

Genomic analysis provides insights into the role of copy number variation in disease, but most methods are not designed to resolve mixed populations of cells. In tumors, where genetic heterogeneity is common1–3, very important information may be lost useful for reconstructing its evolutionary history. Here we show that with flow-sorted nuclei, whole genome amplification (WGA), and next generation sequencing we can accurately quantify genomic copy number within an individual nucleus. We apply single nucleus sequencing (SNS) to investigate tumor population structure and evolution in two breast cancer cases. Analysis of 100 single cells from a polygenomic tumor revealed three distinct clonal subpopulations that likely represent sequential clonal expansions. Additional analysis of 100 single cells from a monogenomic primary tumor and its liver metastasis suggested that a single clonal expansion formed the primary tumor and seeded the metastasis. In both primary tumors, we also identified an unexpectedly abundant subpopulation of genetically diverse ‘pseudodiploid’ cells that do not travel to the metastatic site. In contrast to gradual models of tumor progression, our data indicate that tumors grow by punctuated clonal expansions with few persistent intermediates.
In SNS we isolate nuclei by flow-sorting and amplify DNA using whole genome amplification (WGA) for massively parallel sequencing (Supplementary Fig. 1). We achieve low coverage (~6%) of the genome of a single cell, sufficient to quantify copy number from sequence read depth. Several features of our data analysis were designed for SNS and differ from previous methods4–6 for measuring copy number from sequencing data. In contrast to using fixed intervals to calculate copy number, we use variable length bins but with uniform expected unique counts, which correct for biases that have been reported7–9 in WGA (Supplementary Figure 2; see Methods). For each single cell, we typically achieve a mean read density of 138 per bin (SEM±5.55, n=200). Over-replicated loci called ‘pileups’ that have been previously reported in WGA10–12, do occur in our data but not at recurrent locations in different cells (Supplementary Fig. 3). Pileups are sufficiently randomly distributed and sparse so as not to affect counting at the resolution we have chosen (54 kb). Assuming that single cells will have discrete copy number states, we segment the variable bins and calculate integer copy number profiles (Supplementary figure 4; see Methods).
To validate our method, we compared the sequence counting profile of DNA from a single SK-BR-3 cell (Fig. 1a) with DNA from one million cells (Fig. 1b). The major amplifications (RD2, TPD52, ERBB2, BCAS1) and deletions (DCC) are detected in both profiles, as are much more abundant but less dramatic small changes in copy number. To demonstrate how reproducible small differences are, we assessed data for a complex region on chromosome 8q13.2-q24.23 that contains more than thirty segments with differing copy number. These data were reproducible in both a single cell (Fig. 1c) and a million cell sample (Fig. 1d). We also compared the sequence read profiles from several single cells and from a million cells to each other and to the profile of array CGH from bulk DNA (Supplementary Fig. 5). In all instances the profiles showed very high (r2 > 0.85) correlation. The reproducibility and variation between single cell copy number profiles was also investigated by comparing seven single cells from a culture of SK-BR-3 and seven from normal human fibroblasts. These data are displayed as heat maps (Fig. 1e–f), which show that some genomic variation exists between cells. The diploid fibroblast cultures showed no random events; we observed only a few consistent events at levels expected for heritable copy number variations.
Fig. 1. Comparison of SK-BR-3 Single Cells to Millions.
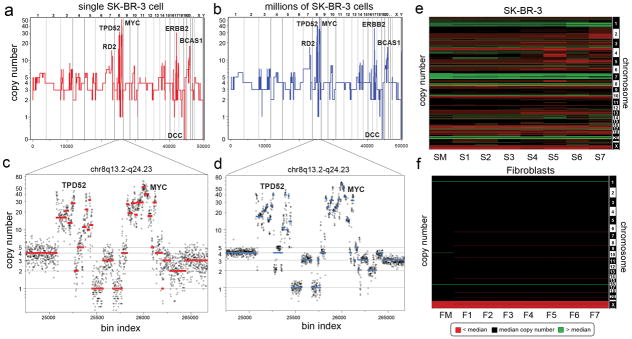
(a) The integer copy number profile for a single SK-BR-3 cell is shown compared to (b) a sequence count profile using millions of cells. (c–d) A region on chromosome 8q13.2-q24.23 is plotted showing the integer copy number profile (in red or blue) and a ratio of raw bin counts in grey for (c) a single cell, and (d) a million cells (e) A heatmap of SK-BR-3 copy number profiles comparing a million cell sample (SM) to seven single cells (S1–S7). (f) A heatmap of SKN1 normal fibroblast profiles comparing a million cell sample (FM) to seven single cells (F1–F7).
We next selected two high grade (III), triple negative (ER−, PR−, Her2−) ductal carcinomas (T10, T16P) and a paired metastatic liver carcinoma (T16M) to study tumor population structure and infer tumor evolution by single cell analysis. T10 was selected to study primary tumor growth, because it was previously shown13 to be genetically heterogeneous (polygenomic), and T16P was selected because it was classified as genetically homogeneous (monogenomic).
