

Abstract
Anopheles mosquitoes are the vectors of several human diseases including malaria. In many malaria endemic areas, several species of Anopheles coexist, sometimes in the form of related sibling species that are morphologically indistinguishable. Determining the size and organization of Anopheles populations, and possible ongoing gene flow among them is important for malaria control and, in particular, for monitoring the spread of insecticide resistance alleles. However, these parameters have been difficult to evaluate in most Anopheles species due to the paucity of genetic data available. Here, we assess the extent of contemporary gene flow and historical variations in population size by sequencing and de novo assembling the genomes of wild-caught mosquitoes from four species of the Anopheles punctulatus group of Papua New Guinea. Our analysis of more than 50 Mb of orthologous DNA sequences revealed no evidence of contemporary gene flow among these mosquitoes. In addition, investigation of the demography of two of the An. punctulatus species revealed distinct population histories. Overall, our analyses suggest that, despite their similarities in morphology, behaviour and ecology, contemporary sympatric populations of An. punctulatus are evolving independently.
Keywords: Anopheles, genomics, inbreeding, malaria, population genetics
Introduction
Anopheles mosquitoes are the vectors of several important human infectious diseases including malaria, lymphatic filariasis and arboviruses (Krzywinski & Besansky 2003). Anopheles mosquitoes are found on all continents, with the exception of Antarctica, and represent more than 500 species that are often organized in sibling species complexes. The African Anopheles gambiae complex has been extensively studied and revealed a complex history of ancient introgression, recent speciation and ongoing gene flow among sibling species (Lawniczak et al. 2010; Neafsey et al. 2010, 2015; Riehle et al. 2011; Lee et al. 2013; Clarkson et al. 2014; Weetman et al. 2014; Fontaine et al. 2015). Unfortunately, most non-African Anopheles species have been much less studied and remained poorly understood.
Papua New Guinea (PNG) has some of the highest rates of malaria (WHO 2013) and lymphatic filariasis (Bockarie & Kazura 2003) in the world. There are at least 38 species of Anopheles recognized in PNG and the South Pacific, classified in 5 complexes or groups (Beebe et al. 2013). Among them, the 13 species of the Anopheles punctulatus group account for the majority of the Anopheles mosquitoes and include the main vectors of malaria and lymphatic filariasis: Anopheles punctulatus s.s., Anopheles farauti s.s. (or An. farauti 1), Anopheles hinesorum (or An. farauti 2), Anopheles farauti 4 and Anopheles koliensis. Many of these species are morphologically indistinguishable (Bryan 1973b; Foley et al. 1993; Cooper et al. 2002) and have large overlaps in their geographical distributions. While these species differ in their larval habitat preferences, with, for example, An. farauti s.s. typically using natural habitats near the coast while An. punctulatus s.s. prefers human-made inland habitats, larvae from multiple species are often found in the same habitat (Beebe & Cooper 2002). Similarly, these species show differences in feeding preferences (e.g. An. punctulatus s.s. being more anthropophilic than An. farauti s.s.), but adult Anopheles are still frequently captured together (Benet et al. 2004; Burkot et al. 2013).
Initially, species of the An. punctulatus group were defined based on forced mating experiments in the laboratory that yielded nonviable or sterile F1 hybrids (Bryan 1973a,b, 1974). Further molecular studies confirmed these initial species classifications (Foley et al. 1993; Beebe & Saul 1995; Beebe et al. 1999). In particular, recent analyses of complete mitochondrial genome sequences revealed that species of the An. punctulatus group diverged from each other several millions years ago (Logue et al. 2013). This study also suggested that these species were much more distantly related than, for example, species of the An. gambiae complex in Africa (Fontaine et al. 2015) for which instances of gene flow between species have been reported (Lee et al. 2013; Clarkson et al. 2014; Weetman et al. 2014). However, recent observations suggested that introgression may be occurring between two An. punctulatus species in southern New Guinea (Ambrose et al. 2012). Analysis of multiple genetic loci would enable to rigorously assess contemporary gene flow among the members of the An. punctulatus group, but these analyses are problematic in most Anopheles species due to the lack of sufficient genetic data.
Understanding the amount of gene flow among sympatric Anopheles species, as well as the structure and diversity of their populations, is critical for malaria control as these demographic parameters will, for example, influence whether insecticide resistance alleles could spread between populations or whether they would have to arise multiple times independently. Here, we circumvent the lack of genomic data for species of the An. punctulatus group by sequencing and de novo assembling the genomes from single wild-caught mosquitoes. Using this genome-scale data, we rigorously evaluate the possibility of contemporary gene flow among these four An. punctulatus species using phylogenetic and population-genetic approaches. We also examine the distribution of the genetic heterozygosity for two of the mosquitoes sequenced at the highest coverage and reconstruct the demographic history of their respective species.
Material and methods
Samples
Wild-caught mosquitoes were collected in the Madang province by the Entomology unit of the Papua New Guinea Institute of Medical Research (PNGIMR) as previously described (Henry-Halldin et al. 2011, 2012). We extracted genomic DNA from individual mosquitoes with DNeasy blood and tissue kits (Qiagen®) according to the supplemental protocol for purification of insect DNA with one modification: each mosquito was initially placed in a 2 mL tube with one 5-mm steel bead and 180 μL of PBS and homogenized by high-speed shaking at 15 Hz for 90 s with a Qiagen TissueLyser II instrument. We extracted DNA from a total of 16 mosquitoes, 8 of which had a total DNA yield of ~2 μg and were further analysed. We determined species identity using a PCR-based assay targeting species-specific nucleotide differences in the ribosomal internal transcribed spacer 2 (ITS2) (Henry-Halldin et al. 2011). For genome sequencing, we selected high-quality DNA extracted from individual mosquitoes of the following species: An. punctulatus s.s. (AP), An. farauti s.s. (AF s.s.), An. farauti 4 (AF4) and An. koliensis (AK).
Whole-genome sequencing and assembly
We sheared 2 μg of genomic DNA from each individual mosquito into 250–300 bp fragments using a Covaris S2 instrument (http://covarisinc.com) and prepared sequencing libraries as previously described (Logue et al. 2013). We then sequenced each library on an Illumina GAIIx or HiSeq 2000 instrument (Table S1, Supporting information). All sequences are available through the NCBI Sequence Read Archive (Accession Number SRP042363).
We de novo assembled each of the four genomes independently. First, we mapped reads from each sample onto its previously assembled mitochondrial genome (Logue et al. 2013) using Bowtie (Langmead et al. 2009) and removed these reads from further analyses. We then corrected the remaining reads for sequencing errors using the program QUAKE with a k-mer size of 17 and default parameters (Kelley et al. 2010). Finally, each sample was de novo assembled using the program ABySS (version 1.3.6) (Simpson et al. 2009). We determined the optimal k-mer for each sample by using various k-mers (ranging from 17 to 71) and selecting the k-mer that produced the highest N50 value and total assembly size closest to that of An. gambiae (260 MB). The best assemblies were obtained with a k-mer of 51 for AF4 and AP, and 31 for AK and AF s.s.
