass than Caucasians at the same BMI (5). Based on the risk of developing metabolic syndrome, the definition of obesity in Japan is BMI > 25 kg/m2, not BMI > 30 kg/m2 (6). Although ~5% of Japanese males in their 30s and 40s are obese according to the World Health Organization’s definition (BMI > 30 kg/m2), this number increases to ~35% if the Japanese definition of obesity (BMI > 25 kg/m2) is used (Figure 1) (7). Therefore, the true number of people at health risk due to “obesity” around the world is somewhere between 600 million (BMI > 30 kg/m2) and 2.5 billion (BMI > 25 kg/m2).
Figure 1.

Age-dependent distribution of Japanese with BMI > 30 kg/m2 (solid bar) and BMI > 25 kg/m2 (open bar) in 2014. (Based on data from the Institute for Health Metrics and Evaluation (IHME). Overweight and Obesity Viz. Seattle, WA, USA: IHME, University of Washington, 2014. Available from: http://vizhub.healthdata.org/obesity).
Obesity is commonly discussed in the context of metabolic syndrome and other diseases that result from obesity. However, the prevalence of obesity is positively associated with age, and the global emergence of overweight and obesity is compounded by simultaneous aging of the population (8, 9). Obesity itself is a risk factor for age-associated diseases (10–13); it accelerates cellular processes in a manner similar to aging and shortens lifespan (14–17). Therefore, understanding gradual declines in quality of health through aging and obesity is as important as simply understanding the consequences of obesity.
Although age-associated weight gain is important for understanding gradual declines in quality of health, fewer studies have addressed the mechanisms of these declines than have investigated diet-induced obesity, partly because the former process is much harder to study than the latter. Although it is difficult to completely separate the contributions to weight gain of age and diet, not everyone in the world eats the high-calorie diet employed in studies of diet-induced obesity; a significant proportion of the population gains weight during middle age. Therefore, outreach strategies targeting age-associated weight gain are expected to impact the health of many people.
Do different mechanisms underpin weight gain associated with age and weight gain caused by diet, or do common mechanisms underlie both processes? In order to tackle the obesity crisis, we must understand how body weight is regulated, as well as how the intricate system that regulates energy homeostasis could be disrupted through both aging and diet.
Brief Overview of Homeostatic Control of Body Weight and Energy Balance by the Central Nervous System
The hypothalamus is the center of homeostatic control of body weight and energy balance. It integrates energy information conveyed from the periphery by nutrients and hormones through two pathways (neural and humoral) and regulates energy intake and expenditure (Figure 2).
Figure 2.
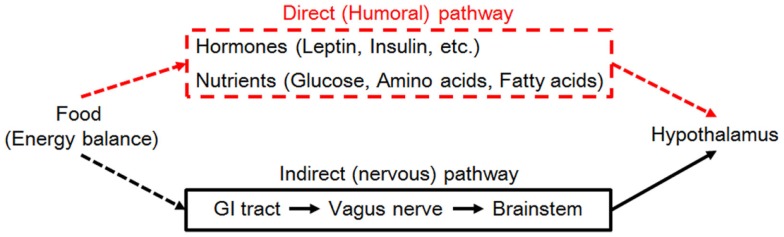
Neural signaling (right, black lines) and hormonal signaling (left, red lines) from the periphery are required for nutrient/energy sensing by the hypothalamus (blue) to regulate body weight. Abbreviation: GI, gastrointestinal.
The neural pathway senses peripheral energy status via vagal afferents innervating the gastrointestinal tract and hepatic portal veins. Nutritional information gathered by vagal afferents is sent to the solitary tract of the brainstem and subsequently into the hypothalamus. The humoral pathway consists of direct input of nutrients and hormones to the primary center for body-weight control: the arcuate nucleus of the hypothalamus (ARC). The ARC is located close to the median eminence, which provides humoral factors with access to the central nervous system. Humoral factors can enter the cerebrospinal fluid (CSF) in the third ventricle and gain access to ARC neurons because the tanycytes lining the third ventricle adjacent to the ARC are permissive to these nutrient cues (18).
Once nutrients and hormones gain access to ARC neurons, they can directly affect neuronal activity. Two major subtypes of neurons located in the ARC are proopiomelanocortin (POMC) neurons and Agouti-related peptide (AgRP) neurons; both types of neurons receive signals containing information about energy status (19). POMC neurons promote weight loss and AgRP neurons promote weight gain. These neurons provide similar projections to secondary centers for body-weight control, such as the paraventricular nucleus of the hypothalamus and the lateral hypothalamus (Figure 3). Activity of the neurons in these secondary centers may be regulated partly through the melanocortin type 4 receptors, since POMC neurons secrete the agonist α-melanocyte-stimulating hormone (made by processing the POMC peptide) and AgRP is an inverse agonist for melanocortin type 4 receptors. AgRP neurons also use the neurotransmitters neuropeptide Y and GABA to suppress target-neuron activity, including that of POMC neurons in the ARC. POMC neurons, AgRP neurons, and melanocortin type 4 receptors comprise the central melanocortin system.
Figure 3.

Humoral factors leptin and insulin, that convey nutrient cues, act on the central melanocortin system (POMC and AgRP neurons) in the ARC to regulate body weight.
Leptin is an adipokine critical to the regulation of energy homeostasis (20). Intracellular leptin signaling is initiated by leptin binding to the long form of leptin receptor (LepRb), which has the intracellular domain, causing a conformational changes in LepRb and promoting the phosphorylation and activation of Jak2 (21). Activated Jak2 phosphorylates three tyrosine residues in the cytoplasmic domain of LepRb (Y985, Y1077, and Y1138), leading to further signal transduction involving STAT3, STAT5, and the ERK and PI3K/AKT pathways. LepRb is expressed in several brain nuclei, including the ARC, the paraventricular nucleus of the hypothalamus, and the dorsomedial, lateral, and ventromedial regions of the hypothalamus (22). Mutations in leptin, leptin receptor, and the central melanocortin system are the most common mutations in the monogenic form of obesity in humans (23). A homozygous loss-of-function mutation in the leptin receptor (the db mutation) causes hyperphagia and obesity, leading to diabetes in mice (24, 25).
Insulin is a pancreatic hormone that plays a crucial role in lowering blood glucose levels. It also contributes to the regulation of body weight. Although insulin acts on peripheral tissues (such as liver, skeletal muscle, and adipose tissue) to cause anabolic effects (to cause weight gain), insulin act as a catabolic anorexigenic signal in the central nervous system (to cause weight loss) (26). The insulin receptor is expressed widely in the central nervous system (27). Insulin binding causes the insulin receptor to recruit insulin receptor substrate (IRS) proteins. IRS-2 is the main mediator of insulin intracellular responses in the brain (28, 29). IRS protein activation leads to activation of PI3K and subsequent activation of AKT. Although the glucose-lowering effect of insulin is mediated by the insulin-sensitive glucose transporter GLUT4 in the periphery, glucose transport into most neurons is GLUT3-dependent; transport into the glia and brain endothelial cells is GLUT1-dependent (30). Therefore, insulin is not needed for glucose transport into most brain cells. Instead, insulin in the brain is important for the central regulation of energy homeostasis and glucose homeostasis.