T10 was macro-dissected into 12 sectors to preserve anatomical information, and nuclei were flow-sorted from six sectors (S1–S6) for SNS (Fig. 2a). FACS analysis showed four major distributions of ploidy: a hypodiploid fraction (F1) exclusive to sectors 1–3; a diploid 2N fraction (F2) in all sectors; and two sub-tetraploid fractions (F3 and F4) in sectors 4–6. We selected 100 single cells from multiple sectors and ploidy fractions for sequencing and calculation of integer copy number profiles (Supplementary Table 1).
Fig. 2. Analysis of 100 Single Cells from Polygenomic Breast Tumor.

(a) T10 was macro-dissected into 12 sectors, and nuclei were isolated from six sectors and flow-sorted by ploidy. FACS profiles show four distributions of ploidy (F1–F4) which were gated to isolate 100 single cells. (b) Neighbor-joining tree of integer copy number profiles showing four major branches of evolution (c) Phylogenetic tree of consensus profiles show the common ancestors and evolutionary distance between subpopulations. Integer copy number profiles from single cells are displayed below, and pie charts indicate the percentage of cells that constitute each subpopulation.
Breast tumors are typically mixtures of cancer cells with normal tissue, stroma, and infiltrating leukocytes. By histopathology, T10 was assessed to contain 63% normal and 37% tumor cells and noted to be heavily infiltrated with leukocytes. Most of the diploid nuclei from F2 had flat genome profiles, characteristic of normal cells. Nearly two-thirds (31/47) of these diploid profiles showed narrow deletions in the T-cell receptor loci or one or more immunoglobulin variable region loci, consistent with infiltration by immunocytes (data not shown). Of the remaining sixteen nuclei from F2, twelve showed no discernable aberrations, but four nuclei exhibited aberrant profiles with diverse chromosome gains and losses. Each of these ‘pseudodiploid’ nuclei profiles appeared unrelated to the others or to those of the major tumor cell populations found in fractions F1, F3 and F4.
To determine population substructure we calculated pair-wise distances between the 100 integer copy number profiles, and built a tree using neighbor-joining14 (Fig. 2b). The 100 profiles clustered into four subpopulations (D+P, H, AA and AB) regardless of their sector of origin. The D+P subpopulation contains predominantly flat diploid (D) profiles, but also pseudodiploid (P) cells that have diverged by varying degrees from the diploids. The three major ‘advanced’ tumor subpopulations (H, AA and AB) are highly clonal with complex genomic rearrangements, and together comprise slightly less than half the cells of the tumor. These cells were isolated from the hypodiploid (F1) and two sub-tetraploid (F3 and F4) ploidy fractions, respectively. We had previously identified these subpopulations by profiling millions of cells by array CGH13, but we could not determine if they were composite mixtures of different tumor clones. By SNS we can now see that each subpopulation is composed of cells that share highly similar copy number profiles, likely representing three clonal expansions. Each subpopulation (H, AA and AB) is clearly related to the others by many shared genomic alterations, but they have also diverged and developed distinct attributes (for example, a massive 50-fold amplification of the KRAS oncogene in AB). The H cells display the characteristic ‘sawtooth’ pattern15 comprising broad chromosomal deletions (Fig. 2c). They are anatomically segregated in the sectors S1–S3 of the tumor, whereas the AA and AB clones are intermixed and occupy sectors S4–S6.
To understand the relationship between subpopulations, we clustered profiles by chromosome breakpoints (which are directly related to the steps by which tumor cells diverge). We identified 657 copy number breakpoints and used them to build a phylogenetic tree, which closely resembles the structure of the neighbor-joining tree based on copy number (Supplementary Fig. 6). We also applied biclustering16 to construct a heat map of breakpoints, and ordered it based on the copy number tree to show which breakpoints were common or divergent between the major subpopulations (Supplementary Fig. 7a). Although there is considerable variation within each subpopulation, no obvious further population substructure was evident. To estimate the common ancestors, we constructed a phylogenetic lineage using the consensus breakpoint patterns from the major tumor subpopulations (Fig. 2c). This lineage shows that the n1 common ancestor diverged a significant distance from the diploid cells, but that the distance between n1 and n2 is very small. By contrast, the divergence of the subpopulations after n1 and n2 is very large, with AB showing the greatest phylogenetic distance from the diploids. We thus infer that the three subpopulations emerged when the tumor was much smaller.
We investigated a second tumor to determine whether these findings extend. We isolated 52 cells from a primary breast tumor (T16P) and 48 cells from its associated liver metastasis (T16M). Each tumor was macro-dissected into 6 sectors, three of which were flow-sorted (Fig. 3a–b). Both T16M and T16P showed diploid peaks (F1) and a single aneuploid tetraploid peak (F2) of roughly equal cell count in all sectors (Supplementary Table 2), consistent with histological sections showing approximately 50% tumor and 50% normal (stromal) cells with low leukocyte infiltration in both samples. To explore population substructure we again constructed neighbor-joining trees from the integer copy number profiles, combining the primary and metastasis cells (Fig. 3c). We again observed numerous pseudodiploid cells, but a single subpopulation of aneuploid cells very diverged from the diploid population. As for T10, the 12 pseudodiploid cells from T16P displayed diverse genomic lesions with no clear relationships to each other or to he main tumor lineage. Of the 24 normal diploids in the primary, two had deletions of the T-cell receptor. There were no pseudodiploid cells among the 26 diploid cells from the metastasis.