To assess sequence coverage, we mapped the original uncorrected reads onto each assembly using Bowtie2 (version 2.1.0) (Langmead & Salzberg 2012). We then calculated the average sequence coverage for each contig >1000 bp. We collapsed overlapping contigs by first aligning each genome assembly to itself using BLAT (Kent 2002) and (i) discarding any contig that completely aligned to another by only keeping the larger of the two; (ii) discarding all contigs where two or more contigs aligned to the same end of another; and (iii) merging contigs that overlapped by at least 500 bp and were 95% identical. The latter criteria are derived from in silico analyses of the An. gambiae genome that showed that <5% of all genomic DNA sequences would be incorrectly or ambiguously mapped using these cut-offs. When merging contigs, any nucleotide differences between them were masked. To further optimize our assemblies, we removed putative paralogous sequences by filtering out contigs with unusually high coverage (top 3%) and all contigs with less than half of the expected mean coverage. In each final assembly, we used the base corresponding to the most frequently sequenced nucleotide at each position, if the coverage was >10 ×, and masked any position sequenced by <10 reads.
Assembly comparison and alignments
We aligned the assembled contigs from all four An. punctulatus species using the Threaded Blockset Aligner (TBA) program (Blanchette et al. 2004). Briefly, we used the lastz program in the TBA package to generate pairwise alignments for all assemblies and kept only alignment blocks that were at least 500 bp in length and had 80% nucleotide identity. Nonunique alignment blocks were discarded. Finally, a multiple alignment containing all four species assemblies was generated using the TBA program.
Phylogenetic analyses
We used three methods to determine species relationships. First, we generated a distance matrix of the total number of pairwise differences using all alignment blocks >500 bp and containing all four species (we refer to these alignment blocks as ‘loci’). Second, we analysed all alignment blocks >5 kb (accounting for 12.2 Mb) using RAxML (Stamatakis 2014) with the default parameters and reconstructed the most likely phylogeny. Third, we determined the species relationships for each locus independently and reconstructed unrooted trees using the maximum-likelihood approach implemented in the program PhyML (Guindon & Gascuel 2003). In PhyML, we used the following parameters: the general time-reversible model of nucleotide substitution with invariant sites, estimated nucleotide frequencies and the approximate likelihood ratio test (aLRT) with a chi-square distribution to assess the statistical support for each tree. To robustly assess the phylogeny of An. punctulatus species, we only examined loci >1000 bp and whose trees were supported by an aLRT score >0.9.
Identification of possible introgression candidates using sequence divergence
To identify loci possibly indicative of introgression, we searched for trees with unusual short branch lengths separating two of the four species. For each tree, we calculated the ratio of the two shortest adjacent external branches to the sum of all four external branches. We then considered, as candidate loci for introgression, any tree for which the ratio was in the 0.01th quantile of the cumulative distribution (Fig. S1, Supporting information). We characterized putative protein-coding genes in each locus by blasting the contig DNA sequence against the NCBI nucleotide database using blastn and retrieved the annotation when the best nucleotide blast hit was to an An. gambiae gene and was at least 70% identical.
Identification of single nucleotide polymorphisms (SNPs) in orthologous regions of the An. punctulatus s.s. and An. farauti 4 genomes
To obtain a first genome-wide perspective on the genetic diversity of AP and AF4, we mapped all sequencing reads from AP and AF4 onto their corresponding contig assembly using Bowtie2 (Langmead & Salzberg 2012) (treating paired-end reads as unpaired). We restricted our analyses to loci >1000 bp and only called SNPs at positions sequenced at high quality (Q ≥ 20) and high coverage (80–200 X in AF4 and 25–80 X in AP, Fig. S2, Supporting information). We considered a site to be variable if >20% of the reads carried the minor allele (see Fig. S3, Supporting information). We defined as a shared polymorphism any orthologous position that was variable in both AP and AF4 and for the same two alleles.
Number of shared polymorphisms expected between species under different population histories
We calculated the expected number of shared polymorphisms between AP and AF4 under different demographic models using the coalescent simulation program ms (Hudson 2002). We estimated the scaled population mutation rate, θ, from the observed heterozygosity and the population recombination rate, ρ, using mlRho (Haubold et al. 2010). We simulated 22 394 loci (i.e. the number of loci analysed for AF4 and AP in our study after filtering, see above) and assigned to each locus a θ value corresponding to the value observed in one of our alignment blocks. We used two different models in our simulations, one without gene flow and one with varying amounts of gene flow. The first model represented two species that diverged in the past from a common ancestor and afterwards evolved independently (i.e. the isolation model with no gene flow between diverged species, see (Sousa & Hey 2013) for a review). For this isolation model, we performed 1000 simulations for each locus. The second model investigated the consequences of varying amount and age of gene flow on the number of shared polymorphisms between the two species. For this model, we simulated each locus 10 000 times, each time randomly selecting values of gene flow (4Nem) and time since the most recent gene flow (in 4Ne generations): for each simulation, we randomly assigned a number of lineages originating from the introgressing population (i.e. 4Nem) by drawing from a uniform distribution between 1 and 5; the time since the most recent gene flow was randomly drawn from a uniform distribution between 0 and 0.5 (scaled in 4Ne generations). In all simulations, we modelled gene flow as continuous for either 100 or 1000 generations and occurring at a constant rate. For facilitating the interpretation of the results, we also included the results scaled using a mutation rate of 2.8 × 10−9 per base pair per generation (Dixit et al. 2014; Keightley et al. 2014) and assuming 12–20 generations per year, as often used for An. gambiae and Aedes aegypti (Chandre et al. 2000; Beserra et al. 2006; Barbosa et al. 2011).
For each simulation, we used a modified version of the software msstats (github.com/rossibarra/msstats) to calculate the total number of shared polymorphisms and the total number of unique polymorphisms within each species across all loci. We then grouped simulations into bins according to the random values of the parameters used for 4Nem (binned in 1.0 increments) and most recent gene flow (binned in 0.1 increments). Each bin typically contained ~500–1000 simulations. We then calculated p-values by calculating the number of simulations producing more shared polymorphisms than observed in the actual data.
Characterization of the historical demography of An. farauti 4 and An. punctulatus s.s
To reconstruct the demographic history of both species of Anopheles from whole-genome data, we used the pairwise sequential markovian coalescent (PSMC) model (Li & Durbin 2011). We divided the consensus DNA sequences of each species into 10-bp nonoverlapping bins and marked each bin as homozygous or heterozygous based on whether it contained at least one SNP. We used bins of 10 bp to account for the high genetic diversity observed in Anopheles, although increasing the size of the bins did not qualitatively change the results. To improve the accuracy of inferring historical recombination events, we excluded all contig sequences shorter than 5000 bp (41–49% of all contigs, 11–19% of all bases) resulting in a total of 117–130 Mb of DNA sequences analysed. Note that in this analysis, each contig is treated as a separate accession (i.e. they are not artificially concatenated). We used the recommended parameters, only adjusting for the higher heterozygosity and recombination rate in Anopheles compared to humans. The settings of the piecewise constant and maximum time (-p and –t options) were adjusted to maintain >20 ancestral recombination events in each time epoch (Li & Durbin 2011). Thus, max time was set to 15 for AP and 20 for AF4 and the number of piecewise parameters was left as default. The ratio of θ to q was set to 1, based on preliminary estimation using mlRho (Haubold et al. 2010). To estimate variance, we applied a bootstrapping approach by splitting genomic sequences into smaller segments and then randomly sampling these segments with replacement (Li & Durbin 2011). We performed a total of 100 bootstraps for each of the samples run in PSMC.