Both leptin and insulin regulate POMC neurons and AgRP neurons at multiple levels. They regulate the transcription of Pomc and AgRP via their downstream target transcription factors STAT3 and FoxO1, respectively, and promote the expression of anorexigenic POMC while suppressing orexigenic AgRP (31, 32). FoxO1 also regulates the expression of carboxypeptidase E, which is necessary for processing POMC into α-melanocyte-stimulating hormone (an agonist of melanocortin type 4 receptors) (33). Leptin and insulin also regulate the activities of POMC neurons and AgRP neurons (34). For example, both leptin and insulin act on POMC neurons to increase sympathetic activity to adipose tissues and to promote the browning of white fat (35). Therefore, these two hormonal signals of satiety regulate transcription, peptide processing, the activity of central melanocortin neurons, and energy and glucose homeostasis.
Three Layers of Leptin/Insulin Resistance in the Central Regulation of Body Weight
The anorexigenic hormone leptin needs to enter the central nervous system and reach target neurons in order to suppress food intake and to stimulate energy expenditure. Leptin action encounters three barriers: the blood–CSF barrier, leptin uptake by the target neuron, and intracellular leptin-signaling resistance.
First, leptin must exit the bloodstream and gain access to the CSF. The short isoform of the leptin receptor is highly expressed in cerebral microvessels and choroid plexuses, and is considered to be the main receptor by which leptin crosses the blood–brain barrier and the blood–CSF barrier (36). Megalin expressed by choroid plexuses can also act as a receptor to transport leptin from the blood to the CSF (37). The fenestrated endothelium of median eminence microvessels and the tight junctions between tanycytes together compose the blood–CSF barrier adjacent to the ARC (18). VEGF-A expression in tanycytes modulates the properties of this barrier, and ERK signaling in tanycytes promotes leptin transport across tanycytes to the CSF (38, 39). Once leptin enters the CSF/brain, it must find its target receptor (LepRb), but gliosis can interfere with the diffusion of leptin to LepRb on the target neurons (40). Finally, intracellular leptin signaling can be down-regulated by several molecules, such as PTP-1B, TC-PTP, SOCS3, and endospanin 1, which act on JAK2, STAT3, LepRb, and LepRb endocytosis, respectively (41, 42).
Although the source of central insulin is debated, peripheral administration of insulin raises insulin concentrations within the CSF (43), indicating that peripheral insulin also serves as a satiety signal for the central nervous system by crossing the blood–brain barrier (44). Kinetic studies indicate that plasma-to-CSF transport of insulin involves a saturable mechanism (45, 46); this transport is decreased in several insulin-resistant states (47–49). Megalin is implicated in insulin transport, at least across renal tubular epithelial cells (50), which suggest that megalin may also work at the blood–CSF barrier as a carrier for insulin. Both PTP-1B and SOCS3 have been reported to cause insulin resistance at the level of the insulin receptor and IRS proteins, respectively (26). Therefore, common mechanisms are involved in central resistance to leptin and insulin (Figure 4).
Figure 4.

Common mechanisms for central leptin/insulin resistance.
Diet-Induced Obesity and Aging Cause Central Resistance to Leptin and Insulin
Diet-induced obesity causes central leptin resistance first by affecting the central access of leptin and later by causing leptin resistance within the central nervous system (51). Obesity is associated with decreased leptin transport across the blood–brain barrier in rats (36) and in humans (52), which explains why obese humans have low CSF leptin levels despite having high-serum leptin levels. Within the central nervous system, diet-induced obesity does not uniformly cause leptin resistance. Diet-induced obesity caused by intake of a chronic high-fat diet mainly induces cellular leptin resistance in the ARC and the ventral tegmental area, but not in the lateral hypothalamus, ventromedial hypothalamus, and dorsomedial hypothalamus (53). Diet-induced obesity also induces astrogliosis (along with activation of microglia), which prevents circulating metabolic feedback factors, such as leptin from accessing neurons (54). Aging also impairs the central response to leptin (55–59) by impairing central leptin access and cellular leptin signaling (60). Aging is associated with down-regulation of megalin expression (37), decreased leptin uptake in the hypothalamus due to decreased expression of leptin receptor mRNA (61), decreased levels of leptin receptor protein in the hypothalamus (62), and increased PTP1B levels in the hypothalamus (63).
The central insulin response is also attenuated by aging and diet-induced obesity (64–67). Insulin levels in the CSF are paradoxically low- compared to high-serum insulin levels in obese humans (68) and in genetically induced and diet-induced obese animals (49, 69). Consumption of a high-fat diet triggers hypothalamic angiopathy (70); endothelial cells of brain microvessels in obese fa/fa rats exhibit reduced insulin binding, leading to reduced internalization of the insulin–insulin receptor complex in rat brain endothelial cells (71). Expression of the insulin receptor decreases with aging in the central nervous system, especially in the hypothalamus, cerebral cortex, and hippocampus in rats (72). Central administration of insulin fails to reduce food intake under conditions of obesity (65, 67, 73), and intranasal application of insulin to the human brain improves peripheral insulin sensitivity in lean but not in obese men (74), indicating that obesity also causes central insulin resistance.
Therefore, previous studies indicate that both central leptin resistance and central insulin resistance are induced by diet-induced obesity and aging at multiple levels. Does a common mechanism underlie these phenomena?
Hypothalamic Inflammation as a Culprit for Central Leptin/Insulin Resistance
Hypothalamic inflammation is induced during diet-induced obesity both in rodents and in humans (75). Although intracellular fatty-acid sensing within the hypothalamus is important for the regulation of energy balance (76), excessive amounts of fatty acid in the diet cause hypothalamic inflammation and lead to obesity. Consumption of a diet rich in saturated fatty acids promotes inflammation, gliosis, and neuronal stresses in the mediobasal hypothalamus, along with inflammatory activation of microglia. Depleting microglia from the mediobasal hypothalamus of mice blocks the inflammation and neuronal stresses induced by dietary saturated fatty acids and improve leptin sensitivity (77). Saturated fatty acids can activate toll-like receptor 2- and 4-dependent signaling, leading to induction of the pro-inflammatory signaling mediated by JNK and NF-κB (78, 79). Excess amounts of free fatty acids, glucose, and amino acids due to over-nutrition induce endoplasmic reticulum (ER) stress and oxidative stress, which also promote the activation of pro-inflammatory signaling and defective autophagy (79). These metabolically induced pro-inflammatory changes in the hypothalamus cause defective intracellular leptin and insulin signaling, leading to central leptin/insulin resistance (80–84) (Figure 5).