Fig. 3. Analysis of 100 Single Cells from a Monogenomic Breast Tumor and its Liver Metastasis.

(a–b) Primary breast tumor T16P was macro-dissected and 52 nuclei were isolated from three sectors for FACS showing two distributions of ploidy (F1 and F2). (b) Liver metastasis T16M was macro-dissected and 48 nuclei were isolated from three sectors for FACS also showing two ploidy distributions (F1 and F2). (c) Neighbor-joining tree of combined integer copy number profiles from the primary and the metastatic tumors. (d) Comparison of primary and metastatic aneuploid consensus copy number profiles.
These data suggest that the primary tumor mass formed by a single clonal expansion of an aneuploid cell, and that one of the cells from this expansion subsequently seeded the metastatic tumor with little further evolution. There are no branches of the tree corresponding to cells intermediate between the aneuploid subpopulation and the diploid root. Although closely related, the primary and metastatic aneuploid cells cleanly separate using the Euclidean metric (Fig. 3c), suggesting the two populations have not mixed since seeding the metastasis. The differences in the profiles that distinguish the primary and metastatic tumor populations are in the degree of copy number change rather than breakpoints (Fig. 3d). In a hierarchical tree created from breakpoints alone, we cannot cleanly separate primary from metastatic aneuploid cells (Supplementary Fig. 6b). Moreover, when we calculate common breakpoints in the single cell profiles and apply biclustering to ordered samples (Supplementary Fig. 7b), a large number of breakpoints are common to both populations and no breakpoints cleanly distinguish them. By these analyses, no further population substructure is evident.
In contrast to the clear clonal relationships among aneuploid subpopulations, pseudodiploid cells are unusual in showing remarkable genomic heterogeneity (Fig. 4). Pseudodiploid profiles are characterized by nonrecurring copy number changes (including whole chromosome arms) that are not shared between any two pseudodiploid cells, nor with the corresponding tumor profiles (Fig. 4e). These data suggest that unlike the aneuploid cells, pseudodiploids do not undergo clonal expansions in the tumor. Nevertheless, they comprise a substantial proportion of the diploid gated cells: 8% in T10 (4/47) and 33% in T16P (12/36), or approximately 4% and 24% of the tumor mass, respectively. In contrast, the 18 profiles from single nuclei of normal adjacent breast tissue are all flat (Fig. 4a). The relative abundance of pseudodiploid cells in primary tumors indicates that they may emerge from an ongoing aberrant process that generate genomic diversity in the tumor.
Fig. 4. Genetically Diverse Pseudodiploid Cells in the Diploid Fractions of Tumors.

(a–d) Hematoxylin and eosin stained tissues sections are displayed in the upper panels with normal (N) and tumor (T) cells percentages indicated. Lower rows display bin counts and copy number profiles of single cells isolated from the 2N gated ploidy distributions, and the total number of cells analyzed is indicated below each column. The columns are: (a) normal breast tissue cells; (b) pseudodiploid cells in T10; (c) pseudodiploid cells in T16P; and (d) diploid-gated nuclei from T16M. (e) Bin counts and copy number profiles of single cells from the major aneuploid tumor subpopulations.
In principle, we can learn about DNA sequence mutations from SNS data. However, the sparse sequence coverage makes this analysis problematic. By combining data from multiple cells, belonging to well defined subpopulations, we can perform global and regional analysis at the many nucleotide positions where sufficient numbers of sequence reads overlap. When examined this way, losses of heterozygosity are unequivocally significant, and map in large contiguous genomic blocks that correlate well with copy number loss (Supplementary Fig. 8 and Supplementary Table 3). The extensive LOH detected in all of the T10 subpopulations and in T16 suggests that both cancers passed through a hypodiploid stage.
Our study demonstrates that we can obtain robust high-resolution copy number profiles by sequencing a single cell and that by examining multiple cells from the same cancer, we can make inferences about the evolution and spread of cancer. Moreover, the identification of pseudodiploid cells shows that these methods can identify cell types previously undetectable by other methods. Our findings are consistent with previous findings17 using bulk DNA, which suggests that copy number profiles in primary tumors are highly similar to the metastases. Thus the metastatic cells emerge from a main advanced expansion, and not from an earlier intermediate or a completely different subpopulation. This is consistent with recent deep-sequencing studies of primary-met pairs, all suggesting that metastatic cells arise late in tumor development18–19.