As the PSMC method relies on the genomic distribution of heterozygous sites, it can only be used when both alleles are called with high confidence. We assessed the influence of sequence coverage on the PSMC results by performing two separate runs for the AF4 mosquito: one with our original coverage (131 X) and a second sequence that was down-sampled to the same coverage as AP (51 X).
All PSMC results were plotted in R statistical software using the R package ggplot2. Unscaled results in θ and pairwise sequence divergence were calculated according to the PSMC manual (github.com/lh3/psmc). Results were scaled using a mutation rate of 2.8 × 10−9 per base pair per generation (Dixit et al. 2014; Keightley et al. 2014) and assuming 12–20 generations per year (Chandre et al. 2000; Beserra et al. 2006).
Results
Genomic sequencing and de novo assembly of four wild-caught mosquitoes of the An. punctulatus group
We sequenced the genomes of four individual mosquitoes, An. punctulatus (AP), An. koliensis (AK), An. farauti s.s (AF s.s.) and An. farauti 4 (AF4), collected in the Madang province of Papua New Guinea (PNG). The Madang province, located on the northern coast of PNG, has one of the highest malaria prevalence in the country (WHO 2013) and has been the focus of many malaria control campaigns since the 1970s (Cattani et al. 1986). In this province, four of the five main malaria vector species (AP, AK, AFss and AF4) coexist and often share the same habitats (Cooper et al. 2002). From each mosquito, we generated ~37–170 million read pairs resulting in 34–131 × coverage (Table 1). We de novo assembled each genome independently and initially obtained 389 743–927 145 contigs with a total assembly size of 193–293 Mb and an N50 of 3691–13 469 (Table S2, Supporting information) (for reference, the genome size of An. gambiae is 260 Mb). Analysis of the distribution of the average contig coverage (Fig. S4A-D, Supporting information) revealed a bimodal distribution with some of the contigs displaying half the expected coverage (based on the overall sequencing effort and the expected size of Anopheles genomes). We hypothesized that these contigs included redundant sequences misassembled due to high genetic heterozygosity (i.e. each parental chromosome was assembled separately), as has previously been observed for other diploid organisms (Takeuchi et al. 2012; Zhang et al. 2012; Zheng et al. 2013). We subsequently merged these sequences together (see Material and methods for details), which eliminated most of the redundancy (Fig. S4E–H, Supporting information). Our final assemblies accounted for 62.9–74.0% of the reads generated and contained 14 407–41 925 contigs with an N50 of 4664–16 229 and a final assembly size of 146.2–161.6 Mb (Table 1). Thus, from single wild-caught mosquitoes, we were able to de novo assemble DNA sequences representing roughly two-thirds of their expected genome size (based on the size of the An. gambiae genome).
Table 1.
Summary of the Anopheles punctulatus species genome assemblies
| Sample | Expected coverage* | Size (kb) | # of contigs | % assembled | N50 | Max (bp) | Median (bp) |
|---|---|---|---|---|---|---|---|
| An. farauti 4 (AF4) | 131× | 146 386 | 14 407 | 63.1 | 16 229 | 331 681 | 6280 |
| An. punctulatus s.s. (AP) | 50× | 146 190 | 20 775 | 62.9 | 10 258 | 97 012 | 5136 |
| An. koliensis (AK) | 39× | 151 327 | 41 925 | 74 | 4664 | 76 110 | 2557 |
| An. farauti s.s. (AF s.s.) | 34× | 161 555 | 27 543 | 72.3 | 8767 | 79 463 | 3975 |
Based on size of An. gambiae genome – 260 Mb.
The best assembly, based on the N50 statistic, was obtained for the AF4 species, which derived from the sample with the highest sequence coverage (Table 1). To evaluate whether the increased sequencing depth was responsible for the better assembly, we randomly subsampled AF4 reads to a coverage similar to that obtained for AP. We then used this subset of reads to independently de novo assemble the genome of AF4. The lower sequence coverage did not reduce the assembly quality of AF4 and yielded similar assembly statistics as those obtained using all reads (Table S3, Supporting information). As de novo genome assembly algorithms sometimes fail to collapse homologous chromosomes into a single DNA sequence due to genetic heterozygosity, we hypothesized that the differences in assembly quality among the four An. punctulatus species was, at least partially, due to differences in genetic heterogeneity among species and that AF4 probably had a lower nucleotide diversity than the other An. punctulatus species assembled here.
Analyses of phylogenetic trees does not provide evidence of introgression
We compared the assemblies of the four species and produced a total of 47 181 four-sequence alignments (or loci) containing 82 651 073 nucleotide positions (or 31.8% of the An. gambiae reference genome) (Figure 1A and B). In this dataset, we identified 11 925 951 variable positions. The number of pairwise differences between species varied from 4 947 209 to 7 053 973 (6% to 8.5% divergence, Table 2) with AF s.s. and AK being most closely related. Maximum-likelihood analysis of all alignment blocks >5 kb (accounting for 12.2 Mb of DNA sequences) yielded a similar phylogeny with AF s.s. and AK grouping together.
Fig. 1.
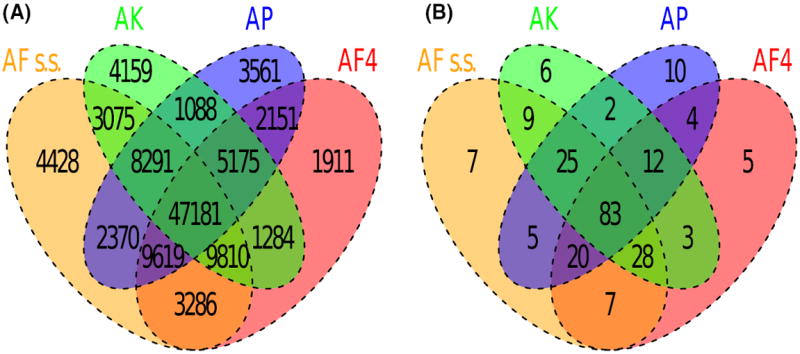
Multiple-alignment statistics. The Venn diagrams summarize (A) the number of alignment blocks >500 bp generated for all species combinations, and (B) the total number of aligned base pairs represented in these blocks (in millions).
Table 2.
