

Summary
The shared exon 2 of the p14ARF-p16INK4a locus is frequently mutated in human cancers. However, in contrast to the exon 1β-encoded N-terminal half of ARF, the function of the exon 2-encoded C-terminal half of ARF has been elusive. Here, we report that the mitochondrial protein p32/C1QBP binds the ARF C terminus. We show that p32 is required for ARF to localize to mitochondria and induce apoptosis, and that ARF mutations specifically disrupting p32 binding can impair both of these functions. Wild-type ARF, but not a p32-binding-deficient ARF mutant, localizes to mitochondria, reduces mitochondrial membrane potential, and sensitizes cells to p53-induced apoptosis. These findings provide a potential explanation for the frequent human cancer mutations targeting the ARF C terminus.
Introduction
The mammalian p14ARF-p16INK4a locus (p19Arf in the mouse, ARF and p16 hereafter) is frequently mutated in human cancers at an overall frequency of approximately 40% (Kamb et al., 1994; Nobori et al., 1994; Ruas and Peters, 1998). This extraordinarily high incidence of mutation is believed to result from the gene's unusual organization. The locus contains three exons, designated 1α, 2, and 3, that encode the CDK inhibitor p16. About 20 kb upstream from exon 1α, the locus contains an additional exon, 1β, from which a second transcript is initiated, encoding ARF. The ARF transcript skips exon 1α and joins the common exon 2 in such a fashion that ARF and p16 exist in alternate reading frames with respect to the shared exon 2 (Sherr, 2001).
Via distinct mechanisms, ARF and p16 activate the p53- and pRb-mediated tumor suppression pathways, respectively. Inactivation of both pathways by loss-of-function mutations represents a frequent, and perhaps obligatory, event in the development of most human cancers (Vogelstein et al., 2000). Targeted deletion in mice of the exon 2 shared by ARF and p16 was shown to result in accelerated tumor development (Serrano et al., 1996). Homozygous deletion of either ARF (exon 1β) (Kamijo et al., 1997) or p16 (exon 1α) (Krimpenfort et al., 2001; Sharpless etal., 2001) alone predisposes micetotumor development, indicating that both genes are bona fide tumor suppressors. Enforced ARF expression causes cell-cycle arrest at both the G1 and G2 phases in a p53-dependent fashion (Quelle et al., 1995) by binding directly to and interfering with the p53 negative-regulator MDM2 (Kamijo et al., 1998; Pomerantz et al., 1998; Stott et al., 1998; Zhang et al., 1998). In addition to MDM2, many other ARF-interacting proteins have been identified, including E2Fs, DNA topoisomerase I, Pex19p, spinophilin/neurabin II, a type 1 protein-phosphatase-binding protein, B23/NPM, ARF-BP1, c-Myc, and CtBP (reviewed recently by Sherr, 2006).
Endogenous ARF expression is known to be elevated by oncogenic Myc, Ras, E2F1, E1A, and v-Abl—all of which activate p53-dependent cell-cycle arrest—leading to the proposal that ARF mediates a p53-dependent cell-cycle arrest checkpoint in response to oncogenic signals (Sherr, 1998). In addition, ARF plays an important role in apoptotic cell death. ARF regulates p53-mediated apoptosis that is induced by adenovirally expressed E1A and c-Myc (de Stanchina et al., 1998; Zindy et al., 1998). Furthermore, apoptosis is increased in mouse embryo fibroblast (MEF) cells engineered to overexpress c-Myc, E1A, or E2F1; however, this effect is not seen in ARF null MEF cells, and reintroducing ARF into the ARF null cells sensitizes them to apoptosis induced by these oncogenes, confirming a critical role in this process for ARF. ARF can also induce apoptosis in cells lacking p53, although with slow kinetics, indicating that ARF also has a p53-independent apoptotic function (Eymin et al., 2003; Hemmati et al., 2002).
The shared exon 2 of the human p14ARF-p16INK4a locus suffers frequent point mutations in cancer that are predicted to affect the protein sequences of p16 and ARF either individually or simultaneously, and most of these point mutations are clustered at the 5′ portion of exon 2, which is shared by p16 and ARF. Of 661 cancer-derived alterations compiled for the entire exon 2 (Greenblatt, 2006; Greenblatt et al., 2003; Ruas and Peters, 1998; Sharpless, 2005), 560 (85%), including 504 sporadic and 56 germline mutations, are clustered in the 5′ portion shared by ARF and p16 (see diagram in Figure 4B). Among these, 118 (21%) are frame-shifting small deletions and insertions that truncate the C-terminal sequences of both ARF and p16, 260 (46%) are point mutations that simultaneously affect the sequence of both proteins, and 39 (7%) are point mutations that affect the protein sequence of ARF, but not p16. The presence of these cancer-associated mutations in the ARF C terminus suggests a potential tumor suppressor function for this region, yet a generally held view is that ARF is not functionally affected by these C-terminal mutations, because ectopic expression of the exon 1β-encoded N-terminal domain of ARF is sufficient to fulfill the known biological roles of the full-length ARF protein, such as association with MDM2 and activation of p53 to cause cell-cycle arrest. It has been shown that some exon 2 mutations could affect the function of ARF's nucleolar localization signal, which is located at the C-terminal half of human ARF (Zhang and Xiong, 1999). Nevertheless, the perception remains that only p16, but not ARF, is functionally affected by the exon 2 mutations. Why, then, the 5′ portion of exon 2 shared by p16 and ARF is preferentially subjected to cancer-associated mutations, particularly those that are ARF specific, remains unexplained.
Figure 4. Cancer-Derived ARF C-Terminal Mutations Impair ARF-p32 Binding.
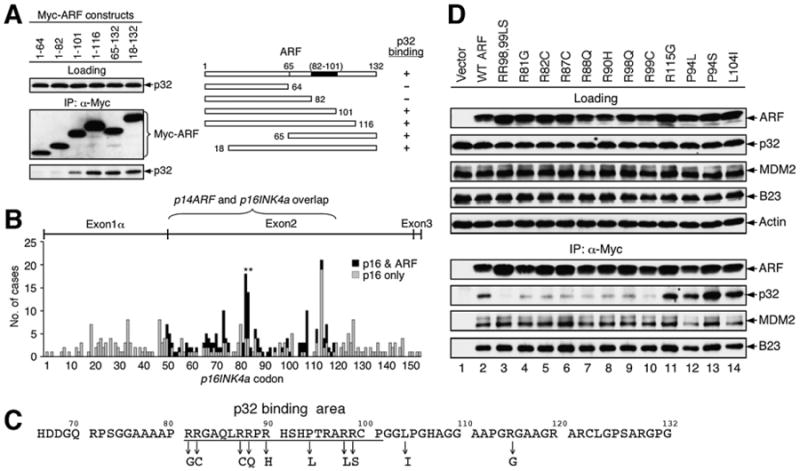
(A) Mapping of the p32 binding domain on human ARF. Extracts from U2OS cells transfected with plasmids encoding various ARF deletion mutants were immunoprecipitated with anti-Myc antibody and blotted with anti-Myc and anti-p32 antibodies. A diagram of human ARF is shown with the indicated p32-binding site (82–101).
(B) Cancer-derived mutations in the p14ARF/p16INK4a gene locus are clustered at the 5′ portion of exon 2. The reported single missense mutations that affect the p16 sequence alone (gray bar) or both p16 and ARF (black bar) are shown in regard to the amino acid sequence of p16 (Greenblatt, 2006). Hotspot mutations at Arg98 and Arg99 of human ARF, overlapping with His83 and Asp84 in p16, are indicated with asterisks.
(C) Amino acid sequence of the exon 2-encoded human ARF C terminus. Individual mutations analyzed in (D) are indicated with downward arrows.
(D) Cancer-derived ARF C-terminal Arg mutations impair p32 binding. U2OS cells were transiently transfected with indicated plasmids expressing various Myc-tagged ARF proteins. Cell extracts were immunoprecipitated with anti-Myc antibody and blotted with the indicated antibodies.
In this study, we have identified a binding partner for the C terminus of ARF: the mitochondria-anchored protein p32 (a.k.a. C1QBP, gC1qR, HABP1). Importantly, the ARF-p32 interaction is disrupted by human cancer-derived point mutations in exon 2 of ARF, including mutations that affect only ARF but not p16. We show that p32 is a critical mediator for both ARF mitochondrial localization and ARF-induced, p53-dependent apoptosis. These findings provide a previously unknown apoptotic function of ARF in the mitochondria and a potential explanation for ARF-specific exon 2 mutations in human cancer.