Figure 5.

Hypothalamic inflammation and central leptin/insulin resistance.
Pro-inflammatory signals in the hypothalamus are also involved in the aging of mice. Aging activates microglia and NF-κB signaling in the hypothalamus, inhibiting these reactions can improve age-dependent declines in body function in mice (85). Therefore, hypothalamic inflammation, specifically microglial activation and induction of NF-κB signaling, is involved in age-dependent and diet-induced obesity, which are accompanied by central leptin/insulin resistance.
Hypothalamic SIRT1 Ameliorates Central Leptin Resistance and Central Insulin Resistance
SIRT1 is an NAD+-dependent protein deacetylase (86) and an energy-sensing molecule responsible for promoting healthy longevity through caloric restriction (87). Caloric restriction prevents aging-associated central leptin resistance in rats (88, 89). In peripheral tissues, SIRT1 has been demonstrated to promote fatty-acid oxidation and to improve insulin sensitivity (90). Single nucleotide polymorphisms in human SIRT1 are linked to both adult obesity (91–94) and childhood obesity (95), indicating that SIRT1 may regulate body weight.
Several groups have reported genetic manipulation of SIRT1 in the hypothalamus and analyzed the effects of this manipulation on energy and glucose homeostasis. Genetic loss of Sirt1 in anorexigenic POMC neurons causes leptin resistance and decreases energy expenditure, whereas increasing SIRT1 levels in POMC neurons improves leptin sensitivity and ameliorates the decreased energy expenditure caused by an insulin-resistant form of FOXO1 in mice (96–98). Genetic loss of Sirt1 in orexigenic AgRP neurons causes weight loss due to decreased food intake via reductions in the firing ability of AgRP neurons in mice (99). Increasing SIRT1 levels in AgRP neurons also decreases food intake and body weight due to improved nutrient/hormone sensing in mice (97). Genetic manipulation of Sirt1 in steroidgenic factor 1 (SF1)-positive ventromedial hypothalamic neurons alters sensitivity to leptin and orexin-A, and modulates glucose uptake by skeletal muscle via the sympathetic nervous system (100). Therefore, SIRT1 stimulates energy expenditure, suppresses food intake, and regulates glucose homeostasis in POMC neurons, AgRP neurons, and ventromedial hypothalamic SF1-positive neurons, respectively, partly through improved nutrient/hormone sensing.
How does SIRT1 improve central leptin/insulin sensitivity? SIRT1 can down-regulate proteins that promote leptin resistance, such as PTP1B, TC-PTP, and SOCS3 (97, 101). It also promotes insulin sensitivity by suppressing PTP1B [which also contributes to insulin resistance (101)], promoting IRS2 function (102), and down-regulating the transcription factor FOXO1 by promoting its degradation (98, 103). Down-regulation of NF-κB signaling may also improve central leptin/insulin sensitivity because SIRT1 suppresses inflammatory reactions by suppressing the p65RelA subunit of NF-κB (104). SIRT1 levels are reported to decrease as microglia age, and microglial SIRT1 deficiency plays a causative role in aging-mediated memory deficits in mice (105). However, the actions of microglial SIRT1 in the context of obesity, induced by aging or by diet, remain elusive. Other candidates for SIRT1’s downstream effects on central leptin/insulin sensitivity are autophagy and ER stress, since disruption of autophagy and increased ER stress are both linked to hypothalamic leptin resistance and perturbed energy homeostasis (80, 82, 83, 106, 107); SIRT1 regulates both autophagy and ER stress through substrates, such as LC3 (108), Atg5, Atg7, Atg8 (109), and XBP1s (110). However, these hypotheses need to be tested experimentally.
SIRT1 can influence epigenetic regulation by directly modifying histones (86) and by affecting DNA methylation through DNMT1 (111) and MeCP2 (112). Thus, SIRT1 can modify the global transcriptional landscape through epigenetic regulation and the targeting of numerous transcription factors and co-factors (90). Therefore, unidentified molecules may also play roles in the improvement of central leptin/insulin sensitivity by SIRT1.
Potential Role of Hypothalamic SIRT1 in Weight Gain and Aging
Overall, SIRT1 in the hypothalamus improves energy and glucose homeostasis and central leptin/insulin sensitivity. Because there is no single reliable marker for SIRT1 function in vivo, here I discuss changes in NAD+ content and SIRT1 protein levels during aging and diet-induced obesity in the hypothalamus.
NAD+ content in the brain decreases with age in humans and in mice (113, 114). In mice, hypothalamic NAD+ content is significantly decreased in diet-induced obesity (after 4 weeks of a high-fat, high-sucrose diet) and in genetically induced obesity (with a homozygous db mutation) (97). Nicotinamide phosphoribosyltransferase (NAMPT), an essential enzyme in the NAD+ biosynthetic pathway that converts nicotinamide into nicotinamide mononucleotide, exists in two forms (intracellular and extracellular) in mammals (115). The brain expresses intracellular NAMPT at very low levels, and NAMPT levels decrease with aging in multiple organs (116, 117). The extracellular form of NAMPT is secreted from adipose tissue to affect NAD+ concentrations in the hypothalamus. Systemic injection of a NAMPT-neutralizing antibody decreases hypothalamic NAD+ content (118). Adipose tissue-specific Nampt knockout mice exhibit reduced plasma levels of extracellular NAMPT as well as reduced NAD+ concentrations in the hypothalamus; in contrast, adipose tissue-specific Nampt knock-in mice display increased plasma levels of extracellular NAMPT and increased hypothalamic NAD+ content (118). Therefore, although the brain relies on circulating extracellular NAMPT and nicotinamide mononucleotide for NAD+ biosynthesis, the supplies of these precursors decrease with age. Supplementation with these precursors effectively prevents diet-induced obesity and aging-induced diabetes in mice (116, 119), and therefore, this supplementation is currently under exploration as a strategy for counteracting aging and obesity in humans.
Restoring SIRT1 protein levels is a less-explored approach to this goal. SIRT1 levels within the ARC decrease with age (97, 120) and after 4 weeks of consumption of a high-fat, high-sucrose diet in mice (97). Increasing SIRT1 protein levels specifically in POMC neurons or in AgRP neurons via genetic approaches was sufficient to prevent age-associated weight gain by stimulating energy expenditure and by suppressing food intake through improved leptin sensitivity (97). However, feeding mice a high-fat, high-sucrose diet reduced both endogenous SIRT1 protein and overexpressed SIRT1 protein in ARC, and eliminated the beneficial effects of genetic overexpression of SIRT1. Therefore, along with increasing SIRT1 activity by providing more NAD+ or a pharmacological activator, increasing the effective enzyme concentration per se also yields benefits. To enable such a SIRT1 “booster” approach, the mechanisms responsible for the decline in ARC SIRT1 levels during diet-induced obesity and/or aging must be identified.