Number of pairwise nucleotide differences (lower diagonal) and per cent divergence (upper diagonal) among the Anopheles punctulatus mosquito species sequenced
| AF s.s. | AF4 | AK | AP | |||
|---|---|---|---|---|---|---|
| AF s.s | — | 8.2% | 6.0% | 8.3% | ||
| AF4 | 6 758 632 | — | 8.2% | 8.1% | ||
| AK | 4 947 209 | 6 808 445 | — | 8.5% | ||
| AP | 6 883 739 | 6 685 658 | 7 053 973 | — |
We also reconstructed a phylogenetic tree separately for each of the 31 312 loci >1000 bp (see Material and methods for details on the filtering criteria) using the maximum-likelihood model implemented in the program PhyML (Guindon & Gascuel 2003). 30 907 (98.7%) of the trees (accounting for 70 921 162 bp) were supported with an aLRT score >0.9 and were further analysed. 99.7% of these well-supported trees also grouped AF s.s. and AK together, while 0.17% and 0.08% of the trees supported two different tree topologies, grouping AF s.s with, respectively, AP and AF4. Note that, for all trees, the internal branch only represented a small proportion of the total branch length, indicating an old and rapid radiation of the An. punctulatus species from a common ancestor (Fig. S5, Supporting information).
Recent (i.e. contemporary) introgression of a given locus from one species to another would result in a tree with unusually short branches between these two species. We therefore calculated for each tree the proportion of the total branch length accounted for by the two most closely related species. We selected as putative introgression candidates the 1% loci with the shortest distances (proportionally) between the two closest tips (see Material and methods for details). These 306 loci accounted for 490 052 bp (or 0.19% of the genome) and contained 172 predicted protein-coding genes (Table S4, Supporting information). Most of these introgression candidates had short branches separating AF s.s. and AK (302 of 306 trees). This observation could indicate that introgression preferentially occurred between these two species, which, based on our phylogenetic analysis, are the most closely related. However, it is also possible that our analysis identified loci at the tail of the distribution of the tree topologies rather than true biological outliers (Fig. S6, Supporting information). Further investigations of these candidates revealed that the number of nucleotide differences between AF s.s. and AK was proportional to the number of differences between AF4 and AP (Fig. S7, Supporting information), indicating that these trees with short branches separating the two closest species were likely the results of unusual substitution rates affecting the entire tree (e.g. selection or low mutation rate) rather than only two of the four branches as we would expect with recent gene flow. Overall, our phylogenetic analyses of a third of the genome of four An. punctulatus species did not reveal any evidence of recent introgression among them.
Analyses of shared polymorphisms between AF4 and AP does not support recent gene flow between these species
In addition to divergence, gene flow also influences the patterns of diversity and, in particular, can lead to shared DNA polymorphisms between species. Here, we restricted our analysis of genetic diversity to the AF4 and AP mosquitoes as the lower sequence coverage in the other species sequenced hampered our ability to reliably call SNPs. Of 51 610 847 orthologous nucleotides sequenced at >20X in both AF4 and AP, we identified 164 081 (0.32%) heterozygous sites in the AF4 mosquito and 318 375 (0.62%) in the AP mosquito (Table 3). Subsampling of the AF4 data to the same coverage as the AP mosquito did not qualitatively change these findings (Table S5, Supporting information), indicating that the higher heterozygosity in AP was not caused by differences in sequence coverage but reflected genuine biological differences. The lower genetic diversity observed in AF4 compared to that of AP is also consistent with the higher assembly quality obtained for this species (see above). Overall, across more than 50 Mb of DNA sequences, only 467 nucleotide positions were polymorphic for the same alleles in both AP and AF4 (Table 3).
Table 3.
Summary of the genetic diversity in Anopheles farauti 4 and Anopheles punctulatus s.s.
| Length | Heterozygous sites in AF4 | Heterozygous sites in AP | Divergence | # of shared polymorphisms | |
|---|---|---|---|---|---|
| Total | 51 610 847 | 164 081 (0.32%) | 318 375 (0.62%) | 4 056 848 (7.86%) | 467 |
| Per locus | 2305 [1000–17 080] | 7.32 [0–86] | 14.22 [0–140] | 181.2 [22–1551] | 0.02 [0–3] |
| Per Kb | NA | 0.007 [0–0.086] | 0.014 [0–0.14] | 0.18 [0.02–1.55] | 2.1E-5 [0–0.003] |
[] represent the minimum and maximum values observed.
At least three nonexclusive mechanisms could generate shared polymorphisms among species: incomplete lineage sorting, gene flow or sequencing errors. To determine how many shared alleles would be expected due to incomplete lineage sorting, we simulated the evolutionary history between two deeply diverged species, AF4 and AP, under a coalescent model and calculated the resulting number of shared polymorphic sites. In all simulations, the sum of shared sites was always 0, indicating that, under a constant population size model without gene flow, we would not expect any shared sites. To discriminate between gene flow and sequencing errors, we first compared the distribution of these shared polymorphisms throughout the genome: if the shared polymorphisms resulted from recent gene flow, we would expect them to be clustered in specific loci (i.e. those that have been recently exchanged). Instead, shared polymorphisms were distributed throughout the entire genome sequence, with an average of 0.02 shared polymorphisms per locus and no locus carried more than three shared polymorphisms (Table 3). This observation suggested that many of the shared polymorphisms could be due to sequencing errors.
To estimate the maximum amount of gene flow consistent with the number of shared polymorphisms observed (under the conservative assumption that all of these resulted from gene flow), we determined the number of shared polymorphisms that would be expected with various amounts of gene flow between AF4 and AP. We simulated under the coalescent two populations of constant size that, at a given time and continuing for 1000 generations, have a specific amount of gene flow (see Material and methods for details). We varied both the amount of gene flow and the most recent time of gene flow and determined which parameter values were consistent with the number of shared sites observed in our data. We binned the simulations based on the coalescent time since the last gene flow event (in 4Ne generations) and the rate of gene flow per generation (in 4Nem). This analysis enabled us to identify introgression parameters that can be excluded given the observed number of shared polymorphisms (Fig. 2). For example, we could exclude gene flow involving more than one individual per generation (4Nem = 2) if it occurs later than 0.13 4Ne generations ago (~11 000 years ago, assuming 12 generations per year) as these parameters produced significantly more shared polymorphisms than we observed in the data.
Fig. 2.
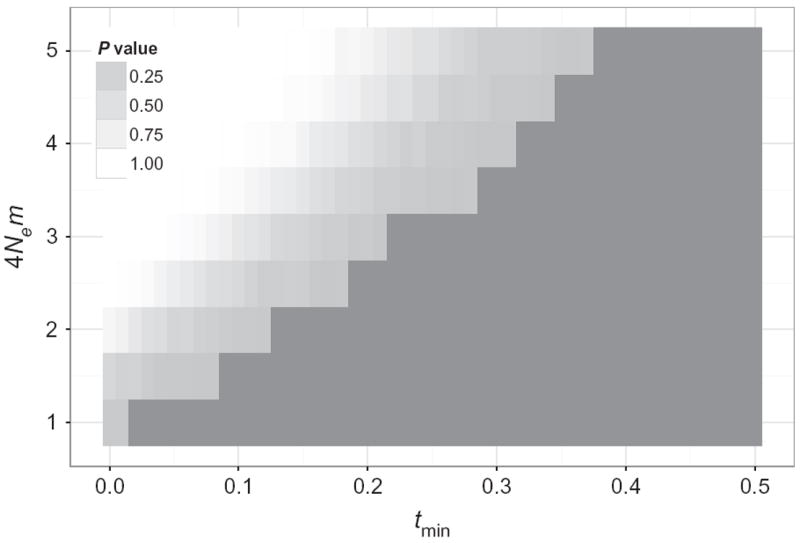
Amount and age of gene flow that can be excluded given the number of shared polymorphisms observed. The figure shows the probability that introgression would lead to significantly more shared polymorphisms than observed based on the amount of gene flow (y-axis, in 4Nem) and time since the last gene flow (x-axis, in 4Ne generations). The white surface in the graph represents combinations of parameters that are incompatible with the data observed. Note that this analysis assumes that all shared polymorphisms were genuine (i.e. not sequencing errors) and therefore likely overestimates possible gene flow.