Results
ARF Interacts with Mitochondrial Protein p32
To better understand the biological functions of ARF, we sought to identify ARF-associated peptides by carrying out a large-scale coimmunoprecipitation (co-IP) following adenovirus-mediated overexpression of human ARF in cells, utilizing an antibody specific for the human ARF protein. A number of polypeptides were copurified with ARF protein from the Ad-ARF infected cells, but not from Ad-GFP infected control cells (Figure 1A). One of the peptides identified by mass spectrometric analysis was the mitochondrial protein p32. p32 (a.k.a. C1QBP for globular head component factor C1q binding protein, gC1qR for receptor for C1q, and HABP1 for hyaluronan-binding protein 1) was originally copurified with the pre-mRNA splicing factor SF2/ASF in HeLa cells (Krainer et al., 1991) and was subsequently shown to be expressed as a pro-protein of 282 residues whose first 73 residues contain a mitochondrial targeting signal and are cleaved during maturation (Ghebrehiwet et al., 1994). p32 has been shown to be a receptor for globular head component factor C1q (Ghebrehiwet et al., 1994) and to play a role in RNA splicing (Petersen-Mahrt et al., 1999) and oxidative phosphorylation (Muta et al., 1997). It is localized predominantly in the mitochondrial matrix (Dedio et al., 1998; Muta et al., 1997) and was recently implicated in mediating cellular apoptotic response (Kamal and Datta, 2006; Sunayama et al., 2004). We confirmed a direct physical interaction between ARF and p32 by a coupled in vitro transcription/translation (IVT) assay using 35S-labeled p32 and ARF proteins (Figure 1B). A direct and specific interaction between ARF and p32 was further confirmed by a GST pulldown assay (data not shown). The in vivo ARF-p32 interaction was demonstrated by co-IP of endogenous ARF with ectopically expressed Myc-p32 in H1299 cells (p53 negative, ARF positive) (Figure 1C), and reciprocally of endogenous p32 with ectopically expressed ARF in U2OS cells (ARF negative) (Figure 1D). Furthermore, we were able to show the ARF-p32 association in Raji cells, which express endogenous ARF and p32, under unstressed physiological conditions (Figure 1E). Finally, we found that the physiological ARF-p32 interaction was enhanced by treating cells with a low dose(5 nM) of actinomycin D, which is known to delocalize ARF from the nucleolus to the nucleoplasm (Lindstrom et al., 2000), suggesting that the interaction takes place preferentially outside of the nucleolus (Figure 1F). Together, these data indicate that p32 interacts with ARF both in vitro and in vivo under physiological conditions.
Figure 1. ARF Interacts with p32.
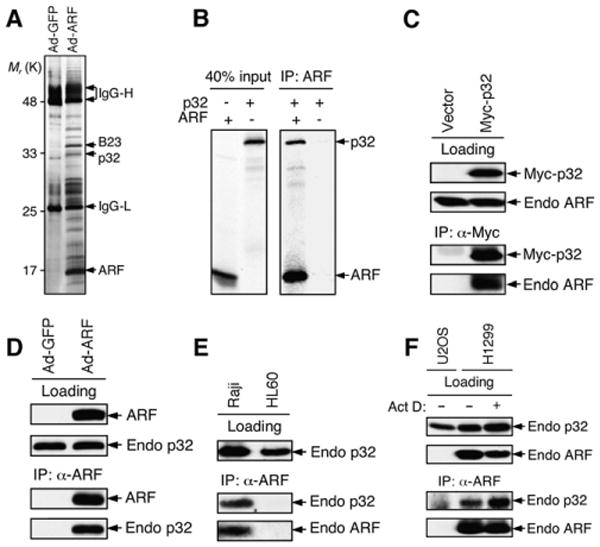
(A) Identification of ARF binding proteins by mass spectrometry. Extracts from U2OS cells infected with adenovirus expressing GFP or ARF were immunoprecipitated with an anti-ARF antibody, resolved by SDS-PAGE, and visualized by silver staining. Peptide bands unique to samples infected with Ad-ARF were subjected to mass spectrometry. ARF, p32, B23 (a known ARF-binding protein), and IgG heavy and light chains (IgG-H and IgG-L) are indicated.
(B) Direct interaction between p32 and ARF. Full-length p32 and ARF were in vitro translated in the presence of 35S-methionine. Equal amounts of p32 protein were incubated with (+) or without (−) ARF followed by immunoprecipitation with an anti-ARF antibody. The resulting ARF immunoprecipitates were separated by SDS-PAGE and visualized by autoradiography.
(C) Coimmunoprecipitation (co-IP) of endogenous ARF with ectopically expressed p32. H1299 cells (p53-negative, ARF-positive) were transfected with Myc-p32 plasmid for 2 days, and cell extracts were immunoprecipitated with anti-Myc antibody. Western blotting was performed with anti-Myc and anti-ARF antibodies. Loading control represents 10% of cell lysate used for co-IP.
(D) Co-IP of endogenous p32 with ectopically expressed ARF. Extracts from U2OS cells infected with Ad-ARF were immunoprecipitated with anti-ARF antibody. Western blotting was performed with anti-ARF and anti-p32 antibodies. Loading control represents approximately 10% of cell lysate used for co-IP.
(E) Co-IP of ARF with p32from Raji cells. Cell extracts from Raji (Burkitt's lymphoma, expressing endogenous ARF and p32) cells were immunoprecipitated with anti-ARF antibody and blotted with anti-p32 and anti-ARF antibodies. HL60 (leukemia, ARF-negative) cells served as a negative control.
(F) Increased ARF-p32 interaction following actinomycin D treatment. H1299 cells were treated with (+) or without (−) 5 nM actinomycin D for 36 hr. Cell extracts were immunoprecipitated with anti-ARF antibody and blotted with anti-p32 and anti-ARF antibodies. Extracts from U2OS (ARF-negative) cells served as a negative control. Loading control represents approximately 10% of cell lysate used for co-IP.
p32 Is Critical for a Subset of ARF to Localize to Mitochondria
The findings that p32 is predominantly a mitochondrial protein while ARF is usually in the nucleolus raise a question about the sub-cellular compartment in which the two proteins might interact or colocalize. To address this question, we fractionated cell lysates into cytoplasmic (Cyto) and mitochondrial (Mito) fractions and examined the existence and the relative abundance of ARF and p32 in each of the fractions. We chose two human cancer cell lines (H1299 and HCC1937)and a MEF cell strain(p53 null) for the study, based on the feature that they all express a relatively high level of ARF protein due to their p53-negative status. We tested several fractionation protocols and found that a combination of a hypo-tonic buffer, digitonin, and Triton X-100 (see Experimental Procedures) yields the cleanest cytoplasmic and mitochondrial fractions, which do not contain detectable contaminations from each other or from nuclear proteins (Figure 2A). With this fractionation scheme, we detected a subset of total ARF protein in the mitochondrial fractions from each of the cells (Figure 2A). On the other hand, p32 was found exclusively in the mitochondria, as previously reported (Dedio et al., 1998; Muta et al., 1997). These evidences show that ARF and p32 colocalize in the mitochondria and suggest that the ARF-p32 interaction might be important for ARF's mitochondrial localization.
Figure 2. p32 Is Critical for a Subset of ARF to Localize to Mitochondria.
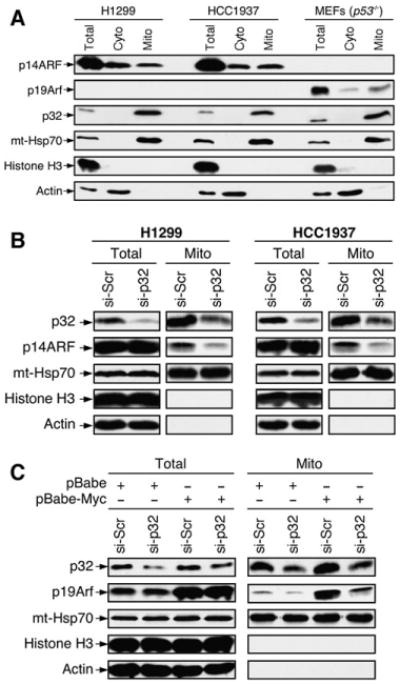
(A) Mitochondrial localization of endogenous ARF. p53-negative H1299 and HCC1937 cells, and p53−/− MEFs were either prepared as SDS lysates (Total) or separated into cytoplasmic (Cyto) and mitochondrial (Mito) fractions. Equal amounts of protein were loaded in each lane. The membrane was blotted with antibodies to p14ARF, p19Arf, p32, mt-Hsp70 (mitochondrial heat shock protein 70, a mitochondrial protein marker), Histone H3 (a nuclear protein marker), and Actin (a cytoplasmic protein marker).