Diet similarly affected ARC SIRT1 protein levels when SIRT1 was expressed from the endogenous Sirt1 locus and when coding-sequence Sirt1 cDNA (without any 5′-UTR or 3′-UTR) was expressed from the endogenous Rosa26 promoter (97). These data indicate that the diet-induced reduction in ARC SIRT1 levels is unlikely to be driven by transcriptional or post-transcriptional regulation, but is likely mediated by post-translational regulation. SIRT1 degradation in vitro and in vivo could be regulated by the ubiquitin-proteasome system in the hypothalamus (121) and possibly in other tissues (122–124). Although exposure to a 4-week, high-fat, high-sucrose diet was sufficient to decrease ARC SIRT1 levels in mice, neither a high-fat diet nor a high-sucrose diet alone was as effective (unpublished observation). Furthermore, the effect was not as clearly evident after 2 weeks of a high-fat, high-sucrose diet (unpublished observation). Therefore, declines in ARC SIRT1 levels may not directly result from nutrients within the diet, but rather may be due to metabolic changes within the hypothalamus or in the periphery caused by chronic over-nutrition. Pro-inflammatory signaling could underlie the declines in ARC SIRT1 levels during aging and during diet-induced obesity because inflammatory changes occur during aging and during chronic over-nutrition.
Conclusion
Central leptin and insulin resistance, hallmarks of disrupted nutrient/energy sensing by the hypothalamus, are common mechanisms for weight gain caused by aging and diet. Hypothalamic SIRT1 can improve these disruptions by acting on several targets that cause central leptin/insulin resistance. Meanwhile, aging and diets that promote weight gain suppress hypothalamic SIRT1 function by affecting the levels of both SIRT1 and NAD+, which is required for SIRT1 activity. Restoration of hypothalamic SIRT1 function can prevent age-associated weight gain in mice, indicating that improving hypothalamic SIRT1 function at multiple levels via supplementation with NAD+ intermediates, SIRT1 activators, and a SIRT1 “booster” may enable novel treatments of weight gain related to metabolic syndromes as well as weight gain caused by aging (Figure 6).
Figure 6.

Schematic of potential strategies to ameliorate hypothalamic SIRT1 dysfunction caused by aging- and diet-induced obesity.
Conflict of Interest Statement
The author declares that the research in this review was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank Dr. Tadahiro Kitamura, Director of the Metabolic Signaling Research Center at the Institute for Molecular and Cellular Regulation, Gunma University, Maebashi, Japan, for his general support for research involving hypothalamic SIRT1. I also thank lab members for their support. This work was funded by a grant-in-aid for scientific research (C, 25460310) to TS by Ministry of Education, Culture, Sports, Science and Technology (MEXT) Japan.
References
- 1.Organization WH. Obesity and Overweight (2015). Available from: http://www.who.int/mediacentre/factsheets/fs311/en/
- 2.Maskarinec G, Erber E, Grandinetti A, Verheus M, Oum R, Hopping BN, et al. Diabetes incidence based on linkages with health plans: the multiethnic cohort. Diabetes (2009) 58(8):1732–8. 10.2337/db08-1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiu M, Austin PC, Manuel DG, Shah BR, Tu JV. Deriving ethnic-specific BMI cutoff points for assessing diabetes risk. Diabetes Care (2011) 34(8):1741–8. 10.2337/dc10-2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jensen CC, Cnop M, Hull RL, Fujimoto WY, Kahn SE. Beta-cell function is a major contributor to oral glucose tolerance in high-risk relatives of four ethnic groups in the U.S. Diabetes (2002) 51(7):2170–8. 10.2337/diabetes.51.7.2170 [DOI] [PubMed] [Google Scholar]
- 5.Lear SA, Humphries KH, Kohli S, Chockalingam A, Frohlich JJ, Birmingham CL. Visceral adipose tissue accumulation differs according to ethnic background: results of the Multicultural Community Health Assessment Trial (M-CHAT). Am J Clin Nutr (2007) 86(2):353–9. [DOI] [PubMed] [Google Scholar]
- 6.Examination Committee of Criteria for ‘Obesity Disease’ in Japan, Japan Society for the Study of Obesity. New criteria for ‘obesity disease’ in Japan. Circ J (2002) 66(11):987–92. 10.1253/circj.66.987 [DOI] [PubMed] [Google Scholar]
- 7.IHME. Global Burden of Diseases 2013 (2015). Available from: http://vizhub.healthdata.org/obesity
- 8.Doak CM, Wijnhoven TM, Schokker DF, Visscher TL, Seidell JC. Age standardization in mapping adult overweight and obesity trends in the WHO European region. Obes Rev (2012) 13(2):174–91. 10.1111/j.1467-789X.2011.00943.x [DOI] [PubMed] [Google Scholar]
- 9.Kelly T, Yang W, Chen CS, Reynolds K, He J. Global burden of obesity in 2005 and projections to 2030. Int J Obes (Lond) (2008) 32(9):1431–7. 10.1038/ijo.2008.102 [DOI] [PubMed] [Google Scholar]
- 10.Kawachi I. Physical and psychological consequences of weight gain. J Clin Psychiatry (1999) 60(Suppl 21):5–9. [PubMed] [Google Scholar]
- 11.Khaodhiar L, McCowen KC, Blackburn GL. Obesity and its comorbid conditions. Clin Cornerstone (1999) 2(3):17–31. 10.1016/S1098-3597(99)90002-9 [DOI] [PubMed] [Google Scholar]
- 12.Lean ME, Han TS, Seidell JC. Impairment of health and quality of life using new US federal guidelines for the identification of obesity. Arch Intern Med (1999) 159(8):837–43. 10.1001/archinte.159.8.837 [DOI] [PubMed] [Google Scholar]
- 13.Thompson D, Edelsberg J, Colditz GA, Bird AP, Oster G. Lifetime health and economic consequences of obesity. Arch Intern Med (1999) 159(18):2177–83. 10.1001/archinte.159.18.2177 [DOI] [PubMed] [Google Scholar]
- 14.Calle EE, Thun MJ, Petrelli JM, Rodriguez C, Heath CW, Jr. Body-mass index and mortality in a prospective cohort of U.S. adults. N Engl J Med (1999) 341(15):1097–105. 10.1056/NEJM199910073411501 [DOI] [PubMed] [Google Scholar]
- 15.Ahima RS. Connecting obesity, aging and diabetes. Nat Med (2009) 15(9):996–7. 10.1038/nm0909-996 [DOI] [PubMed] [Google Scholar]