Overall, our analyses suggested that there was no significant contemporary gene flow between AF4 and AP and that the small number of shared polymorphisms observed likely resulted from ancient gene flow or, perhaps more likely, from sequencing errors.
Demographic history of An. farauti 4 and An. punctulatus s.s
We used the pairwise sequential markovian coalescent (PSMC) model (Li & Durbin 2011) to estimate the demographic history of AP and AF4. Due to inherent uncertainty in estimating mutation rates and generation time for Anopheles mosquitoes, we present the results as θ and pairwise sequence divergence (Fig. 3; the results scaled in effective population size (Ne) and generations are shown in Fig. S8, Supporting information). Our analyses showed that the two species had similar population sizes from 15 000 000 to until about 600 000 generations in the past, after which time their population histories diverged (Fig. S8, Supporting information). At that point, the AP population increased in size while the AF4 population initially declined before a short period of expansion followed by a final and rapid decline in population size (Fig. 3).
Fig. 3.
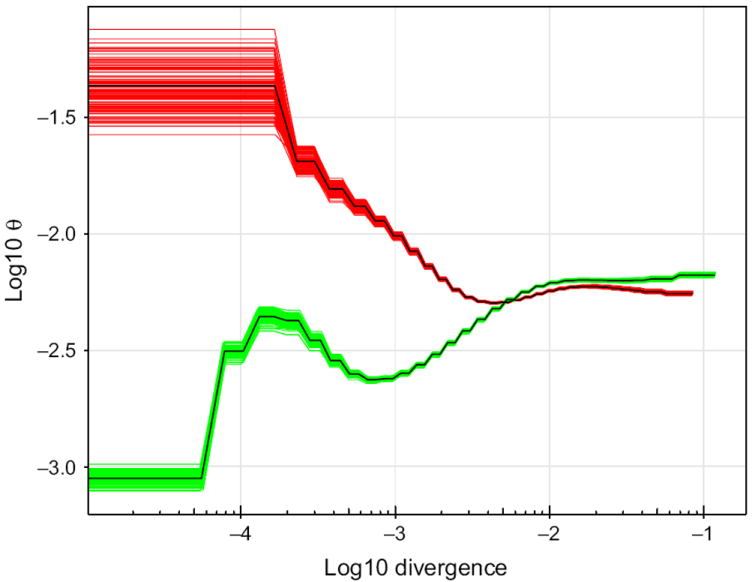
Demographic histories of AF4 and AP. The figure shows the estimated historical variations in effective population size for AF4 (green) and AP (red) based on PSMC analyses. The composite green (AF4) and red (AP) lines represent 100 bootstrap replicates. The y-axis represents the population mutation rate (in log10 of θ), and the x-axis represents the log10 pairwise sequence divergence.
Bootstrap analysis showed little variance associated with all population size estimates except for the most recent time periods as expected due to the limited number of recent coalescent events that can be inferred from a single genome sequence (Li & Durbin 2011). Note also that estimates of population sizes in the very recent past (<30 000 generations ago) might be influenced by the fragmentation of our assembly: a small proportion of the contigs analysed might be shorter than the maximum identical by descent (IBD) tracks expected, increasing the statistical uncertainty of very recent estimates. More importantly, PSMC has been shown to be sensitive to false heterozygosities identified in sequence data. We thus down-sampled AF4 to the same coverage as AP to test whether the differences in population history between AP and AF4 could be artificially caused by the differences in sequence coverage and our ability to correctly identify SNPs. The down-sampled AF4 sequence contained more heterozygous sites than the original sequence, reflecting the difficulty in accurately identifying polymorphisms with lower coverage, but the overall pattern of the AF4 population history remained similar and distinct from that of AP (Fig. S9, Supporting information).
Discussion
If malaria eradication is to be successful, it is important that we better understand how insecticide resistance could spread across mosquito populations. Unfortunately, given the limited genetic data available for most mosquito species, there are insufficient numbers of nuclear loci available to rigorously evaluate gene flow between populations and species. This is especially problematic for non-African Anopheles mosquitoes that have been under studied, though reference genomes for several important Anopheles species have been recently published (Neafsey et al. 2015). In particular, detailed analyses of gene flow relying on the genome-wide pattern of diversity, that have been extremely informative for An. gambiae (Lee et al. 2013; Clarkson et al. 2014), are impossible to conduct for most of the other Anopheles species as we currently lack both a high-quality reference genome sequence and enough polymorphic genetic markers distributed throughout the genome. Alternatively, investigations of the genome-wide patterns of divergence between species and analyses of the distribution of the genetic heterozygosity within a single diploid individual could provide rigorous assessments of gene flow and can be performed using single genome sequences. In this study, we sequenced and de novo assembled the genomes of four important malaria vectors from the An. punctulatus group of PNG to rigorously test for recent (i.e. contemporary) introgression among these species.
Evolutionary relationship of An. punctulatus
Our phylogenetic analysis, based on 71 Mb of nuclear sequence, revealed that the An. punctulatus species are separated by very long external branches and short internal branches supporting our previous findings that they rapidly diverged from each other millions of years ago (Logue et al. 2013). The tree topology obtained in this study highlights different relationships among species than we previously described based on the mitochondrial genome alone (Logue et al. 2013). However, while our previous study was based on a single, nonrecombining, locus that captures a single history, here we independently reconstructed here the species relationships from 30 907 nuclear loci, representing different histories. We therefore believe that the species relationships described in this study, which are consistent with previous, though not statistically well-supported, phylogenies (Foley et al. 1998; Beebe et al. 1999, 2000) recapitulate the true relationships among An. punctulatus mosquitoes.