(B) Downregulation of p32 by siRNA inhibits ARF mitochondrial localization. H1299 and HCC1937 cells were transiently transfected with a scrambled siRNA (si-Scr) or a p32 siRNA (si-p32) for 3 days. The cells were then isolated for mitochondrial fractions and analyzed by western blotting with indicated antibodies.
(C) Downregulation of p32 by siRNA inhibits localization of oncogenic Myc-induced mouse p19Arf to mitochondria. Freshly prepared wild-type MEFs were infected with retrovirus carrying either no insert (pBabe) or oncogenic c-Myc (pBabe-Myc) and selected with puromycin for 3 days. The cells were then transiently transfected with scrambled siRNA (si-Scr) or p32 siRNA (si-p32) for 3 days. Total cell lysates and mitochondrial fractions were prepared and analyzed by western blotting with indicated antibodies.
To study the role of p32 in ARF's localization to the mitochondria, we knocked down p32 in H1299 and HCC1937 cells by siRNA, fractionated the cell lysates, and examined the level of mitochondrial ARF by western blotting. In both H1299 and HCC1937 cells, transfection of the p32 siRNA led to approximately 70%–80% reduction of endogenous p32. This amount of p32 reduction did not affect the total level of ARF expression, nor did it apparently affect the growth properties of the cells (data not shown). Importantly however, we observed a significant reduction of the level of ARF in the mitochondria, and the reduction was roughly in proportion with the level by which p32 was reduced (Figure 2B). Similar results were also seen in U2OS cells (p53 positive, ARF negative) with ectopically expressed ARF (Figure S1A available online) and in primary MEFs with onco-gene-induced ARF (Figure 2C). Furthermore, coexpression of ARF and p32 significantly increased the level of ARF protein in the mitochondrial fraction without changing the total ARF level (Figure S1B), indicating that a high level of p32 facilitates ARF's mitochondrial localization. Thus, these findings indicate that p32 is critical for a subset of ARF protein to localize to the mitochondria, and this can occur both in unstressed ARF-expressing tumor cells and in oncogene-challenged normal fibroblast cells.
p32 Is Critical for ARF-Induced Apoptosis
To elucidate the functional significance of mitochondrial ARF and its interaction with p32, we wanted to determine whether p32 plays any role in ARF-induced cell-cycle arrest and/or apoptosis. We carried out our study first in U2OS cells because the cells undergo both growth arrest and apoptosis upon ectopic ARF overexpression (Hemmati et al., 2002; Kim et al., 2004; Yarbrough et al., 2002). We first reduced p32 expression by transfection of p32 siRNA before infecting the cells with adenovirus expressing ARF. We then examined cell death by microscopic observation. Adenovirus-mediated ARF overexpression yielded extensive cell death in U2OS cells; after 2 days of infection, only about 25% of cells remained attached to the control plate transfected with scrambled siRNA (Figure 3A, open bar, and Figure S2, panel 2). In contrast, in samples transfected with p32 siRNA, more than 70% of cells survived ARF overexpression and remained attached to the plate (Figure 3A, filled bar, and Figure S2, panel 4). We next examined the cells by flow cytometry analysis. Knocking down p32 resulted in a significant reduction in ARF-induced apoptosis (measured by percentage of cells in sub-G1 phase) but had virtually no effect on ARF-induced S phase reduction (Figure 3B), indicating that p32 is important only in mediating ARF-induced apoptosis, but not growth arrest. Consistent with this notion, western blotting analysis indicated that a reduction of p32 attenuated ARF-mediated PARP (poly ADP ribose polymerase) cleavage—a characteristic of cells undergoing irreversible apoptosis—but did not affect ARF's induction of p53 (Figure 3C).
Figure 3. p32 Is Critical for ARF-Induced Apoptosis.
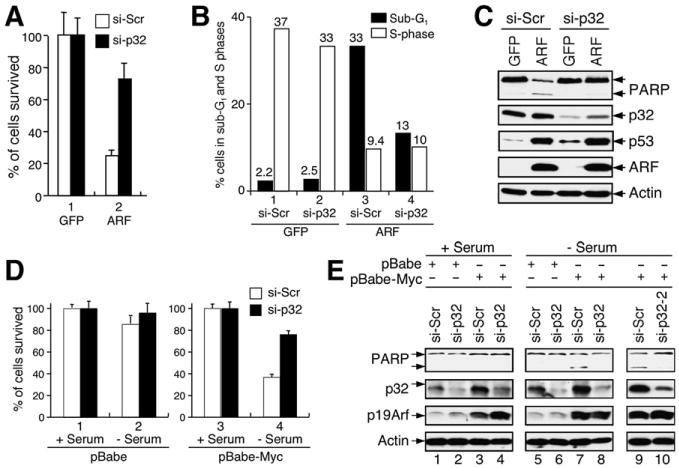
(A) p32 depletion causes resistance of cells to ARF-induced apoptosis. U2OS cells were transfected for 24 hr with indicated siRNA and then infected for 2 days with adenovirus expressing either GFP or ARF. Cells that survived ARF overexpression were counted, and the percentages of surviving cells relative to GFP-expressing control cells were plotted. Error bars represent the mean ± SD of four independent experiments.
(B) p32 depletion attenuates ARF-induced apoptosis. U2OS cells were treated as described in (A) and collected for flow cytometry analysis of cell-cycle profiles. The percentages of cells in S and sub-G1 phases are shown.
(C) p32 depletion causes resistance of cells to ARF-induced PARP cleavage. U2OS cells were transfected with siRNA and infected with adenoviruses as described above. Cell extracts were analyzed by western blotting with antibodies to indicated proteins.
(D) p32 depletion causes resistance of wild-type MEFs to c-Myc-induced apoptosis. Freshly prepared MEFs were infected with retrovirus carrying either no insert (pBabe) or oncogenic c-Myc (pBabe-Myc) and were selected with puromycin for 3 days. The cells were then transfected with siRNA for 2 days and subsequently cultured with (+) or without (−) serum for 24 hr. The percentages of cells surviving the treatment were quantified, and the results were plotted. Error bars represent the mean ± SD of four independent experiments.
(E) A portion of the MEFs described in (D) was analyzed for p32 knockdown, p19Arf expression, and PARP cleavage by western blotting. Note that a second p32 siRNA oligos (si-p32-2) gives rise to identical results as the first p32 siRNA (si-p32).
To further analyze the apoptotic role of p32 under a more physiological condition, we tested whether inhibition of p32 expression could affect Myc-induced ARF-dependent apoptosis in primary MEFs. For this purpose, early passage (P1–P3) wild-type MEFs were engineered to stably express either pBabe vector (control) or pBabe-Myc. Apoptosis was subsequently triggered by culturing near-confluent cells in serum-free medium for 24 hr, and the cells surviving the treatment (those that remained attached to the plate) were counted. As shown in Figure 3D, in samples transfected with scrambled siRNA (si-Scr), a combination of Myc overexpression and withdrawing serum led to cell death in a majority of the cell population, with only approximately 30% of cells remaining attached to the plate (Figure 3D, column 4, open bar). In contrast, in samples transfected with p32 siRNA (si-p32), more than 70% of cells survived the treatment (Figure 3D, column 4, filled bar), indicating that Myc-induced cell death in the primary MEFs depends on p32. To further demonstrate that the reduction in cell death was due to inhibition of apoptosis, we analyzed PARP cleavage in the MEFs by western blotting. pBabe-mediated Myc overexpression resulted in activation of ARF in the cells (Figure 3E, ARF panel). A combination of Myc overexpression and withdrawing serum resulted in PARP cleavage (Figure 3E, lanes 7), and this was relegated by depletion of p32 (Figure 3E, lanes 8). To rule out a potential off-target effect of the p32 siRNA, we used a second pair of p32 siRNA oligomers (si-p32-2), which produced identical results (Figure 3E, lanes 9 and 10). Together, these results indicate that p32 is a critical mediator for ARF-induced apoptosis but is perhaps irrelevant to ARF-induced cell-cycle arrest.