- 16.Peto R, Whitlock G, Jha P. Effects of obesity and smoking on U.S. life expectancy. N Engl J Med (2010) 362(9):855–6; author reply 6–7. 10.1056/NEJMc1000079 [DOI] [PubMed] [Google Scholar]
- 17.Russell SJ, Kahn CR. Endocrine regulation of ageing. Nat Rev Mol Cell Biol (2007) 8(9):681–91. 10.1038/nrm2234 [DOI] [PubMed] [Google Scholar]
- 18.Mullier A, Bouret SG, Prevot V, Dehouck B. Differential distribution of tight junction proteins suggests a role for tanycytes in blood-hypothalamus barrier regulation in the adult mouse brain. J Comp Neurol (2010) 518(7):943–62. 10.1002/cne.22273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasaki T, Kitamura T. Roles of FoxO1 and Sirt1 in the central regulation of food intake. Endocr J (2010) 57(11):939–46. 10.1507/endocrj.K10E-320 [DOI] [PubMed] [Google Scholar]
- 20.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature (1998) 395(6704):763–70. 10.1038/27376 [DOI] [PubMed] [Google Scholar]
- 21.Myers MG, Jr, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab (2010) 21(11):643–51. 10.1016/j.tem.2010.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wada N, Hirako S, Takenoya F, Kageyama H, Okabe M, Shioda S. Leptin and its receptors. J Chem Neuroanat (2014) 6(1–62):191–9. 10.1016/j.jchemneu.2014.09.002 [DOI] [PubMed] [Google Scholar]
- 23.Farooqi S, O’Rahilly S. Genetics of obesity in humans. Endocr Rev (2006) 27(7):710–8. 10.1210/er.2006-0040 [DOI] [PubMed] [Google Scholar]
- 24.Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature (1996) 379(6566):632–5. 10.1038/379632a0 [DOI] [PubMed] [Google Scholar]
- 25.Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell (1996) 84(3):491–5. 10.1016/S0092-8674(00)81294-5 [DOI] [PubMed] [Google Scholar]
- 26.Vogt MC, Bruning JC. CNS insulin signaling in the control of energy homeostasis and glucose metabolism - from embryo to old age. Trends Endocrinol Metab (2013) 24(2):76–84. 10.1016/j.tem.2012.11.004 [DOI] [PubMed] [Google Scholar]
- 27.Havrankova J, Roth J, Brownstein M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature (1978) 272(5656):827–9. 10.1038/272827a0 [DOI] [PubMed] [Google Scholar]
- 28.Araki E, Lipes MA, Patti ME, Bruning JC, Haag B, III, Johnson RS, et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature (1994) 372(6502):186–90. 10.1038/372186a0 [DOI] [PubMed] [Google Scholar]
- 29.Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature (1994) 372(6502):182–6. 10.1038/372182a0 [DOI] [PubMed] [Google Scholar]
- 30.McEwen BS, Reagan LP. Glucose transporter expression in the central nervous system: relationship to synaptic function. Eur J Pharmacol (2004) 490(1–3):13–24. 10.1016/j.ejphar.2004.02.041 [DOI] [PubMed] [Google Scholar]
- 31.Kitamura T, Feng Y, Kitamura YI, Chua SC, Jr, Xu AW, Barsh GS, et al. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med (2006) 12(5):534–40. 10.1038/nm1392 [DOI] [PubMed] [Google Scholar]
- 32.Kim MS, Pak YK, Jang PG, Namkoong C, Choi YS, Won JC, et al. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci (2006) 9(7):901–6. 10.1038/nn1731 [DOI] [PubMed] [Google Scholar]
- 33.Plum L, Lin HV, Dutia R, Tanaka J, Aizawa KS, Matsumoto M, et al. The obesity susceptibility gene Cpe links FoxO1 signaling in hypothalamic pro-opiomelanocortin neurons with regulation of food intake. Nat Med (2009) 15(10):1195–201. 10.1038/nm.2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, et al. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest (2008) 118(5):1796–805. 10.1172/JCI32964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dodd GT, Decherf S, Loh K, Simonds SE, Wiede F, Balland E, et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell (2015) 160(1–2):88–104. 10.1016/j.cell.2014.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burguera B, Couce ME, Curran GL, Jensen MD, Lloyd RV, Cleary MP, et al. Obesity is associated with a decreased leptin transport across the blood-brain barrier in rats. Diabetes (2000) 49(7):1219–23. 10.2337/diabetes.49.7.1219 [DOI] [PubMed] [Google Scholar]
- 37.Dietrich MO, Spuch C, Antequera D, Rodal I, de Yebenes JG, Molina JA, et al. Megalin mediates the transport of leptin across the blood-CSF barrier. Neurobiol Aging (2008) 29(6):902–12. 10.1016/j.neurobiolaging.2007.01.008 [DOI] [PubMed] [Google Scholar]
- 38.Balland E, Dam J, Langlet F, Caron E, Steculorum S, Messina A, et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab (2014) 19(2):293–301. 10.1016/j.cmet.2013.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langlet F, Levin BE, Luquet S, Mazzone M, Messina A, Dunn-Meynell AA, et al. Tanycytic VEGF-A boosts blood-hypothalamus barrier plasticity and access of metabolic signals to the arcuate nucleus in response to fasting. Cell Metab (2013) 17(4):607–17. 10.1016/j.cmet.2013.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jastroch M, Morin S, Tschop MH, Yi CX. The hypothalamic neural-glial network and the metabolic syndrome. Best Pract Res Clin Endocrinol Metab (2014) 28(5):661–71. 10.1016/j.beem.2014.02.002 [DOI] [PubMed] [Google Scholar]
- 41.St-Pierre J, Tremblay ML. Modulation of leptin resistance by protein tyrosine phosphatases. Cell Metab (2012) 15(3):292–7. 10.1016/j.cmet.2012.02.004 [DOI] [PubMed] [Google Scholar]
- 42.Seron K, Couturier C, Belouzard S, Bacart J, Monte D, Corset L, et al. Endospanins regulate a postinternalization step of the leptin receptor endocytic pathway. J Biol Chem (2011) 286(20):17968–81. 10.1074/jbc.M111.224857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blazquez E, Velazquez E, Hurtado-Carneiro V, Ruiz-Albusac JM. Insulin in the brain: its pathophysiological implications for states related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front Endocrinol (2014) 5:161. 10.3389/fendo.2014.00161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwartz MW, Bergman RN, Kahn SE, Taborsky GJ, Jr, Fisher LD, Sipols AJ, et al. Evidence for entry of plasma insulin into cerebrospinal fluid through an intermediate compartment in dogs. Quantitative aspects and implications for transport. J Clin Invest (1991) 88(4):1272–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banks WA, Jaspan JB, Kastin AJ. Selective, physiological transport of insulin across the blood-brain barrier: novel demonstration by species-specific radioimmunoassays. Peptides (1997) 18(8):1257–62. 10.1016/S0196-9781(97)00198-8 [DOI] [PubMed] [Google Scholar]