There are many gradual models for tumor progression, including clonal evolution20, the mutator phenotype21,22, and stochastic progression23. While we have examined only two cancers in depth, both display a pattern of tumor growth which we call ‘punctuated clonal evolution’, borrowing a term from species evolution used to explain gaps in the fossil record24. Explicitly, the tumor subpopulations are each distant from their root, without observable intermediate branching. In contrast to gradual models, this pattern reflects the sudden emergence of a tumor cell whose rate of effective population growth dramatically exceeds its rate of genomic evolution.
METHODS
Samples
The frozen ductal carcinoma T10 (CHTN0173) was obtained from the Cooperative Human Tissue Network, and T16P and T16M were obtained from Asterand (Detroit, MI) Pathology shows that both tumors were poorly differentiated and high grade (III) as determined by the Bloom-Richardson score, and triple-negative (ER−, PR− and Her2/Neu−) as determined by immunohistochemistry. The cell lines used in this study include a normal male immortalized skin fibroblast (SKN1) and a breast cancer cell line (SK-BR-3). Normal breast tissue was obtained from Dr. Hanina Hibshoosh from Columbia University.
Single Nucleus Sequencing (SNS)
Nuclei were isolated from cell lines and from the frozen tumor using an NST-DAPI buffer (800 mL of NST [146 mM NaCl, 10 mM Tris base at pH 7.8, 1 mM CaCl2, 21 mM MgCl2, 0.05% BSA, 0.2% Nonidet P-40]), 200 mL of 106 mM MgCl2, 10 mg of DAPI, and 0.1% DNase-free RNase A. The frozen tumor was first macro-dissected into 12 sectors of equal size using surgical scalpels and nuclei were isolated from six sectors for FACS by finely mincing a tumor sector in a Petri dish in 1.0–2.0 mL of NST-DAPI buffer using two no. 11 scalpels in a cross-hatching motion. The cell lines were lysed directly in a culture plate using the NST-DAPI buffer, after first removing the cell culture media. All nuclei suspensions were filtered through 37-μm plastic mesh prior to flow-sorting.
Single Nuclei were sorted by FACS using the BD Biosystems Aria II flow cytometer by gating cellular distributions with differences in their total genomic DNA content (or, ploidy) according to DAPI intensity. First a small amount of prepared nuclei from each tumor sample was mixed with a diploid control sample (derived from a lymphoblastoid cell line of a normal person) to accurately determine the diploid peak position within the tumor and establish FACS collection gates. Before sorting single nuclei, a few thousand cells were sorted to determine the DNA content distributions for gating. A 96-well plate was prepared with 10ul of lysis solution in each well from the Sigma-Aldrich GenomePlex© WGA4 kit. Single nuclei were deposited into individual wells in the 96-well plate along with several negative controls in which no nuclei were deposited.
Whole genome amplification was performed on single flow-sorted nuclei as described in the Sigma-Aldrich GenomePlex WGA4 kit (cat # WGA4-50RXN) protocol. WGA fragments from the frozen breast tumor and SK-BR-3 single cells were used directly for Single-read library construction using the Illumina Genomic DNA Sample Prep Kit (cat # FC-102-1001) and following standard protocol with a gel purification size range of 300-250bp. WGA fragments from the fibroblast cell line were first sonicated using the Diagenode Bioruptor© using the following program: 2 times, 7 minutes with 30 seconds high on/off mode in ice cold water. Sonication removes a specific 28bp adapter sequence that is added on during WGA, and improves the total number of sequencing reads per lane.
Single-read libraries from single nuclei were sequenced on individual flow-cell lanes using the Illumina GA2 analyzer for 76 cycles. Data was processed using the Illumina GAPipeline-1.3.2 to 1.6.0 Sequence reads were aligned to the human genome (HG18/NCBI36) using the Bowtie alignment software44 with the following parameters: ‘bowtie –S –t –m 1 –best –strata –p16’ to report only top scoring unique mappings for each sequence read. To eliminate PCR duplicates, we removed sequences with identical start coordinates.
Read Depth Counting in Variable Bins
Copy number is calculated from read density, by dividing the genome into an ‘bins’ and counting the number of unique reads in each bin. In previous copy number studies read density was calculated using bins with uniform fixed length 16–19. In contrast we use bins of variable length, that adjust size depending on the mappability of sequences to regions of the human genome. In regions of repetitive elements, lower numbers of reads are expected and thus the bin size is increased. To determine interval sizes we simulated sequence reads by sampling 200 million sequences of length 48 from the human reference genome (HG18/NCBI36) and introduced single nucleotide errors with a frequency encountered during Illumina sequencing. These sequences were mapped back to the human reference genome using Bowtie15 with unique parameters as described above. We assigned a number of bins to each chromosome based on the proportion of simulated reads mapped. We then divided each chromosome into bins with an equal number of simulated reads. This resulted in 50009 genomic bins with no bins crossing chromosome boundaries. The median genomic length spanned by each bin is 54kb. For each cell the number of reads mapped to each variable length bin was counted. This variable binning efficiently reduces false deletion events when compared to uniform length fixed bins as shown in Supplementary Fig. 2b and 2c. For a single cell we typically measure 138 sequence reads per bin.