Human Cancer-Derived ARF C-Terminal Mutations Impair p32 Binding
To better understand the nature of ARF-p32 binding, we identified the protein sequence in ARF that mediates its interaction with p32. We constructed a series of Myc-tagged ARF deletion mutants, transiently expressed them in U2OS cells, and determined the interaction of each mutant protein with endogenous p32 by an IP-western assay. Notably, unlike other ARF-interacting proteins such as MDM2 and B23 that interact with the exon 1β-encoded N terminus of ARF, p32 interacted with ARF via a sequence between residues 82 and 101 (aa numbers corresponding to human ARF) in the exon 2-encoded C-terminal half of the ARF protein (Figure 4A). Intriguingly, human cancer-derived small mutations are frequently observed in exon 2 of the ARF-INK4a gene locus, and these mutations are clustered at the 5′ portion of exon 2 that is shared by ARF and p16 (Figure 4B). Previous studies have shown that p32 interacts with several viral and cellular proteins at sequence domains rich in arginine (Arg) (Figure S3) (Beatch et al., 2005; Hall et al., 2002; Luo et al., 1994; Nikolakaki et al., 1996; Wang et al., 1997). The ARF protein is rich in Arg (24 Arg among 132 residues in human ARF; 18%), especially within the area mapped for p32 binding, where 8 out of the 20 residues (40%) are Arg (Figure 4C). Cancer mutations affecting each of the eight Arg residues within the p32 binding area have been reported, and many of these mutations are ARF specific (i.e., not affecting p16 due to alternative reading frames). To determine whether any of these Arg mutations might affect the ARF-p32 interaction, we constructed ARF mutants according to reported Arg mutations and tested them for p32 binding in transiently transfected cells. Remarkably, each of the Arg mutations within the p32-binding domain significantly affected the ARF-p32 interaction (Figure 4D, lanes 3–10), whereas non-Arg mutations (P94L, P94S, L104I) and an Arg mutation (R115G) outside of the p32-binding domain did not(Figure 4D,lanes 11–14). None of the Arg mutations disrupted the interaction of ARF with MDM2 or B23, suggesting that the impairment of ARF-p32 binding by the Arg mutations is p32 specific (Figure 4D, MDM2 and B23 panels). Hence, these data show that the ARF-p32 interaction specifically can be impaired by cancer-derived mutations targeting the ARF C terminus.
p32-Binding-Deficient ARF Mutants Are Attenuated in Apoptotic Function
To study how the mutations disrupting p32 binding could affect ARF function, we constructed two adenoviruses expressing ARF mutants (RR98,99LS and R98Q, designated RR and R), according to hotspot mutations found in human patients (Figure 4B, shown with asterisks), and compared growth arrest and apoptosis in U2OS cells expressing the mutant ARF. Consistent with findings from our earlier experiments (Figure 3) and others (Hemmati et al., 2002; Kim et al., 2004; Yarbrough et al., 2002), over-expression of wild-type ARF caused both cell-cycle arrest and apoptosis in U2OS cells: the cell population in S phase (Figure 5A, open bar) was reduced from approximately 28% to 11%, while the population in sub-G1 increased from 1% to about 24% (Figure 5A, filled bar). Notably, however, although overexpression of each ARF mutant (RR and R) resulted in an equivalent level of cell-cycle arrest as that caused by wild-type ARF expression, the mutant ARF caused only a moderate increase in apoptosis (Figure 5A, filled bar), despite expression of the mutant proteins at equivalent or higher levels compared to wild-type ARF (see inset in Figure 5A). These data indicate that the ARF C-terminal mutants are impaired selectively in their apoptotic function. To further investigate ARF's apoptotic function, we examined PARP cleavage in cells overexpressing the ARF mutants. As determined by immunostaining of ARF, increasing amounts of moi (multiplicity of infection) result in higher levels of ARF expression in each of the infected cells (data not shown). As shown in Figure 5B, even though PARP cleavage could be seen in cells infected with adenoviruses expressing each of the ARF proteins, including the mutants, the extent of PARP cleavage was evidently lesser in cells expressing the ARF mutants than in cells expressing wild-type ARF (Figure 5B). In contrast, the induction of p53 by ARF was unaffected by these Arg mutations (Figure 5B, p53 panel). These results, together with the cell-cycle studies shown in Figure 5A, suggest that ARF's apoptotic function is selectively affected by cancer-associated C-terminal Arg mutations that disrupt its binding with p32.
Figure 5. p32-Binding-Deficient ARF Mutants Are Attenuated in Apoptotic Function.
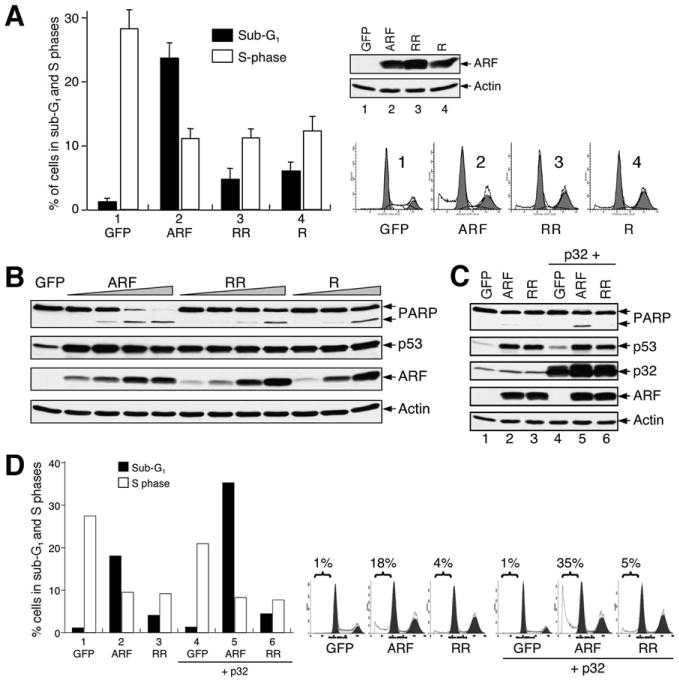
(A) ARF C-terminal Arg mutations impair ARF's apoptotic function. U2OS cells were infected with adenoviruses expressing either wild-type ARF (ARF) or p32-binding-deficient ARF RR98,99LS (RR) and ARF R98Q (R) mutants for 2 days. Cells (both floating and attached) were collected for flow cytometry analysis of cell-cycle distribution. The fraction of cells in sub-G1 phase (filled bar) and S phase (open bar) and the level of ARF protein expression (inset) are shown. Cell-cycle profiles are also shown to the right. Error bars represent the mean ± SD of three independent experiments. p < 0.01 for pairwise comparisons of cells in sub-G1 infected by wild-type ARF and each of the mutants.
(B) ARF C-terminal Arg mutations impair ARF-mediated PARP cleavage, but not p53 induction. U2OS cells treated as described in (A) were analyzed for PARP cleavage and p53 induction by western blotting.
(C) p32 enhances ARF-induced PARP cleavage. Cells were infected with adenoviruses expressing the indicated proteins for 2 days and then harvested to be analyzed for PARP cleavage. Note that p32-enhanced PARP cleavage was seen only with wild-type ARF, but not the p32-binding-deficient mutant ARF.
(D) p32 enhances ARF-induced apoptosis but not ARF-induced cell-cycle arrest. U2OS cells were infected with adenoviruses expressing the indicated proteins for 2 days and harvested to determine the percentages of cells in sub-G1 and S phase fractions by flow cytometry analysis.
To investigate whether p32 could augment ARF's apoptotic activity, we ectopically expressed p32 in U2OS cells along with ARF or the ARF mutants and examined PARP cleavage by western blotting. ARF overexpression was adjusted to a low level so that it would not by itself cause apoptosis in the cells. Coexpression of p32 with wild-type ARF increased PARP cleavage (Figure 5C, lane 5); in contrast, the p32-binding-deficient ARF mutant was unresponsive to p32 overexpression, and PARP cleavage was not observed (Figure 5C, lane 6). Of note, overexpression of p32 did not stabilize endogenous p53, nor did it by itself induce PARP cleavage (Figure 5C, lane 4). The ability of p32 to augment ARF's apoptotic function was further examined by flow cytometry analysis. Consistently, p32 enhanced ARF-induced apoptosis specifically, but not cell-cycle arrest, and this synergistic effect was not observed when p32 was coexpressed with the p32-binding-deficient ARF mutant (Figure 5D). Together, these data indicate that p32 augments ARF-induced apoptosis specifically, and this function of p32 requires ARF-p32 physical association.
Wild-Type ARF, but Not a p32-Binding-Deficient ARF Mutant, Reduces Mitochondrial Membrane Potential and Synergizes with p53 to Induce Apoptosis
To explore possible mechanisms by which the p32-binding-deficient ARF mutant is impaired in its apoptotic function, we examined whether the ARF-p32 physical interaction is required in order for ARF to localize to the mitochondria. We expressed both the wild-type ARF and the p32-binding-deficient ARF (RR) mutant in cells and examined the level of each ARF protein present in the mitochondrial fraction by western blotting. Using the same fractionation scheme and H1299 cells as shown in Figure 2, we were able to obtain mitochondrial fractions containing no detectable nuclear or cytoplasmic proteins. We found that ectopically expressed wild-type ARF could be readily detected in the mitochondria, while the RR mutant, although expressed at the same level, was much less efficient in localizing to the mitochondria (Figure 6A). A similar observation was also made in U2OS cells (data not shown). This finding is consistent with the idea that the ARF-p32 interaction is required to mobilize ARF to the mitochondria.