- 46.Schwartz MW, Sipols A, Kahn SE, Lattemann DF, Taborsky GJ, Jr, Bergman RN, et al. Kinetics and specificity of insulin uptake from plasma into cerebrospinal fluid. Am J Physiol (1990) 259(3 Pt 1):E378–83. [DOI] [PubMed] [Google Scholar]
- 47.Baura GD, Foster DM, Kaiyala K, Porte D, Jr, Kahn SE, Schwartz MW. Insulin transport from plasma into the central nervous system is inhibited by dexamethasone in dogs. Diabetes (1996) 45(1):86–90. 10.2337/diabetes.45.1.86 [DOI] [PubMed] [Google Scholar]
- 48.Begg DP, Mul JD, Liu M, Reedy BM, D’Alessio DA, Seeley RJ, et al. Reversal of diet-induced obesity increases insulin transport into cerebrospinal fluid and restores sensitivity to the anorexic action of central insulin in male rats. Endocrinology (2013) 154(3):1047–54. 10.1210/en.2012-1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaiyala KJ, Prigeon RL, Kahn SE, Woods SC, Schwartz MW. Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes (2000) 49(9):1525–33. 10.2337/diabetes.49.9.1525 [DOI] [PubMed] [Google Scholar]
- 50.Orlando RA, Rader K, Authier F, Yamazaki H, Posner BI, Bergeron JJ, et al. Megalin is an endocytic receptor for insulin. J Am Soc Nephrol (1998) 9(10):1759–66. [DOI] [PubMed] [Google Scholar]
- 51.Lin S, Thomas TC, Storlien LH, Huang XF. Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. Int J Obes Relat Metab Disord (2000) 24(5):639–46. 10.1038/sj.ijo.0801209 [DOI] [PubMed] [Google Scholar]
- 52.Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Opentanova I, Goldman WH, et al. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet (1996) 348(9021):159–61. 10.1016/S0140-6736(96)03173-X [DOI] [PubMed] [Google Scholar]
- 53.Matheny M, Shapiro A, Tumer N, Scarpace PJ. Region-specific diet-induced and leptin-induced cellular leptin resistance includes the ventral tegmental area in rats. Neuropharmacology (2011) 60(2–3):480–7. 10.1016/j.neuropharm.2010.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Horvath TL, Sarman B, Garcia-Caceres C, Enriori PJ, Sotonyi P, Shanabrough M, et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc Natl Acad Sci U S A (2010) 107(33):14875–80. 10.1073/pnas.1004282107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scarpace PJ, Matheny M, Moore RL, Tumer N. Impaired leptin responsiveness in aged rats. Diabetes (2000) 49(3):431–5. 10.2337/diabetes.49.3.431 [DOI] [PubMed] [Google Scholar]
- 56.Scarpace PJ, Matheny M, Shek EW. Impaired leptin signal transduction with age-related obesity. Neuropharmacology (2000) 39(10):1872–9. 10.1016/S0028-3908(00)00014-9 [DOI] [PubMed] [Google Scholar]
- 57.Shek EW, Scarpace PJ. Resistance to the anorexic and thermogenic effects of centrally administrated leptin in obese aged rats. Regul Pept (2000) 92(1–3):65–71. 10.1016/S0167-0115(00)00151-8 [DOI] [PubMed] [Google Scholar]
- 58.Ma XH, Muzumdar R, Yang XM, Gabriely I, Berger R, Barzilai N. Aging is associated with resistance to effects of leptin on fat distribution and insulin action. J Gerontol A Biol Sci Med Sci (2002) 57(6):B225–31. 10.1093/gerona/57.6.B225 [DOI] [PubMed] [Google Scholar]
- 59.Petervari E, Rostas I, Soos S, Tenk J, Miko A, Furedi N, et al. Age versus nutritional state in the development of central leptin resistance. Peptides (2014) 56:59–67. 10.1016/j.peptides.2014.03.011 [DOI] [PubMed] [Google Scholar]
- 60.Scarpace PJ, Tumer N. Peripheral and hypothalamic leptin resistance with age-related obesity. Physiol Behav (2001) 74(4–5):721–7. 10.1016/S0031-9384(01)00616-3 [DOI] [PubMed] [Google Scholar]
- 61.Fernandez-Galaz C, Fernandez-Agullo T, Campoy F, Arribas C, Gallardo N, Andres A, et al. Decreased leptin uptake in hypothalamic nuclei with ageing in Wistar rats. J Endocrinol (2001) 171(1):23–32. 10.1677/joe.0.1710023 [DOI] [PubMed] [Google Scholar]
- 62.Scarpace PJ, Matheny M, Tumer N. Hypothalamic leptin resistance is associated with impaired leptin signal transduction in aged obese rats. Neuroscience (2001) 104(4):1111–7. 10.1016/S0306-4522(01)00142-7 [DOI] [PubMed] [Google Scholar]
- 63.Morrison CD, White CL, Wang Z, Lee SY, Lawrence DS, Cefalu WT, et al. Increased hypothalamic protein tyrosine phosphatase 1B contributes to leptin resistance with age. Endocrinology (2007) 148(1):433–40. 10.1210/en.2006-0672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Garcia-San Frutos M, Fernandez-Agullo T, De Solis AJ, Andres A, Arribas C, Carrascosa JM, et al. Impaired central insulin response in aged Wistar rats: role of adiposity. Endocrinology (2007) 148(11):5238–47. 10.1210/en.2007-0543 [DOI] [PubMed] [Google Scholar]
- 65.Ikeda H, West DB, Pustek JJ, Figlewicz DP, Greenwood MR, Porte D, Jr, et al. Intraventricular insulin reduces food intake and body weight of lean but not obese Zucker rats. Appetite (1986) 7(4):381–6. 10.1016/S0195-6663(86)80006-X [DOI] [PubMed] [Google Scholar]
- 66.Carvalheira JB, Ribeiro EB, Araujo EP, Guimaraes RB, Telles MM, Torsoni M, et al. Selective impairment of insulin signalling in the hypothalamus of obese Zucker rats. Diabetologia (2003) 46(12):1629–40. 10.1007/s00125-003-1246-x [DOI] [PubMed] [Google Scholar]
- 67.Clegg DJ, Benoit SC, Reed JA, Woods SC, Dunn-Meynell A, Levin BE. Reduced anorexic effects of insulin in obesity-prone rats fed a moderate-fat diet. Am J Physiol Regul Integr Comp Physiol (2005) 288(4):R981–6. 10.1152/ajpregu.00675.2004 [DOI] [PubMed] [Google Scholar]
- 68.Kern W, Benedict C, Schultes B, Plohr F, Moser A, Born J, et al. Low cerebrospinal fluid insulin levels in obese humans. Diabetologia (2006) 49(11):2790–2. 10.1007/s00125-006-0409-y [DOI] [PubMed] [Google Scholar]