Integer Copy Number Quantification
Single cells will have integer copy number states that we can infer from sequence read counts, as follows. Unique sequence reads are counted in variable bins (Supplemental Fig. 4a) and segmented using the Kolmogorov-Smirnov (KS) statistic (Supplemental Fig. 4b). To estimate the integer differences of copy number states, we calculate Gaussian kernel smoothed density plots using Splus (MathSoft, Inc.), showing the difference between median bin counts for all pair-wise combinations of different segments (Supplemental Fig. 4c–e) The uniform steps between groups are very apparent, and is a general property of single cell data. We then convert our KS-segmented data into profiles of integer copy number as follows. We take the differential bin count of the second peak, denoted by an asterisk in Supplemental Fig. 4a, to represent a copy number “increment” of 1. We then divide every bin count in the profile by the increment and round to infer the integer copy number. We show in Supplemental Fig. 4f–g how closely the segmentation profile agrees with the integer copy number profile. However, for diploid or near diploid cells there are few to no steps from which to observe the increment, and we use a different method, taking the increment as the median bin count on the autosomes divided by two.
Gene Annotations
Amplifications and deletions identified in the single cell copy number profiles were annotated to identify UCSC genes. Cancer genes were identified using a compiled database from the cancer gene consensus and the NCI cancer gene index (Sophic Systems Alliance Inc., Biomax Informatics A.G).
Neighbor-joining Trees of Copy Number Profiles
Integer copy number profiles of single cells were used to calculate Neighbor-joining trees using a Euclidean distance metric with Matlab (Mathworks). Branches were flipped to orient nodes within subpopulations and trees were rooted using the last common diploid node.
Common Breakpoint Detection
Breakpoints are defined as bins with a copy number different than the previous bin in genome order. A transition from a lower copy number to a higher copy number (in genome order) is considered to be a different event than the opposite transition. To find breakpoint regions we count each breakpoint in each cell and the immediately neighboring bins. A contiguous set of bins with counts greater than 1 is designated a breakpoint region. This results in a set of common breakpoint regions. Each cell is then scored for the occurrence of each of these events, a one meaning the cell has a copy number transition of that type (low to high or high to low) in that genomic region and a zero meaning no copy number transition of that type in that region.
Hierarchical Tree of Chromosome Breakpoints
We used chromosome breakpoints patterns to build a neighbor-joining tree. To eliminate breakpoints events with a high standard deviation, we limited our analysis to breakpoint regions covering no more than seven adjacent bins (N = 657). Using a Euclidean metric, we calculated a distance matrix from the binary chromosome breakpoint patterns identified in the single cells using Matlab (Mathworks). From this distance matrix we constructed a tree using average-linkage.
Heatmap of Chromosome Breakpoints
The heatmap is based on the same set of breakpoints used to build the neighbor-joining tree. Blue indicates the presence of an event, and white means no event. The columns are ordered as in the tree. The rows are ordered to show clearly which of the subsets of the four main groups in the tree share which events. The groups are ordered by subpopulation. A four dimensional binary vector represents each of the 16 possible subsets of these groups (subset vector). Each breakpoint is represented by a four dimensional vector of the percent of cells in each group having an event at that breakpoint (the “breakpoint vector”). The angle from each breakpoint vector to each subset vector is computed as well as the length of each projection vector. If the length of the projection vector is less than 0.05 the breakpoint vector is assigned to the empty (0,0,0,0) subset, otherwise it is assigned to the subset vector with the smallest angle to the breakpoint vector. The rows are ordered by subset vector in the following order: (1,1,1,1), (0,0,0,1), (0,0,1,0), (0,1,0,0), (1,0,0,0), (0,0,1,1), (0,1,0,1), (1,0,0,1), (0,1,1,0), (1,0,1,0), (1,1,0,0), (0,1,1,1), (1,0,1,1), (1,1,0,1), (1,1,1,0), (0,0,0,0). Within each subset the rows are in descending order by the number of cells in that subset having an event and then in ascending order by the number of cells not in that subset having an event.
Analysis of LOH Point Mutations in Tumor Subpopulations
PCR duplicates were removed from mapped sequence reads and bases with a quality score below 30 were excluded from analysis. We then determined the set of observed nucleotide types for each cell sequenced from the T10 and T16P and T16M tumors and every position in the genome. For each subpopulation we classified a position as the observed nucleotides only if one or two nucleotide types were each observed in five or more cells in the subpopulation. For each grouping of subpopulations DH, DA, if a classification was made in every subpopulation in the group, we translated the classifications into the generic nucleotides (a,b) based upon the order in which they were seen in the group, from left to right. We counted the resulting classifications of positions for each group by class, and determined whether long blocks of identical classifications along a chromosome were expected by chance. To establish the significance of our classification counts we repeated our analysis 100 times with randomly permuted cell labels within each group of subpopulations. We eliminated any effects from differing subpopulation size in a separate set of runs of the same analysis, each with 24 randomly selected cells in every subpopulation.