Figure 6. Wild-type ARF, but Not a p32-Binding-Deficient ARF Mutant, Localizes to Mitochondria and Reduces Mitochondrial Membrane Potential.
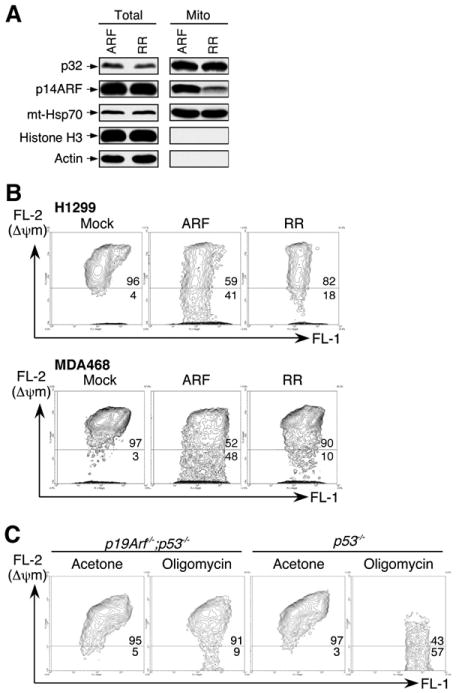
(A) A p32-binding-deficient ARF mutant is impaired in mitochondrial localization. p53-negative H1299 cells were infected with adenovirus expressing either wild-type ARF (ARF) or a p32-binding-deficient ARF mutant (RR). Total SDS lysates and mitochondrial fractions were prepared 2 days after infection and analyzed by western blotting with antibodies to p32, ARF, mt-Hsp70, HistoneH3, and Actin.
(B) Wild-type ARF, but not a p32-binding-deficient ARF (RR) mutant, reduces mitochondrial membrane potential (Δψm). p53-negative H1299 and MDA468 cells were either uninfected (Mock) or infected with adenoviruses expressing wild-type ARF (ARF) or a p32-binding-deficient ARF mutant (RR) for 2 days. Δψm was measured by incubating the cells with JC-1 dye followed by flow cytometry analysis. In nonapoptotic cells (high Δψm), JC-1 exists as a monomer in the cytosol (green, FL-1) and also accumulates as aggregates in the mitochondria, which stains red (FL-2). In apoptotic cells (low Δψm), JC-1 exists in monomeric form and stains the cytosol green. Cells with preserved Δψm are high in both FL-1 and FL-2 (top right of graph). Cells with loss of Δψm are high in FL-1 and low in FL-2 (bottom of graph). Flow cytometry data plots are shown with percentage of total events indicated. Data are representative of three independent experiments.
(C) Endogenous ARF sensitizes cells to mitochondrial stress, leading to reduction of Δψm. Freshly prepared p19Arf−/−;p53−/− and p53−/− MEFs were incubated with acetone (as a control) or 2 μg/ml of oligomycin, an inhibitor of mitochondrial ATP synthase widely used to generate mitochondrial stress in cultured cells. After 48 hr of oligomycin treatment, the cells were collected and analyzed for Δψm by JC-1 dye.
It has been shown that a decrease in the mitochondrial inner membrane potential (Δψm) is associated with early onset of apoptosis (Green and Kroemer, 2004). To study the functional significance of ARF's mitochondrial localization, we examined Δψm in cells in the presence of both wild-type and RR mutant ARF. We chose p53-negative H1299 and MDA468 cells for the experiment in order to avoid ARF-induced, p53-dependent rapid apoptosis and, more importantly, to allow us to examine ARF's function in mitochondria without the interference of p53 activity. The Δψm was analyzed by incubating the cells with JC-1 dye (Cell Technology, Inc., Mountain View, CA) followed by flow cytometry analysis. We adjusted ARF expression to a moderate level by reducing the moi so that it would not cause apoptosis in the cells. Under such conditions, we observed an evident decrease in Δψm in cells expressing wild-type ARF, but much less so in cells expressing the RR mutant ARF (Figure 6B). The same results were also obtained in p53-negative Saos2 and HeLa cells (data not shown). The decrease in Δψm as a consequence of ARF expression was also observed in U2OS (p53-positive) cells (Figure S4), suggesting that ARF's ability to decrease Δψm is a general phenomenon and possibly p53 independent. To assess the physiological role of ARF in mitochondria, we examined Δψm in primary MEFs under mitochondrial stress induced by oligomycin–an inhibitor of mitochondrial ATP synthase (Chappell and Greville, 1961) that is widely used to impose mitochondrial stress in cultured cells. We treated p53 null (p53−/−) and ARF/p53 double null (p19Arf−/−;p53−/−) MEFs with 2 μg/ml of oligomycin and examined their Δψm. In the presence of oligomycin treatment, a reduction of Δψm was observed in both MEFs, without signs of apoptosis. However, the reduction was much more pronounced in p53−/− MEFs than in p19Arf−/−;p53−/− MEFs (Figure 6C). Together, these observations suggest that the presence of ARF in the mitochondria sensitizes cells to mitochondrial stress and leads to loss of Δψm, but not necessarily apoptosis, which might also require the presence of p53.
Previous studies have shown that ARF-induced apoptosis is mostly dependent on the function of p53. In the absence of p53, ARF induces apoptosis only at a nonphysiological high level of expression, and with slow kinetics. Consistent with this notion, we have observed that, in both ARF-overexpressing H1299 cells and oligomycin-treated p53−/− MEFs, even though the loss of Δψm was evident, apoptosis did not occur. Because release of cytochrome c from the mitochondrial intermembrane space into the cytoplasm is considered a key initiative step in the apoptotic process (Jiang and Wang, 2004), we therefore examined whether or not this ARF-induced loss of Δψm leads to cytochrome c release (Figure 7A). We infected p53-negative H1299 and p53-positive U2OS cells with increasing amounts of ARF adenovirus. Two days after infection, cell lysates were collected, soluble cytoplasmic (mitochondria-free) fractions were purified, and the presence of cytochrome c in the cytoplasmic fractions was analyzed by western blotting. Interestingly, even though ARF was expressed to even higher levels (by increasing moi) than those achieved in the Δψm assay, cytochrome c release was not observed in ARF virus-infected H1299 (p53-negative) cells. In contrast, the same level of ARF overexpression in U2OS (p53-positive) cells triggered cytochrome c release, consistent with the observed apoptosis in ARF-overexpressing U2OS cells (Figures 3A–3C). These findings, together with the results from the Δψm assay, suggest that ARF, although ineffective in causing apoptosis by itself, sensitizes cells to p53-induced apoptosis.
Figure 7. ARF Synergizes with p53 to Induce Cytochrome c Release.
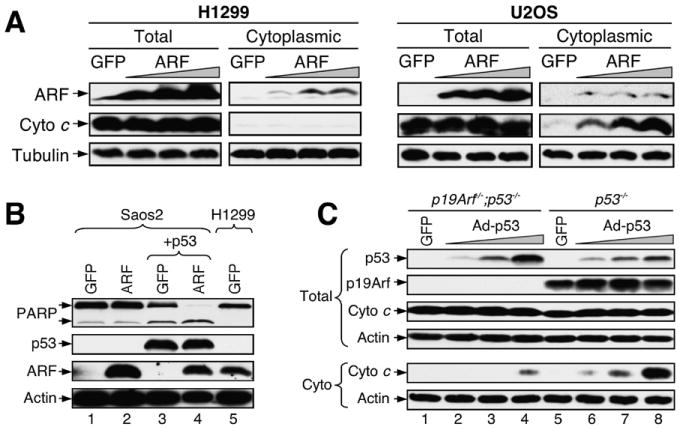
(A) ARF expression did not induce cytochrome c release in H1299 cells. p53-negative H1299 and p53-positive U2OS cells were infected with increasing amounts of adenovirus expressing ARF for 2 days. Total SDS lysates and cytoplasmic fractions were prepared and analyzed by western blotting with indicated antibodies.
(B) ARF synergizes with p53 to induce PARP cleavage. Saos2 (p53-negative) cells were infected with adenovirus expressing GFP, ARF, or p53 as indicated. Cells were analyzed by western blotting with antibodies against the indicated proteins. Note that the level of ectopically expressed ARF in the Saos2 cells is comparable to that of endogenous ARF in the H1299 cells.