- 69.Stein LJ, Dorsa DM, Baskin DG, Figlewicz DP, Porte D, Jr, Woods SC. Reduced effect of experimental peripheral hyperinsulinemia to elevate cerebrospinal fluid insulin concentrations of obese Zucker rats. Endocrinology (1987) 121(5):1611–5. 10.1210/endo-121-5-1611 [DOI] [PubMed] [Google Scholar]
- 70.Yi CX, Gericke M, Kruger M, Alkemade A, Kabra DG, Hanske S, et al. High calorie diet triggers hypothalamic angiopathy. Mol Metab (2012) 1(1–2):95–100. 10.1016/j.molmet.2012.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schwartz MW, Figlewicz DF, Kahn SE, Baskin DG, Greenwood MR, Porte D, Jr. Insulin binding to brain capillaries is reduced in genetically obese, hyperinsulinemic Zucker rats. Peptides (1990) 11(3):467–72. 10.1016/0196-9781(90)90044-6 [DOI] [PubMed] [Google Scholar]
- 72.Zhao WQ, Chen H, Quon MJ, Alkon DL. Insulin and the insulin receptor in experimental models of learning and memory. Eur J Pharmacol (2004) 490(1–3):71–81. 10.1016/j.ejphar.2004.02.045 [DOI] [PubMed] [Google Scholar]
- 73.Hallschmid M, Benedict C, Schultes B, Born J, Kern W. Obese men respond to cognitive but not to catabolic brain insulin signaling. Int J Obes (Lond) (2008) 32(2):275–82. 10.1038/sj.ijo.0803722 [DOI] [PubMed] [Google Scholar]
- 74.Heni M, Wagner R, Kullmann S, Veit R, Mat Husin H, Linder K, et al. Central insulin administration improves whole-body insulin sensitivity via hypothalamus and parasympathetic outputs in men. Diabetes (2014) 63(12):4083–8. 10.2337/db14-0477 [DOI] [PubMed] [Google Scholar]
- 75.Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest (2012) 122(1):153–62. 10.1172/JCI59660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang H, Astarita G, Taussig MD, Bharadwaj KG, DiPatrizio NV, Nave KA, et al. Deficiency of lipoprotein lipase in neurons modifies the regulation of energy balance and leads to obesity. Cell Metab (2011) 13(1):105–13. 10.1016/j.cmet.2010.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Valdearcos M, Robblee MM, Benjamin DI, Nomura DK, Xu AW, Koliwad SK. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep (2014) 9(6):2124–38. 10.1016/j.celrep.2014.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, et al. Reciprocal modulation of toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem (2003) 278(39):37041–51. 10.1074/jbc.M305213200 [DOI] [PubMed] [Google Scholar]
- 79.Cai D. One step from prediabetes to diabetes: hypothalamic inflammation? Endocrinology (2012) 153(3):1010–3. 10.1210/en.2011-2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell (2008) 135(1):61–73. 10.1016/j.cell.2008.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kleinridders A, Schenten D, Konner AC, Belgardt BF, Mauer J, Okamura T, et al. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab (2009) 10(4):249–59. 10.1016/j.cmet.2009.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Meng Q, Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J Biol Chem (2011) 286(37):32324–32. 10.1074/jbc.M111.254417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab (2009) 9(1):35–51. 10.1016/j.cmet.2008.12.004 [DOI] [PubMed] [Google Scholar]
- 84.Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab (2009) 296(5):E1003–12. 10.1152/ajpendo.90377.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang G, Li J, Purkayastha S, Tang Y, Zhang H, Yin Y, et al. Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature (2013) 497(7448):211–6. 10.1038/nature12143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature (2000) 403(6771):795–800. 10.1038/35001622 [DOI] [PubMed] [Google Scholar]
- 87.Fontana L, Partridge L. Promoting health and longevity through diet: from model organisms to humans. Cell (2015) 161(1):106–18. 10.1016/j.cell.2015.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fernandez-Galaz C, Fernandez-Agullo T, Perez C, Peralta S, Arribas C, Andres A, et al. Long-term food restriction prevents ageing-associated central leptin resistance in Wistar rats. Diabetologia (2002) 45(7):997–1003. 10.1007/s00125-002-0851-4 [DOI] [PubMed] [Google Scholar]
- 89.Soos S, Balasko M, Jech-Mihalffy A, Szekely M, Petervari E. Anorexic vs. metabolic effects of central leptin infusion in rats of various ages and nutritional states. J Mol Neurosci (2010) 41(1):97–104. 10.1007/s12031-009-9294-4 [DOI] [PubMed] [Google Scholar]
- 90.Herskovits AZ, Guarente L. SIRT1 in neurodevelopment and brain senescence. Neuron (2014) 81(3):471–83. 10.1016/j.neuron.2014.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell (2006) 127(6):1109–22. 10.1016/j.cell.2006.11.013 [DOI] [PubMed] [Google Scholar]
- 92.Peeters AV, Beckers S, Verrijken A, Mertens I, Roevens P, Peeters PJ, et al. Association of SIRT1 gene variation with visceral obesity. Hum Genet (2008) 124(4):431–6. 10.1007/s00439-008-0567-8 [DOI] [PubMed] [Google Scholar]
- 93.Zillikens MC, van Meurs JB, Rivadeneira F, Amin N, Hofman A, Oostra BA, et al. SIRT1 genetic variation is related to BMI and risk of obesity. Diabetes (2009) 58(12):2828–34. 10.2337/db09-0536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van den Berg SW, Dolle ME, Imholz S, van der AD, van‘t Slot R, Wijmenga C, et al. Genetic variations in regulatory pathways of fatty acid and glucose metabolism are associated with obesity phenotypes: a population-based cohort study. Int J Obes (Lond) (2009) 33(10):1143–52. 10.1038/ijo.2009.152 [DOI] [PubMed] [Google Scholar]
- 95.Kilic U, Gok O, Elibol-Can B, Ozgen IT, Erenberk U, Uysal O, et al. SIRT1 gene variants are related to risk of childhood obesity. Eur J Pediatr (2015) 174(4):473–9. 10.1007/s00431-014-2424-1 [DOI] [PubMed] [Google Scholar]