Supplementary Material
Supplementary Fig. 1 | Overview of the SNS Approach
(a) Cells are lysed and nuclei are isolated. (b) DAPI stained nuclear suspensions are loaded into the FACS machine. (c) Cells are run to measure ploidy, and then subpopulations are gated from which individual nuclei are deposited into single wells on a 96-well plate containing lysis buffer. (d) DNA is fragmented in alkali solution and amplified by WGA resulting in a distribution of fragments from 100–1000bp that can be visualized on an agarose gel. Negative control (NC) reactions are also prepared without any nuclei deposited which show no DNA fragments on agarose gels. (e) WGA amplified DNA is sonicated to remove specific 28bp adapters (f). Illumina single-end libraries are prepared and each library is sequenced on individual flow-cell lanes.
Supplementary Fig. 2 | Fixed vs. Variable Binning of Sequence Read Depth
(a) 54kb uniform length and variable (uniform expected count) bin intervals are plotted on a repetitive region of chromosome 1p, illustrating that variable bin size increases to maintain a constant mean read count. (b–c) Uniform length and variable bins were calculated for a single SK-BR-3 cell and plotted along with an annotation track for repetitive elements below. (b) Chromosome 8q shows similar copy number in both uniform and variable bin profiles, but the uniform bin method erroneously shows the region near the telomere as a large homozygous deletion. (c) Chromosome 15q12-14 shows three erroneous homozygous deletions in a repetitive region of the human genome using the uniform length bin profile, while the variable bin method maintains a constant ground state copy number.
Supplementary Fig. 3 | Pileups and Distributions of WGA Sequencing Reads in Single Cells
(a) Sequence read depth from three single SK-BR-3 cells are plotted for a 2.2mb region on chromosome 8q13.2 illustarting that reads are fairly randomly distributed and non-overlapping between three different single cells. Two regions of over-replicated pileups are shown and marked with asterisks. (b) Read depth from three different single SK-BR-3 cells are also plotted on chromosome 8q13.3 that contains an amplification of the MYC locus, showing an increase in sequence read density.
Supplementary Fig. 4 | Integer Copy Number Determination from Read Density
(a–c, f–g) Integer copy number calculations are shown for a single SK-BR-3 cell. (a) Mapped sequence reads are counted in variable bins of uniform expected read density. (b) Variable bin counts in grey are plotted on a log scale and KS-segments are plotted in blue for a region from chromosome 17–19. (c–e) Gaussian kernel smoothed density plots are shown with asterisks denoting the first increment peak, used for normalization, for (c) an SK-BR-3 cell, (d) a hypodiploid tumor cell from T10, and (e) an aneuploid tumor cell from T10. (f) A normalized KS-segmented profile in blue is compared to the integer copy number profile in red for an SK-BR-3 cell, and (g) this region is shown for chromosome 17 with integer copy number on the ordinate.
Supplementary Fig. 5 | R2 plots of CGH and SNS Profiles from Single Cells and Bulk DNA
(a–d) R2 plots of SK-BR-3 copy number profiles using various platforms and number of cells. (a) Sequence read count of a million cell sample compared to an array CGH profile using million of cells (b) Single cell profile measured by SNS versus a million cell array CGH profile, (c) Single cell profile measured by SNS compared to a million cell sequence read count profile, (d) A single cell profile (cell number 1) compared to another single cell profile (cell number 5) measured by SNS.
Supplementary Fig. 6 | Chromosome Breakpoint trees of T10 and T16
(a–b) Hierarchical clustering was used to construct trees from chromosome breakpoint patterns from single cells (a) hierarchical tree of 100 single cells from T10 (b) hierarchical tree of 100 single cells from T16 combining profiles from both the primary and metastatic tumors.
Supplementary Fig. 7 | Biclustered Heatmaps of Chromosome Breakpoints
(a) T10 heatmap of common chromosome breakpoints that have been biclustered and ordered to correspond to the integer copy number tree (b) T16P and T16M heatmap of common chromosome breakpoints that have been biclustered and ordered according to the integer copy number tree.
Supplementary Fig. 8 | Comparison of LOH and Copy Number Profiles
(a–c) Copy number profiles from tumor subpopulations are plotted with LOH at heterozygous SNPs that were identified by comparing to the diploid subpopulation. Asteriks indicates regions where LOH point mutations and copy number are in disagreement. (a) LOH in the T10 hypodiploid subpopulation. (b) LOH in the T10 aneuploid subpopulations. (c) LOH in the T16P aneuploid subpopulation. (d) Summaries of nucleotide classes and LOH detected in each subpopulation. At a given position, “ab” means two nucleotides are seen each in at least 5 cells, and “a” or “b” mean a single nucleotide was observed, always in at least 5 single cells.
Information and statistics are shown for 100 single cells flow-sorted from the polygenomic breast tumor T10
Information and statistics are shown for 52 cells from the primary breast tumor T16P and 48 cells from its corresponding metastasis T16M.