(C) A low level of p53 induces cytochrome c release in ARF-positive cells. p19Arf−/−;p53−/− and p53−/− MEFs were infected with adenovirus expressing p53 at a low moi. Two days after infection, total cell lysates and cytoplasmic fractions were prepared and analyzed by western blotting.
To test this hypothesis directly, we ectopically expressed ARF and p53 either individually or together in p53-negative Saos2 cells and examined PARP cleavage. We adjusted p53 expression to a low level so that it alone would cause only marginal PARP cleavage in Saos2 cells, and we adjusted ARF expression to a level equivalent to that of endogenous ARF in H1299 cells (see Figure 7B, ARF panel in lanes 4–5). When expressed together, however, the same low levels of ARF and p53 led to extensive PARP cleavage (Figure 7B), indicating that ARF sensitizes cells to p53-induced apoptosis. To study the role of ARF in p53-induced apoptosis in a more physiological setting, we transiently expressed increasing amounts of p53 in p53−/− and p19Arf−/−;p53−/− MEFs and examined cytochrome c release by western blotting. As shown in Figure 7C, in the p19Arf−/−; p53−/− MEFs, a small amount of cytochrome c release was observed only with a high level of p53 expression (lane 4). However, in the p53−/− MEFs, which contain endogenous ARF, cytochrome c release was observed with the lowest level of p53 expression; this effect was even more pronounced with a higher level of p53 (lanes 6–8). These results indicate that an endogenous level of ARF is able to sensitize cells to p53-induced apoptosis.
Discussion
In the present study, we report identification of the mitochondrial protein p32 as an ARF C-terminal domain-associated protein whose association with ARF can be disrupted by human cancer-derived point mutations in exon 2 of the p14ARF-p16INK4a gene locus. We found that both p32 and ARF-p32 binding are required for a subset of ARF protein to mobilize to the mitochondria (Figures 2 and 6). siRNA-mediated downregulation of p32 by up to 80% did not affect cell growth and proliferation noticeably but significantly inhibited apoptosis caused by either ectopic overexpression of ARF in p53-positive tumor cells or induction of endogenous ARF in wild-type MEFs (Figure 3), indicating that p32 is an essential mediator of oncogenic insult-induced, ARF-mediated apoptosis. The identification of p32 as an essential mitochondrial binding partner for ARF and a key modulator of ARF-induced, p53-dependent apoptosis extends the complex relationship between ARF and p53.
We adapted a mitochondrial fractionation protocol from Ramsby and Makowski (1999) and were able to isolate clean mitochondrial fractions (Figure 2A). With this method, we have investigated the physiological function of ARF in the mitochondria. We unexpectedly found that, at least in various p53-negative cancer cells and p53−/− MEFs, a subset of full-length ARF protein is in the mitochondria. Our study indicates that the localization of ARF in the mitochondria reduces Δψm. This function of ARF is likely independent of p53, because it was similarly observed in both p53-positive and p53-negative cells. However, ARF-mediated reduction of Δψm did not lead to apoptosis in p53-negative cells, but did so only in the presence of p53 (Figure 7), indicating that a functional collaboration between ARF and p53 is required for ARF-induced apoptosis to occur. Our data are consistent with a model in which physiological ARF and p53 must collaborate to induce efficient mitochondria-mediated apoptosis (Figure 8). In this model, ARF, following induction by oncogenic insults, inhibits the E3 function of MDM2 via the exon 1β-encoded N terminus, thereby stabilizing and activating p53 in the nucleus. Meanwhile, a subset of ARF interacts with p32 via the exon 2-encoded C terminus and mobilizes to the mitochondria, where it causes a decrease in Δψm. The activation of p53 leads to further permeabilization of the ARF-weakened mitochondrial membrane, resulting in release of mitochondrial contents—such as cytochrome c—and apoptotic cell death. Without p32 and/or mitochondria-localized ARF, p53-induced cell-cycle arrest may still take place normally, but apoptosis will be attenuated. This model is consistent with the findings that downregulation of p32 by siRNA blocks ARF-induced apoptosis specifically, but not cell-cycle arrest (Figure 3), and that the p32-binding-deficient ARF mutants are impaired only in their apoptotic, but not cell-cycle arrest, functions (Figure 5); in both cases, ARF's induction of p53 is unaffected. It has been reported recently that a short isoform of ARF (smARF) translated from an internal methionine localizes to the mitochondria through its interaction with p32 (Reef et al., 2007), and when overexpressed, smARF causes autophagic cell death (Reef et al., 2006). However, we were unable to detect smARF in the mitochondrial fractions using our fractionation protocol, although we did find a small amount (∼1%–5% of total ARF) of smARF in the total cell lysate in various human tumor cell lines (data not shown). The reason for this discrepancy remains to be determined.
Figure 8. A Model for the Function of Mitochondrial ARF.
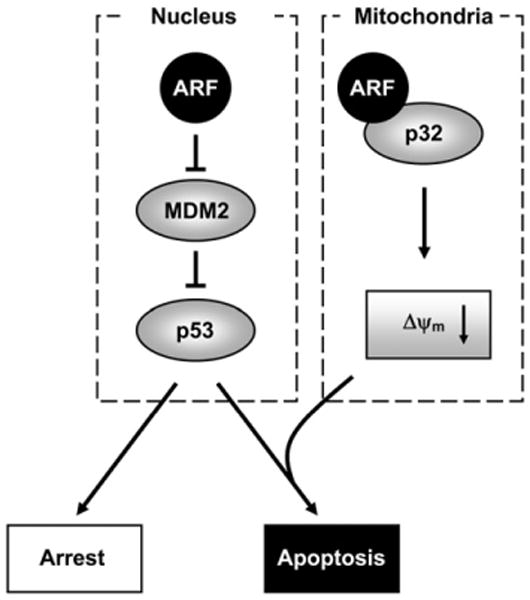
Nuclear ARF stabilizes and activates p53 through its N-terminal interaction with and inhibition of MDM2 (left). A subset of ARF also localizes to the mitochondria through its C-terminal interaction with mitochondrial p32 (right). The mitochondria-localized ARF reduces Δψm and sensitizes cells to p53-induced apoptosis.
Studies of cultured cell lines have shown that cancer-associated mutations in ARF and p53 are general mutually exclusive (Stott et al., 1998). One plausible explanation for this observation, according to the results presented in current study, could be that ARF and p53 may depend on each other to induce apoptosis. For example, a moderate level of p53 activation in the absence of ARF (or mitochondrial ARF in particular) may be sufficient to cause growth arrest, but not apoptosis. The same level of p53, however, could be sufficient to cause apoptosis if ARF is present in the mitochondria and therefore able to reduce Δψm. Thus, the level of p53 activation in response to different environmental cues, together with the status of ARF, could eventually dictate the fate of the cells committing to either growth arrest or apoptosis.
It is interesting to speculate that p32 might be mutated in the course of cancer development, given its role in mediating ARF- and p53-induced apoptosis. However, we have not found reports indicating whether p32 is mutated in human cancer. Instead, it has been shown that p32 is overexpressed in certain cancer cells (Rubinstein et al., 2004). This finding is not surprising because, in addition to its apoptotic role, as we found in this study, p32 also plays important housekeeping roles in RNA splicing (Petersen-Mahrt et al., 1999) and oxidative phosphorylation (Muta et al., 1997). Cancer cells, which are more metabolically active, may require more p32 to sustain fast growth and proliferation. Furthermore, p32 is a downstream mediator of apoptosis for tumor suppressor ARF, which itself is frequently mutated in cancer. Mutations in the upstream tumor suppressor ARF could release the pressure for mutations in the downstream p32.
Based mostly on in vitro overexpression systems, several published studies imply that the ARF N-terminal region encoded by exon 1β is sufficient for all known functions of the ARF protein, leading to the notion that the exon 2-encoded ARF C terminus does not possess any function (Kim et al., 2003; Lohrum et al., 2000; Midgley et al., 2000; Quelle et al., 1997; Stott et al., 1998; Weber et al., 2000; Zhang et al., 1998). Intriguingly, however, data from human cancer studies find that the majority of small mutations in the p14ARF-p16INK4a gene locus cluster in the 5′ portion of exon 2 that is shared by p16 and ARF, including those that are ARF specific, suggesting a tumor-suppressive role for the ARF C terminus. Our findings that ARF-induced apoptosis requires association of p32 with the exon 2-encoded C terminus, and that both the ARF-p32 association and ARF's apoptotic function can be simultaneously disrupted by single point mutations found in human cancers, has assigned a potential tumor-suppressive function to the ARF C terminus. Whether or not our in vitro data recapitulate the functional interaction of ARF and p32 under a truly physiological environment can only be determined definitively by introducing mutations into laboratory animals. Nonetheless, this study has demonstrated a promising role for p32 as a critical mediator of apoptosis, and these findings provide a basis for further investigation into the role of p32 in cancer development.