- 96.Ramadori G, Fujikawa T, Fukuda M, Anderson J, Morgan DA, Mostoslavsky R, et al. SIRT1 deacetylase in POMC neurons is required for homeostatic defenses against diet-induced obesity. Cell Metab (2010) 12(1):78–87. 10.1016/j.cmet.2010.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sasaki T, Kikuchi O, Shimpuku M, Susanti VY, Yokota-Hashimoto H, Taguchi R, et al. Hypothalamic SIRT1 prevents age-associated weight gain by improving leptin sensitivity in mice. Diabetologia (2014) 57(4):819–31. 10.1007/s00125-013-3140-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Susanti VY, Sasaki T, Yokota-Hashimoto H, Matsui S, Lee YS, Kikuchi O, et al. Sirt1 rescues the obesity induced by insulin-resistant constitutively-nuclear FoxO1 in POMC neurons of male mice. Obesity (Silver Spring) (2014) 22(10):2115–9. 10.1002/oby.20838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dietrich MO, Antunes C, Geliang G, Liu ZW, Borok E, Nie Y, et al. Agrp neurons mediate Sirt1’s action on the melanocortin system and energy balance: roles for Sirt1 in neuronal firing and synaptic plasticity. J Neurosci (2010) 30(35):11815–25. 10.1523/JNEUROSCI.2234-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ramadori G, Fujikawa T, Anderson J, Berglund ED, Frazao R, Michan S, et al. SIRT1 deacetylase in SF1 neurons protects against metabolic imbalance. Cell Metab (2011) 14(3):301–12. 10.1016/j.cmet.2011.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sun C, Zhang F, Ge X, Yan T, Chen X, Shi X, et al. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab (2007) 6(4):307–19. 10.1016/j.cmet.2007.08.014 [DOI] [PubMed] [Google Scholar]
- 102.Zhang J. The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J Biol Chem (2007) 282(47):34356–64. 10.1074/jbc.M706644200 [DOI] [PubMed] [Google Scholar]
- 103.Kitamura YI, Kitamura T, Kruse JP, Raum JC, Stein R, Gu W, et al. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab (2005) 2(3):153–63. 10.1016/j.cmet.2005.08.004 [DOI] [PubMed] [Google Scholar]
- 104.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J (2004) 23(12):2369–80. 10.1038/sj.emboj.7600244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cho SH, Chen JA, Sayed F, Ward ME, Gao F, Nguyen TA, et al. SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1beta. J Neurosci (2015) 35(2):807–18. 10.1523/JNEUROSCI.2939-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Coupe B, Ishii Y, Dietrich MO, Komatsu M, Horvath TL, Bouret SG. Loss of autophagy in pro-opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metab (2012) 15(2):247–55. 10.1016/j.cmet.2011.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Quan W, Kim HK, Moon EY, Kim SS, Choi CS, Komatsu M, et al. Role of hypothalamic proopiomelanocortin neuron autophagy in the control of appetite and leptin response. Endocrinology (2012) 153(4):1817–26. 10.1210/en.2011-1882 [DOI] [PubMed] [Google Scholar]
- 108.Huang R, Xu Y, Wan W, Shou X, Qian J, You Z, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell (2015) 57(3):456–66. 10.1016/j.molcel.2014.12.013 [DOI] [PubMed] [Google Scholar]
- 109.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A (2008) 105(9):3374–9. 10.1073/pnas.0712145105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang FM, Chen YJ, Ouyang HJ. Regulation of unfolded protein response modulator XBP1s by acetylation and deacetylation. Biochem J (2011) 433(1):245–52. 10.1042/BJ20101293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Peng L, Yuan Z, Ling H, Fukasawa K, Robertson K, Olashaw N, et al. SIRT1 deacetylates the DNA methyltransferase 1 (DNMT1) protein and alters its activities. Mol Cell Biol (2011) 31(23):4720–34. 10.1128/MCB.06147-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zocchi L, Sassone-Corsi P. SIRT1-mediated deacetylation of MeCP2 contributes to BDNF expression. Epigenetics (2012) 7(7):695–700. 10.4161/epi.20733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci U S A (2015) 112(9):2876–81. 10.1073/pnas.1417921112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Braidy N, Poljak A, Grant R, Jayasena T, Mansour H, Chan-Ling T, et al. Mapping NAD(+) metabolism in the brain of ageing Wistar rats: potential targets for influencing brain senescence. Biogerontology (2014) 15(2):177–98. 10.1007/s10522-013-9489-5 [DOI] [PubMed] [Google Scholar]
- 115.Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab (2007) 6(5):363–75. 10.1016/j.cmet.2007.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab (2011) 14(4):528–36. 10.1016/j.cmet.2011.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu LY, Wang F, Zhang XY, Huang P, Lu YB, Wei EQ, et al. Nicotinamide phosphoribosyltransferase may be involved in age-related brain diseases. PLoS One (2012) 7(10):e44933. 10.1371/journal.pone.0044933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yoon MJ, Yoshida M, Johnson S, Takikawa A, Usui I, Tobe K, et al. SIRT1-mediated eNAMPT secretion from adipose tissue regulates hypothalamic NAD and function in mice. Cell Metab (2015) 21(5):706–17. 10.1016/j.cmet.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab (2012) 15(6):838–47. 10.1016/j.cmet.2012.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lafontaine-Lacasse M, Richard D, Picard F. Effects of age and gender on Sirt 1 mRNA expressions in the hypothalamus of the mouse. Neurosci Lett (2010) 480(1):1–3. 10.1016/j.neulet.2010.01.008 [DOI] [PubMed] [Google Scholar]
- 121.Sasaki T, Kim HJ, Kobayashi M, Kitamura YI, Yokota-Hashimoto H, Shiuchi T, et al. Induction of hypothalamic Sirt1 leads to cessation of feeding via agouti-related peptide. Endocrinology (2010) 151(6):2556–66. 10.1210/en.2009-1319 [DOI] [PubMed] [Google Scholar]
- 122.Ao N, Liu Y, Feng H, Bian X, Li Z, Gu B, et al. Ubiquitin-specific peptidase USP22 negatively regulates the STAT signaling pathway by deubiquitinating SIRT1. Cell Physiol Biochem (2014) 33(6):1863–75. 10.1159/000362964 [DOI] [PubMed] [Google Scholar]
- 123.Huang KP, Chen C, Hao J, Huang JY, Liu PQ, Huang HQ. AGEs-RAGE system down-regulates Sirt1 through the ubiquitin-proteasome pathway to promote FN and TGF-beta1 expression in male rat glomerular mesangial cells. Endocrinology (2015) 156(1):268–79. 10.1210/en.2014-1381 [DOI] [PubMed] [Google Scholar]
- 124.Lin Z, Yang H, Kong Q, Li J, Lee SM, Gao B, et al. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol Cell (2012) 46(4):484–94. 10.1016/j.molcel.2012.03.024 [DOI] [PubMed] [Google Scholar]