Summaries of nucleotide classes and LOH detected in each tumor subpopulation by comparing to the diploid subpopulation. At a given position, “ab” means two nucleotides are seen each in at least 5 cells, and “a” or “b” mean a single nucleotide was observed, always in at least 5 single cells. All nucleotides have a quality score > 30. See methods section for further details.
Acknowledgments
We thank Michael Ronemus, Tara Spencer, Anthony Leotta, Jennifer Meth, Melissa Kramer, Laura Gelley, Elena Ghiban. We also thank Patrick Blake and Nancy Navin at Sophic Systems Alliance Inc. This work was supported by the NCI T32 Fellowship to N.N., and grants to M.W. and J.H. from the Department of the Army (W81XWH04-1-0477), the Breast Cancer Research Foundation, and the Simons Foundation. M.W. is an American Cancer Society Research Professor.
Footnotes
Author Contributions
N.N. designed and performed experiments and analysis, and wrote manuscript. J.K., A.K., L.M., D.L. and P.A. developed analysis programs. J.T., L.R., K.C, J.M, D.E. and A.S. performed experiments. R.M. designed experiments. J.H. and M.W. designed experiments, performed analysis and wrote manuscript.
Database Accession
All data has been deposited into the NCBI Sequence Read Archive (SRA018951.105)
Competing Interests
The authors have no competing interests to declare
References
- 1.Park SY, Gonen M, Kim HJ, Michor F, Polyak K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J Clin Invest. 2010;120:636–644. doi: 10.1172/JCI40724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torres L, et al. Intratumor genomic heterogeneity in breast cancer with clonal divergence between primary carcinomas and lymph node metastases. Breast Cancer Res Treat. 2007;102:143–155. doi: 10.1007/s10549-006-9317-6. [DOI] [PubMed] [Google Scholar]
- 3.Farabegoli F, et al. Clone heterogeneity in diploid and aneuploid breast carcinomas as detected by FISH. Cytometry. 2001;46:50–56. doi: 10.1002/1097-0320(20010215)46:1<50::aid-cyto1037>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 4.Chiang DY, et al. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods. 2009;6:99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;19:1586–1592. doi: 10.1101/gr.092981.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alkan C, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geigl JB, et al. Identification of small gains and losses in single cells after whole genome amplification on tiling oligo arrays. Nucleic Acids Res. 2009;37 doi: 10.1093/nar/gkp526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuhrmann C, et al. High-resolution array comparative genomic hybridization of single micrometastatic tumor cells. Nucleic Acids Res. 2008;36 doi: 10.1093/nar/gkn101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pugh TJ, et al. Impact of whole genome amplification on analysis of copy number variants. Nucleic Acids Res. 2008;36 doi: 10.1093/nar/gkn378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Talseth-Palmer BA, Bowden NA, Hill A, Meldrum C, Scott RJ. Whole genome amplification and its impact on CGH array profiles. BMC Res Notes. 2008;1:56. doi: 10.1186/1756-0500-1-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes S, et al. Use of whole genome amplification and comparative genomic hybridisation to detect chromosomal copy number alterations in cell line material and tumour tissue. Cytogenet Genome Res. 2004;105:18–24. doi: 10.1159/000078004. [DOI] [PubMed] [Google Scholar]
- 12.Huang J, Pang J, Watanabe T, Ng HK, Ohgaki H. Whole genome amplification for array comparative genomic hybridization using DNA extracted from formalin-fixed, paraffin-embedded histological sections. J Mol Diagn. 2009;11:109–116. doi: 10.2353/jmoldx.2009.080143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navin N, et al. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010;20:68–80. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 15.Hicks J, et al. Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res. 2006;16 doi: 10.1101/gr.5460106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prelic A, et al. A systematic comparison and evaluation of biclustering methods for gene expression data. Bioinformatics. 2006;22:1122–1129. doi: 10.1093/bioinformatics/btl060. [DOI] [PubMed] [Google Scholar]
- 17.Liu W, et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med. 2009;15:559–565. doi: 10.1038/nm.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding L, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yachida S, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 21.Loeb LA, Springgate CF, Battula N. Errors in DNA replication as a basis of malignant changes. Cancer Res. 1974;34:2311–2321. [PubMed] [Google Scholar]
- 22.Bielas JH, Loeb KR, Rubin BP, True LD, Loeb LA. Human cancers express a mutator phenotype. Proc Natl Acad Sci U S A. 2006;103:18238–18242. doi: 10.1073/pnas.0607057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heng HH, et al. Stochastic cancer progression driven by non-clonal chromosome aberrations. J Cell Physiol. 2006;208:461–472. doi: 10.1002/jcp.20685. [DOI] [PubMed] [Google Scholar]
- 24.Gould SJ, Eldredge N. Punctuated equilibria comes of age. Nature. 1993;366:223–227. doi: 10.1038/366223a0. [DOI] [PubMed] [Google Scholar]
- 25.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1 | Overview of the SNS Approach
(a) Cells are lysed and nuclei are isolated. (b) DAPI stained nuclear suspensions are loaded into the FACS machine. (c) Cells are run to measure ploidy, and then subpopulations are gated from which individual nuclei are deposited into single wells on a 96-well plate containing lysis buffer. (d) DNA is fragmented in alkali solution and amplified by WGA resulting in a distribution of fragments from 100–1000bp that can be visualized on an agarose gel. Negative control (NC) reactions are also prepared without any nuclei deposited which show no DNA fragments on agarose gels. (e) WGA amplified DNA is sonicated to remove specific 28bp adapters (f). Illumina single-end libraries are prepared and each library is sequenced on individual flow-cell lanes.