Experimental Procedures
Cell Lines, Cell Culture, and Reagents
U2OS, H1299, HCC1937, MDA-MB-468, and Saos2 cells were obtained from ATCC. Raji and HL60 cells were gifts from Dr. Beverly S. Mitchell (Stanford). p19Arf−/−;p53−/− MEFs were a gift from Dr. Norman Sharpless (UNC-Chapel Hill). All cells were cultured in a 37°C incubator with 5% CO2 in DMEM supplied with 10% FBS. After each treatment, cell viability was determined microscopically by trypan blue exclusion, and cell number was counted with a hematocytometer.
Plasmids, Adenoviruses, and Flow Cytometry Analysis
Full-length p32 cDNA was purchased from Open Biosystems (GenBank accession number BC000435), and both untagged and Myc-tagged full-length p32 (aa 1–282) were cloned into a pcDNA3.1 vector (Invitrogen). All cloned constructs were confirmed by direct DNA sequencing. Recombinant adenoviruses carrying untagged p32, ARF, ARF mutants, p53, and GFP were produced using the AdEasy XL Adenoviral Vector System (Stratagene) according to the manufacturer's protocol. The multiplicity of infection (moi) to achieve more than 90% of cells infected by the adenovirus was determined by immunofluorescence staining before carrying out experiments. Retroviruses expressing c-Myc proteins were produced as described previously (Itahana et al., 2003). ARF point mutations were introduced by PCR-based site-directed mutagenesis. For flow cytometry analysis, both floating and attached cells were collected, fixed with 70% ethanol, stained with propidium iodide, and analyzed at the UNC Lineberger Cancer Center Flow Cytometry Facility. The software packages Summit (DakoCytomation) and ModFit LT (Verity) were used for flow cytometry data acquisition, and all flow cytometry data are representative of at least three independent experiments.
Protein Analysis
Rabbit polyclonal anti-p32 antibody was produced using a synthetic peptide (CEERKIQKHKTLPKMSG) corresponding to amino acid residues 92–107 of human p32. Antibodies for cytochrome c (7H8.2C12, BD PharMingen), mtHSP70/GRP75 (H155, Santa Cruz), p19Arf (5-C3-1, Santa Cruz), Histone H3 (9715, Cell signaling), PARP(C2-10, BD PharMingen), B23(Fc-61991, Invitrogen/Zymed), Actin (MAB1501, Chemicon International), and MDM2 (4B11, Calbiochem) were purchased commercially. Antibodies for human ARF (P02; N terminus epitope), p53 (DO1), Tubulin (DM1A+DM1B), and Myc (9E10.3) were purchased from Lab Vision/Neomarker. Rabbit polyclonal antibodies to Myc, human ARF (C terminus epitope), and mouse ARF (C terminus epitope) were gifts from Dr. Yue Xiong (UNC-Chapel Hill). The in vitro transcription/translation (IVT) assay is described elsewhere (Enomoto et al., 2006). Cells were lysed in 0.1% NP-40 buffer for immunoprecipitation and 0.5% NP-40 buffer for straight western blotting unless otherwise stated. For analysis of PARP cleavage, both floating and attached cells were collected.
RNA Interference
The siRNA oligonucleotides targeting p32 mRNA, and the control scrambled oligonucleotides (si-Scr), were synthesized either commercially by Dharmacon (Lafayette, CO) or by using the Silencer siRNA Construction Kit (Ambion, TX). The two targeting sequences for p32 are 5′-GGUUGAAGAACAGGAGCCUdTdT-3′ (si-p32) and 5′-UCACGGUCACUUUCAACATdTdT3′ (si-p32-2) (Sunayama et al., 2004). siRNA oligonucleotides (80 pmol) were transfected using Oligofectamine (Invitrogen) or Lipofectamine (Invitrogen) in a 35 mm dish according to the manufacturer's protocol. All cultures were performed without antibiotics during siRNA transfection. For adenovirus infection after transfection, cells were transfected with siRNA oligonucleotides for 1–2 days, washed with medium, and infected with adenovirus for another 1–2 days.
Cytoplasmic and Mitochondrial Fractionation
Cytoplasmic extracts were prepared using a low concentration (0.15 mg/ml) of digitonin solution adapted from Hemmati et al. (2006). Mitochondrial extracts were prepared using a low concentration (0.5%) of Triton X-100 (Fisher Scientific) solution adapted from Ramsby and Makowski (1999) with slight modifications. Briefly, both attached and floating cells were collected by centrifugation and washed twice with PBS. One-fourth of the cells were lysed with SDS lysis buffer (100 mM Tris-HCl [pH 6.8], 2% SDS, 10% glycerol) for direct SDS-PAGE analysis. The remaining cells were lysed in cold hypotonic buffer (20 mM HEPES [pH 7.4], 10 mM KCl, 2 mM MgCl2, 1 mM EDTA) supplemented with freshly made 1 mM PMSF and 0.15 mg/ml digitonin (Sigma). After incubation on ice for 3 min, cellular debris, including nuclei and mitochondria, were pelleted in a microcentrifuge (12,000 rpm) for 10 min at 4°C. The supernatants were transferred to a fresh tube as the cytoplasmic fraction. The cell pellets were further washed twice with cold hypotonic buffer with 1 mM PMSF and then pelleted by centrifugation (12,000 rpm, 2 min). The digitonin-insoluble pellets (containing mitochondria) were lysed with cold 0.5% Triton X-100/hypotonic buffer with 1 mM PMSF and incubated on ice for 1 min. Subsequently, cellular debris including intact nuclei were pelleted in a microcentrifuge (12,000 rpm) for 5 min at 4°C, and the supernatants were transferred to a fresh tube as the mitochondrial fraction. Both cytoplasmic and mitochondrial fractions were mixed with 5× SDS-PAGE loading buffer before loading onto the gel.
Measurement of Mitochondrial Membrane Potential
Both attached and floating cells were collected and washed twice with culturing medium. Equal concentrations of cells (1 × 106 cells/ml) were incubated for 15 min in culture medium containing 1× JC-1 dye (Cell Technology, Inc., Mountain View, CA) at 37°C in a 5% CO2 incubator according to the manufacturer's instructions. Cells were washed three times with culturing medium and analyzed by flow cytometry in the FL-1 and FL-2 channels. Mitochondrial permeability transition was assessed with decreased red fluorescence (FL-2), which indicates the presence of mitochondria with a lower membrane potential (Δψm).
Supplementary Material
Significance.
Although ARF's cell-cycle arrest function is well understood, little is known about ARF's role in apoptosis, due largely to the lack of specific molecules linking ARF to this process. The current view holds that the C terminus of ARF is dispensable for tumor suppression because overexpression of the ARF N-terminal half alone is sufficient to activate p53. The study reported here identifies mitochondrial p32 as a binding partner of ARF's C terminus and a critical mediator of ARF's apoptotic function in the mitochondria. Importantly, the ARF-p32 interaction and ARF's apoptotic function are simultaneously disrupted by cancer-derived point mutations in the ARF C terminus. Our results provide mechanistic insight into ARF-induced, p53-dependent apoptosis and demonstrate a potential tumor-suppressive function for the ARF C terminus.
Acknowledgments
We thank Hilary Clegg for helpful discussions throughout the project and critical reading of the manuscript and Mohanish Deshmukh, Norman Sharpless, and Yue Xiong for providing invaluable reagents. Y.Z. is a recipient of a Career Award in Biomedical Science from the Burroughs Wellcome Fund, a Howard Temin Award from the National Cancer Institute, and a Scholar Award from the Leukemia and Lymphoma Society. This study was supported by NIH grants and a grant from the Leukemia Research Foundation (to Y.Z.).
Footnotes
Supplemental Data: The Supplemental Data include four supplemental figures and can be found with this article online at http://www.cancercell.org/cgi/content/full/13/6/542/DC1/.