No evidence of contemporary gene flow among An. punctulatus species
Determining whether gene flow is currently occurring between species is critical for designing efficient vector control strategies as gene flow would, for example, increase the spread of insecticide resistance alleles across mosquito populations. Earlier studies based on forced mating experiments between species of the An. punctulatus group revealed that hybrids were sterile or nonviable in the laboratory (Bryan 1973b). However, such studies are complicated to perform and may not accurately represent natural processes. By contrast, population-genetic studies provide an opportunity to test whether gene flow has occurred in the history of the studied populations and can reveal even low amounts of gene flow between two populations. For example, a recent study reported evidence of gene flow between two An. punctulatus mosquito species, An. farauti s.s. and An. hinesorum, in southern New Guinea (Ambrose et al. 2012). However, as this introgression was detected using only a mitochondrial locus, it is impossible to exclude that the mosquitoes studied were not sterile hybrids that would not contribute to the genetic diversity of either species. Our analyses of thousands of independent loci in four of the main An. punctulatus species did not reveal any evidence of recent gene flow. In particular, the analyses of genetic diversity throughout a large fraction of the genome of an AP and AF4 mosquito indicates that, if any gene flow occurred between these two species, it was extremely minute or happened in the distant past. In fact, our simulations showed that migration of more than one individual per generation during the last 0.13 4Ne generations (11 000 years assuming 12 generations per year) could be statistically excluded given our data. While estimates of mutation rates and generation times are still very crude, our study clearly shows that there is no contemporary gene flow occurring among these An. punctulatus species in this location of PNG and that, from a vector control perspective, these species can be considered to be reproductively isolated.
It is, however, important to note the limitations of our population-genetic analyses. First, our models assume constant population sizes, no population structure and no selection. In this regard, the few genetic studies conducted in An. punctulatus mosquitoes have shown limited fine-scale population stratification in mainland PNG (Henry-Halldin et al. 2012; Logue et al. 2013; Seah et al. 2013). In addition, we would expect positive selection (e.g. for insecticide resistance) to increase the signal of gene flow, as the introgressed alleles would provide higher fitness in the presence of insecticides and therefore would be more likely to spread through the populations. Overall, we believe that violations of our model assumptions are unlikely to qualitatively change our findings. Second, our analyses are based on a single mosquito per species. In this regard, it is important to emphasize that our analyses, as they are based on numerous independent loci, investigate possible gene flow throughout the entire genealogy of each sample sequenced and therefore capture a large history of the population. In addition, as the different mosquitoes were collected in the same geographic area, it should increase the probability to detect any gene flow among them. However, it is possible that, if there are important genetic differences between populations from various locations of PNG or the Southwest Pacific, gene flow might occur in other regions of PNG or the Southwest Pacific. This could, for example, explain the differences between our findings and the observations of possible introgression between An. hinesorum and An. farauti s.s. in southern New Guinea (Ambrose et al. 2012). Analysis of additional samples from across Papua New Guinea and involving multiple individuals per species would enable to rigorously test this hypothesis.
Discordant demographic histories of AF4 and AP
Very little is known about the population size, organization and history of the different Anopheles species in PNG. Since we sequenced wild-caught mosquitoes (as opposed to colony mosquitoes that are bred to reduce their genetic heterozygosity), we were able to characterize the genome-wide patterns of genetic diversity in AP and AF4 by cataloguing differences between the two homologous chromosomes carried by each individual. While both species shared a similar demographic history until ~52 000 years ago (630 957 generations assuming 12 generations per year) they then diverged and underwent independent variations in population sizes: the AP population continuously expanded in size, while the AF4 population fluctuated before a recent and dramatic decrease in size. One possible explanation for these observations may be the arrival of humans in PNG ~50 000 years ago (Lilley 1992), which might have favoured the AP species that can breed more successfully in transient bodies of water (e.g. water containers) than AF4 mosquitoes (Charlwood et al. 1986; Cooper et al. 2002). Alternatively, AP mosquitoes seem to be the most anthropophilic of all An. punctulatus species [(Beebe et al. 2013) and references therein] and it is possible that, after the arrival of humans, AP would have had more hosts available for blood meals. In this case, we would also predict that AP would be more impacted by bed nets and insecticides than AF4 mosquitoes. However, it is possible that the apparent differences in population size are in fact caused by population structure rather than genuine changes in the effective population size of AF4. For example, in humans, population divergence can cause an increase in effective population size (Li & Durbin 2011). The final population ‘crash’ in AF4 could therefore possibly reflect the population size of a small subpopulation analysed. Additional sampling for other areas of PNG would enable us to differentiate between these scenarios.
Implications for disease transmission in PNG
Our genomic analyses have important implications for ongoing and future vector control strategies implemented in PNG. The lack of detectable contemporary gene flow suggests that insecticide resistance alleles are unlikely to spread across the An. punctulatus species. This is encouraging for vector control in this region as it will considerably slow down the spread of insecticide resistance: the resistance alleles will need to arise independently in each species. Currently, there are no standing variants for insecticide resistance reported among An. punctulatus mosquitoes (Henry-Halldin et al. 2012) but the widespread deployment of long-lasting insecticide-treated bed nets and indoor residual spraying campaigns (Kazura et al. 2012; Hetzel et al. 2014) in PNG may rapidly change this situation.
Our analyses of the population sizes and their dynamics in AF4 and AP are also interesting from a vector control perspective. The distinct demographic histories observed between AP and AF4 reveal that, historically, these species have responded differently to environmental changes. This suggests that these species may also respond differently to current environmental perturbations such as climate change, or, more importantly, to the deployment of vector control measures. Additionally, as the effective population size of a species directly influences the probability of an advantageous mutation being swept to fixation, the larger effective population size in AP suggests that any insecticide resistance or other advantageous allele would likely reach fixation faster in AP than in AF4.
Conclusion
With a new worldwide effort to eradicate malaria, it is imperative that we better understand the biology of Anopheles mosquitoes. In particular, we need to characterize the extent of current gene flow between morphologically identical Anopheles species that overlap geographically. Population genetics provide powerful methods to identify gene flow but typically require a wealth of genomic resources that are not available for most non-African Anopheles species. Here, we circumvented the paucity of genomic data for members of the An. punctulatus group by sequencing and de novo assembling the genomes of four known malaria vectors from PNG. We found no evidence of recent gene flow between sibling species, suggesting that insecticide resistance is unlikely to spread among these species through exchange of resistance alleles. Analysis of the demography of AF4 and AP revealed distinct population histories between species, suggesting that these species will most likely respond differently to the deployment of vector control measures and other environmental perturbations. Overall, our results provide a first characterization of the population history of species of the An. punctulatus group and illustrate how this approach could be applied to other disease vectors with limited available genomic information.
Supplementary Material
Fig. S1 Identification of putative introgression candidates based on divergence.
Fig. S2 Distribution of the sequence coverage per locus.
Fig. S3 Distribution of the reference allele frequencies.
Fig. S4 Distribution of the sequence coverage per contig for the initial assemblies (A–D) and after removing redundant contigs (E–H).
Fig. S5 Distribution of the internal branch lengths.
Fig. S6 Distribution of the external branch lengths.
Fig. S7 Distribution of nucleotide differences between species.
Fig. S8 Demographic history of AF4 and AP.
Fig. S9 Influence of sequence coverage on the estimated demographic history of AF4.
Table S1 Sequencing summary for each sample.
Table S2 Summary of the initial de novo genome assemblies.
Table S3 Summary of the Anopheles farauti4 assembly after sub-sampling sequencing reads to the coverage of Anopheles punctulatus s.s.
Table S4 Gene annotation of each introgression candidate as determined by the best nucleotide blast hit to Anopheles gambiae genes.