Supplementary Fig. 2 | Fixed vs. Variable Binning of Sequence Read Depth
(a) 54kb uniform length and variable (uniform expected count) bin intervals are plotted on a repetitive region of chromosome 1p, illustrating that variable bin size increases to maintain a constant mean read count. (b–c) Uniform length and variable bins were calculated for a single SK-BR-3 cell and plotted along with an annotation track for repetitive elements below. (b) Chromosome 8q shows similar copy number in both uniform and variable bin profiles, but the uniform bin method erroneously shows the region near the telomere as a large homozygous deletion. (c) Chromosome 15q12-14 shows three erroneous homozygous deletions in a repetitive region of the human genome using the uniform length bin profile, while the variable bin method maintains a constant ground state copy number.
Supplementary Fig. 3 | Pileups and Distributions of WGA Sequencing Reads in Single Cells
(a) Sequence read depth from three single SK-BR-3 cells are plotted for a 2.2mb region on chromosome 8q13.2 illustarting that reads are fairly randomly distributed and non-overlapping between three different single cells. Two regions of over-replicated pileups are shown and marked with asterisks. (b) Read depth from three different single SK-BR-3 cells are also plotted on chromosome 8q13.3 that contains an amplification of the MYC locus, showing an increase in sequence read density.
Supplementary Fig. 4 | Integer Copy Number Determination from Read Density
(a–c, f–g) Integer copy number calculations are shown for a single SK-BR-3 cell. (a) Mapped sequence reads are counted in variable bins of uniform expected read density. (b) Variable bin counts in grey are plotted on a log scale and KS-segments are plotted in blue for a region from chromosome 17–19. (c–e) Gaussian kernel smoothed density plots are shown with asterisks denoting the first increment peak, used for normalization, for (c) an SK-BR-3 cell, (d) a hypodiploid tumor cell from T10, and (e) an aneuploid tumor cell from T10. (f) A normalized KS-segmented profile in blue is compared to the integer copy number profile in red for an SK-BR-3 cell, and (g) this region is shown for chromosome 17 with integer copy number on the ordinate.
Supplementary Fig. 5 | R2 plots of CGH and SNS Profiles from Single Cells and Bulk DNA
(a–d) R2 plots of SK-BR-3 copy number profiles using various platforms and number of cells. (a) Sequence read count of a million cell sample compared to an array CGH profile using million of cells (b) Single cell profile measured by SNS versus a million cell array CGH profile, (c) Single cell profile measured by SNS compared to a million cell sequence read count profile, (d) A single cell profile (cell number 1) compared to another single cell profile (cell number 5) measured by SNS.
Supplementary Fig. 6 | Chromosome Breakpoint trees of T10 and T16
(a–b) Hierarchical clustering was used to construct trees from chromosome breakpoint patterns from single cells (a) hierarchical tree of 100 single cells from T10 (b) hierarchical tree of 100 single cells from T16 combining profiles from both the primary and metastatic tumors.
Supplementary Fig. 7 | Biclustered Heatmaps of Chromosome Breakpoints
(a) T10 heatmap of common chromosome breakpoints that have been biclustered and ordered to correspond to the integer copy number tree (b) T16P and T16M heatmap of common chromosome breakpoints that have been biclustered and ordered according to the integer copy number tree.
Supplementary Fig. 8 | Comparison of LOH and Copy Number Profiles
(a–c) Copy number profiles from tumor subpopulations are plotted with LOH at heterozygous SNPs that were identified by comparing to the diploid subpopulation. Asteriks indicates regions where LOH point mutations and copy number are in disagreement. (a) LOH in the T10 hypodiploid subpopulation. (b) LOH in the T10 aneuploid subpopulations. (c) LOH in the T16P aneuploid subpopulation. (d) Summaries of nucleotide classes and LOH detected in each subpopulation. At a given position, “ab” means two nucleotides are seen each in at least 5 cells, and “a” or “b” mean a single nucleotide was observed, always in at least 5 single cells.
Information and statistics are shown for 100 single cells flow-sorted from the polygenomic breast tumor T10
Information and statistics are shown for 52 cells from the primary breast tumor T16P and 48 cells from its corresponding metastasis T16M.
Summaries of nucleotide classes and LOH detected in each tumor subpopulation by comparing to the diploid subpopulation. At a given position, “ab” means two nucleotides are seen each in at least 5 cells, and “a” or “b” mean a single nucleotide was observed, always in at least 5 single cells. All nucleotides have a quality score > 30. See methods section for further details.