Table S5 Summary of the genetic diversity in Anopheles farauti4 estimated after sub-sampling Anopheles farauti4 reads to the coverage of Anopheles punctulatus s.s.
Acknowledgments
We thank the staff of the Entomology Unit of the Papua New Guinea Institute of Medical Research (PNGIMR) for their help in collecting Anopheles samples. We also thank two anonymous reviewers for their helpful comments and suggested modifications to this manuscript. This work was supported by Cleveland Clinic Funds to DS and the Fogarty International Center (TW007872) and NIAID International Centers of Excellence in Malaria Research Program (5U19AI089686) to PAZ.
Footnotes
P.A.Z. and D.S. designed the research; K.L., S.T.S. and E.R.C. performed the experiments and analysed the data; L.R., P.M.S., P.A.Z. and D.S. contributed new reagents or analytical tools; and K.L., S.T.S., P.A.Z. and D.S. wrote the manuscript.
Supporting information
Additional supporting information may be found in the online version of this article.
Data accessibility
Raw sequencing data: NCBI SRA: SRP042363; DNA sequence assemblies: GenBank Accessions JXWZ00000000-JXXC00000000; SNP information: Dryad doi:10.5061/dryad.16hc8.
References
- Ambrose L, Riginos C, Cooper RD, et al. Population structure, mitochondrial polyphyly and the repeated loss of human biting ability in anopheline mosquitoes from the southwest Pacific. Molecular Ecology. 2012;21:4327–4343. doi: 10.1111/j.1365-294X.2012.05690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa S, Black WCT, Hastings I. Challenges in estimating insecticide selection pressures from mosquito field data. PLoS Neglected Tropical Diseases. 2011;5:e1387. doi: 10.1371/journal.pntd.0001387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beebe NW, Cooper RD. Distribution and evolution of the Anopheles punctulatus group (Diptera: Culicidae) in Australia and Papua New Guinea. International Journal for Parasitology. 2002;32:563–574. doi: 10.1016/s0020-7519(01)00359-9. [DOI] [PubMed] [Google Scholar]
- Beebe NW, Saul A. Discrimination of all members of the Anopheles punctulatus complex by polymerase chain reaction–restriction fragment length polymorphism analysis. American Journal of Tropical Medicine and Hygiene. 1995;53:478–481. doi: 10.4269/ajtmh.1995.53.478. [DOI] [PubMed] [Google Scholar]
- Beebe NW, Ellis JT, Cooper RD, Saul A. DNA sequence analysis of the ribosomal DNA ITS2 region for the Anopheles punctulatus group of mosquitoes. Insect Molecular Biology. 1999;8:381–390. doi: 10.1046/j.1365-2583.1999.83127.x. [DOI] [PubMed] [Google Scholar]
- Beebe NW, Cooper RD, Morrison DA, Ellis JT. A phylogenetic study of the Anopheles punctulatus group of malaria vectors comparing rDNA sequence alignments derived from the mitochondrial and nuclear small ribosomal subunits. Molecular Phylogenetics and Evolution. 2000;17:430–436. doi: 10.1006/mpev.2000.0853. [DOI] [PubMed] [Google Scholar]
- Beebe NW, Russell TL, Burkot TR, Lobo NF, Cooper RD. The Systematics and Bionomics of Malaria Vectors in the Southwest Pacific. In: Manguin PS, editor. Anopheles Mosquitoes – New Insights Into Malaria Vectors. InTech; Rijeka, Croatia: 2013. pp. 357–394. [Google Scholar]
- Benet A, Mai A, Bockarie F, et al. Polymerase chain reaction diagnosis and the changing pattern of vector ecology and malaria transmission dynamics in Papua New Guinea. American Journal of Tropical Medicine and Hygiene. 2004;71:277–284. [PubMed] [Google Scholar]
- Beserra EB, de Castro FP, Jr, dos Santos JW, Santos TdaS, Fernandes CR. Biology and thermal exigency of Aedes aegypti (L.) (Diptera: Culicidae) from four bioclimatic localities of Paraiba. Neotropical Entomology. 2006;35:853–860. doi: 10.1590/s1519-566x2006000600021. [DOI] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Research. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockarie MJ, Kazura JW. Lymphatic filariasis in Papua New Guinea: prospects for elimination. Medical Microbiology and Immunology. 2003;192:9–14. doi: 10.1007/s00430-002-0153-y. [DOI] [PubMed] [Google Scholar]
- Bryan JH. Studies on the Anopheles punctulatus complex. I. Identification by proboscis morphological criteria and by cross-mating experiments. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1973a;67:64–69. doi: 10.1016/0035-9203(73)90322-2. [DOI] [PubMed] [Google Scholar]
- Bryan JH. Studies on the Anopheles punctulatus complex. II. Hybridization of the member species. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1973b;67:70–84. doi: 10.1016/0035-9203(73)90323-4. [DOI] [PubMed] [Google Scholar]
- Bryan JH. Morphological studies on Anopheles punctulatus Donitz complex. Transactions of the Royal Entomological Society London. 1974;125:413–435. [Google Scholar]
- Burkot TR, Russell TL, Reimer LJ, et al. Barrier screens: a method to sample blood-fed and host-seeking exophilic mosquitoes. Malaria Journal. 2013;12:49. doi: 10.1186/1475-2875-12-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattani JA, Tulloch JL, Vrbova H, et al. The epidemiology of malaria in a population surrounding Madang, Papua New Guinea. American Journal of Tropical Medicine and Hygiene. 1986;35:3–15. doi: 10.4269/ajtmh.1986.35.3. [DOI] [PubMed] [Google Scholar]
- Chandre F, Darriet F, Duchon S, et al. Modifications of pyrethroid effects associated with kdr mutation in Anopheles gambiae. Medical and Veterinary Entomology. 2000;14:81–88. doi: 10.1046/j.1365-2915.2000.00212.x. [DOI] [PubMed] [Google Scholar]
- Charlwood JD, Graves PM, Alpers MP. The ecology of the Anopheles punctulatus group of mosquitoes from Papua New Guinea: a review of recent work. Papua New Guinea Medical Journal. 1986;29:19–26. [PubMed] [Google Scholar]
- Clarkson CS, Weetman D, Essandoh J, et al. Adaptive introgression between Anopheles sibling species eliminates a major genomic island but not reproductive isolation. Nature Communications. 2014;5:4248. doi: 10.1038/ncomms5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper RD, Waterson DG, Frances SP, Beebe NW, Sweeney AW. Speciation and distribution of the members of the Anopheles punctulatus (Diptera: Culicidae) group in Papua New Guinea. Journal of Medical Entomology. 2002;39:16–27. doi: 10.1603/0022-2585-39.1.16. [DOI] [PubMed] [Google Scholar]
- Dixit J, Arunyawat U, Huong NT, Das A. Multilocus nuclear DNA markers reveal population structure and demography of Anopheles minimus. Molecular Ecology. 2014;3:5599–5618. doi: 10.1111/mec.12943. [DOI] [PubMed] [Google Scholar]
- Foley DH, Paru R, Dagoro H, Bryan JH. Allozyme analysis reveals six species within the Anopheles punctulatus complex of mosquitoes in Papua New Guinea. Medical and Veterinary Entomology. 1993;7:37–48. doi: 10.1111/j.1365-2915.1993.tb00649.x. [DOI] [PubMed] [Google Scholar]
- Foley DH, Bryan JH, Yeates D, Saul A. Evolution and systematics of Anopheles: insights from a molecular phylogeny of Australasian mosquitoes. Molecular Phylogenetics and Evolution. 1998;9:262–275. doi: 10.1006/mpev.1997.0457. [DOI] [PubMed] [Google Scholar]
- Fontaine MC, Pease JB, Steele A, et al. Mosquito genomics. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science. 2015;347:1258524. doi: 10.1126/science.1258524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Haubold B, Pfaffelhuber P, Lynch M. mlRho - a program for estimating the population mutation and recombination rates from shotgun-sequenced diploid genomes. Molecular Ecology. 2010;19(Suppl 1):277–284. doi: 10.1111/j.1365-294X.2009.04482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry-Halldin CN, Reimer L, Thomsen E, et al. High throughput multiplex assay for species identification of Papua New Guinea malaria vectors: members of the Anopheles punctulatus (Diptera: Culicidae) species group. American Journal of Tropical Medicine and Hygiene. 2011;84:166–173. doi: 10.4269/ajtmh.2011.10-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry-Halldin CN, Nadesakumaran K, Keven JB, et al. Multiplex assay for species identification and monitoring of insecticide resistance in Anopheles punctulatus group populations of Papua New Guinea. American Journal of Tropical Medicine and Hygiene. 2012;86:140–151. doi: 10.4269/ajtmh.2012.11-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetzel MW, Choudhury AA, Pulford J, et al. Progress in mosquito net coverage in Papua New Guinea. Malaria Journal. 2014;13:242. doi: 10.1186/1475-2875-13-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Kazura JW, Siba PM, Betuela I, Mueller I. Research challenges and gaps in malaria knowledge in Papua New Guinea. Acta Tropica. 2012;121:274–280. doi: 10.1016/j.actatropica.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD, Ness RW, Halligan DL, Haddrill PR. Estimation of the spontaneous mutation rate per nucleotide site in a Drosophila melanogaster full-sib family. Genetics. 2014;196:313–320. doi: 10.1534/genetics.113.158758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DR, Schatz MC, Salzberg SL. Quake: qualityaware detection and correction of sequencing errors. Genome Biology. 2010;11:R116. doi: 10.1186/gb-2010-11-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT–the BLAST-like alignment tool. Genome Research. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski J, Besansky NJ. Molecular systematics of Anopheles: from subgenera to subpopulations. Annual Review of Entomology. 2003;48:111–139. doi: 10.1146/annurev.ento.48.091801.112647. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawniczak MK, Emrich SJ, Holloway AK, et al. Widespread divergence between incipient Anopheles gambiae species revealed by whole genome sequences. Science. 2010;330:512–514. doi: 10.1126/science.1195755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Marsden CD, Norris LC, et al. Spatiotemporal dynamics of gene flow and hybrid fitness between the M and S forms of the malaria mosquito, Anopheles gambiae. Proceedings of the National Academy of Sciences USA. 2013;110:19854–19859. doi: 10.1073/pnas.1316851110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley I. Papua New Guinea’s human past: the evidence of archaeology. In: Attenborough R, Alpers M, editors. Human Biology In Pupua New Guinea: The Small Cosmos. Oxford University Press; Oxford: 1992. pp. 150–171. [Google Scholar]
- Logue K, Chan ER, Phipps T, et al. Mitochondrial genome sequences reveal deep divergences among Anopheles punctulatus sibling species in Papua New Guinea. Malaria Journal. 2013;12:64. doi: 10.1186/1475-2875-12-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey DE, Lawniczak MK, Park DJ, et al. SNP genotyping defines complex gene-flow boundaries among African malaria vector mosquitoes. Science. 2010;330:514–517. doi: 10.1126/science.1193036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey DE, Waterhouse RM, Abai MR, et al. Mosquito genomics. Highly evolvable malaria vectors: the genomes of 16 Anopheles mosquitoes. Science. 2015;347:1258522. doi: 10.1126/science.1258522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehle MM, Guelbeogo WM, Gneme A, et al. A cryptic subgroup of Anopheles gambiae is highly susceptible to human malaria parasites. Science. 2011;331:596–598. doi: 10.1126/science.1196759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seah IM, Ambrose L, Cooper RD, Beebe NW. Multilocus population genetic analysis of the Southwest Pacific malaria vector Anopheles punctulatus. International Journal for Parasitology. 2013;43:825–835. doi: 10.1016/j.ijpara.2013.05.004. [DOI] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, et al. ABySS: a parallel assembler for short read sequence data. Genome Research. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa V, Hey J. Understanding the origin of species with genome-scale data: modelling gene flow. Nature Reviews Genetics. 2013;14:404–414. doi: 10.1038/nrg3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Kawashima T, Koyanagi R, et al. Draft genome of the pearl oyster Pinctada fucata: a platform for understanding bivalve biology. DNA Research. 2012;19:117–130. doi: 10.1093/dnares/dss005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weetman D, Steen K, Rippon EJ, et al. Contemporary gene flow between wild An. gambiae s.s and An. arabiensis. Parasites and Vectors. 2014;7:345. doi: 10.1186/1756-3305-7-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. World Malaria Report 2013. World Health Organization; Geneva: 2013. [Google Scholar]
- Zhang G, Fang X, Guo X, et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature. 2012;490:49–54. doi: 10.1038/nature11413. [DOI] [PubMed] [Google Scholar]
- Zheng W, Huang L, Huang J, et al. High genome heterozygosity and endemic genetic recombination in the wheat stripe rust fungus. Nature Communications. 2013;4:2673. doi: 10.1038/ncomms3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Identification of putative introgression candidates based on divergence.
Fig. S2 Distribution of the sequence coverage per locus.
Fig. S3 Distribution of the reference allele frequencies.
Fig. S4 Distribution of the sequence coverage per contig for the initial assemblies (A–D) and after removing redundant contigs (E–H).
Fig. S5 Distribution of the internal branch lengths.
Fig. S6 Distribution of the external branch lengths.
Fig. S7 Distribution of nucleotide differences between species.
Fig. S8 Demographic history of AF4 and AP.
Fig. S9 Influence of sequence coverage on the estimated demographic history of AF4.
Table S1 Sequencing summary for each sample.
Table S2 Summary of the initial de novo genome assemblies.
Table S3 Summary of the Anopheles farauti4 assembly after sub-sampling sequencing reads to the coverage of Anopheles punctulatus s.s.
Table S4 Gene annotation of each introgression candidate as determined by the best nucleotide blast hit to Anopheles gambiae genes.
Table S5 Summary of the genetic diversity in Anopheles farauti4 estimated after sub-sampling Anopheles farauti4 reads to the coverage of Anopheles punctulatus s.s.