References
- Beatch MD, Everitt JC, Law LJ, Hobman TC. Interactions between rubella virus capsid and host protein p32 are important for virus replication. J Virol. 2005;79:10807–10820. doi: 10.1128/JVI.79.16.10807-10820.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell JB, Greville GD. Effects of oligomycin on respiration and swelling of isolated liver mitochondria. Nature. 1961;190:502–504. doi: 10.1038/190502a0. [DOI] [PubMed] [Google Scholar]
- Dedio J, Jahnen-Dechent W, Bachmann M, Muller-Esterl W. The multiligand-binding protein gC1qR, putative C1q receptor, is a mitochondrial protein. J Immunol. 1998;160:3534–3542. [PubMed] [Google Scholar]
- de Stanchina E, McCurrach ME, Zindy F, Shieh SY, Ferbeyre G, Samuelson AV, Prives C, Roussel MF, Sherr CJ, Lowe SW. E1A signaling to p53 involves the p19ARF tumor suppressor. Genes Dev. 1998;12:2434–2442. doi: 10.1101/gad.12.15.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto T, Lindstrom MS, Jin A, Ke H, Zhang Y. Essential role of the B23/NPM core domain in regulating ARF binding and B23 stability. J Biol Chem. 2006;281:18463–18472. doi: 10.1074/jbc.M602788200. [DOI] [PubMed] [Google Scholar]
- Eymin B, Leduc C, Coll JL, Brambilla E, Gazzeri S. p14ARF induces G2 arrest and apoptosis independently of p53 leading to regression of tumours established in nude mice. Oncogene. 2003;22:1822–1835. doi: 10.1038/sj.onc.1206303. [DOI] [PubMed] [Google Scholar]
- Ghebrehiwet B, Lim BL, Peerschke EI, Willis AC, Reid KB. Isolation, cDNA cloning, and overexpression of a 33-kD cell surface glycoprotein that binds to the globular “heads” of C1q. J Exp Med. 1994;179:1809–1821. doi: 10.1084/jem.179.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Greenblatt M. UVM BioDesktop CDKN2a Database Project. 2006 http://biodesktop.uvm.edu/perl/p16.
- Greenblatt MS, Beaudet JG, Gump JR, Godin KS, Trombley L, Koh J, Bond JP. Detailed computational study of p53 and p16: Using evolutionary sequence analysis and disease-associated mutations to predict the functional consequences of allelic variants. Oncogene. 2003;22:1150–1163. doi: 10.1038/sj.onc.1206101. [DOI] [PubMed] [Google Scholar]
- Hall KT, Giles MS, Calderwood MA, Goodwin DJ, Matthews DA, Whitehouse A. The Herpesvirus Saimiri open reading frame 73 gene product interacts with the cellular protein p32. J Virol. 2002;76:11612–11622. doi: 10.1128/JVI.76.22.11612-11622.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmati PG, Gillissen B, von Haefen C, Wendt J, Starck L, Guner D, Dorken B, Daniel PT. Adenovirus-mediated overexpression of p14(ARF) induces p53 and Bax-independent apoptosis. Oncogene. 2002;21:3149–3161. doi: 10.1038/sj.onc.1205458. [DOI] [PubMed] [Google Scholar]
- Hemmati PG, Guner D, Gillissen B, Wendt J, von Haefen C, Chinnadurai G, Dorken B, Daniel PT. Bak functionally complements for loss of Bax during p14(ARF)-induced mitochondrial apoptosis in human cancer cells. Oncogene. 2006;25:6582–6594. doi: 10.1038/sj.onc.1209668. [DOI] [PubMed] [Google Scholar]
- Itahana K, Bhat KP, Jin A, Itahana Y, Hawke D, Kobayashi R, Zhang Y. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol Cell. 2003;12:1151–1164. doi: 10.1016/s1097-2765(03)00431-3. [DOI] [PubMed] [Google Scholar]
- Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- Kamal A, Datta K. Upregulation of hyaluronan binding protein 1 (HABP1/p32/gC1qR) is associated with Cisplatin induced apoptosis. Apoptosis. 2006;11:861–874. doi: 10.1007/s10495-006-5396-4. [DOI] [PubMed] [Google Scholar]
- Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavitgian SV, Stockert E, Day RS, Johnson BE, Skolnick MH. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264:436–440. doi: 10.1126/science.8153634. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel M, Sherr CJ. Functional and physical interaction of the ARF tumor suppressor with p53 and MDM2. Proc Natl Acad Sci USA. 1998;95:8292–8297. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Mitchell M, Fujii H, Llanos S, Peters G. Absence of p16INK4a and truncation of ARF tumor suppressors in chickens. Proc Natl Acad Sci USA. 2003;100:211–216. doi: 10.1073/pnas.0135557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Sgagias M, Deng X, Jung YJ, Rikiyama T, Lee K, Ouellette M, Cowan K. Apoptosis induced by adenovirus-mediated p14ARF expression in U2OS osteosarcoma cells is associated with increased Fas expression. Biochem Biophys Res Commun. 2004;320:138–144. doi: 10.1016/j.bbrc.2004.05.135. [DOI] [PubMed] [Google Scholar]
- Krainer AR, Mayeda A, Kozak D, Binns G. Functional expression of cloned human splicing factor SF2: Homology to RNA-binding proteins, U1 70K, and Drosophila splicing regulators. Cell. 1991;66:383–394. doi: 10.1016/0092-8674(91)90627-b. [DOI] [PubMed] [Google Scholar]
- Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature. 2001;413:83–86. doi: 10.1038/35092584. [DOI] [PubMed] [Google Scholar]
- Lindstrom MS, Klangby U, Inoue R, Pisa P, Wiman KG, Asker CE. Immunolocalization of human p14(ARF) to the granular component of the interphase nucleolus. Exp Cell Res. 2000;256:400–410. doi: 10.1006/excr.2000.4854. [DOI] [PubMed] [Google Scholar]
- Lohrum MAE, Ashcroft M, Kubbutat MHG, Vousden KH. Contribution of two independent MDM2-binding domains in p14ARF to p53 stabilization. Curr Biol. 2000;10:539–542. doi: 10.1016/s0960-9822(00)00472-3. [DOI] [PubMed] [Google Scholar]
- Luo Y, Yu H, Peterlin BM. Cellular protein modulates effects of human immunodeficiency virus type 1 Rev. J Virol. 1994;68:3850–3856. doi: 10.1128/jvi.68.6.3850-3856.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgley CA, Desterro JM, Saville MK, Howard S, Sparks A, Hay RT, Lane DP. An N-terminal p14ARF peptide blocks Mdm2-dependent ubiquitination in vitro and can activate p53 in vivo. Oncogene. 2000;19:2312–2323. doi: 10.1038/sj.onc.1203593. [DOI] [PubMed] [Google Scholar]
- Muta T, Kang D, Kitajima S, Fujiwara T, Hamasaki N. p32 protein, a splicing factor 2-associated protein, is localized in mitochondrial matrix and is functionally important in maintaining oxidative phosphorylation. J Biol Chem. 1997;272:24363–24370. doi: 10.1074/jbc.272.39.24363. [DOI] [PubMed] [Google Scholar]
- Nikolakaki E, Simos G, Georgatos SD, Giannakouros T. A nuclear envelope-associated kinase phosphorylates arginine-serine motifs and modulates interactions between the lamin B receptor and other nuclear proteins. J Biol Chem. 1996;271:8365–8372. doi: 10.1074/jbc.271.14.8365. [DOI] [PubMed] [Google Scholar]
- Nobori T, Mlura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletion of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature. 1994;368:753–756. doi: 10.1038/368753a0. [DOI] [PubMed] [Google Scholar]
- Petersen-Mahrt SK, Estmer C, Ohrmalm C, Matthews DA, Russell WC, Akusjarvi G. The splicing factor-associated protein, p32, regulates RNA splicing by inhibiting ASF/SF2 RNA binding and phosphorylation. EMBO J. 1999;18:1014–1024. doi: 10.1093/emboj/18.4.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, DePinho RA. The INK4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- Quelle DE, Zindy F, Ashmun R, Sherr CJ. Alterative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Quelle DE, Cheng M, Ashmun RA, Sherr CJ. Cancer-associated mutations at the INK4a locus cancel cell cycle arrest by p16INK4a but not by the alternative reading frame protein p19ARF. Proc Natl Acad Sci USA. 1997;94:3436–3440. doi: 10.1073/pnas.94.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsby ML, Makowski GS. Differential detergent fractionation of eukaryotic cells. Analysis by two-dimensional gel electrophoresis. Methods Mol Biol. 1999;112:53–66. doi: 10.1385/1-59259-584-7:53. [DOI] [PubMed] [Google Scholar]
- Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M, Kimchi A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell. 2006;22:463–475. doi: 10.1016/j.molcel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Reef S, Shifman O, Oren M, Kimchi A. The autophagic inducer smARF interacts with and is stabilized by the mitochondrial p32 protein. Oncogene. 2007;26:6677–6683. doi: 10.1038/sj.onc.1210485. [DOI] [PubMed] [Google Scholar]
- Ruas M, Peters G. The p16INK4a/CDK2A tumor suppressor and its relatives. Biochim Biophys Acta. 1998;1378:F115–F177. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- Rubinstein DB, Stortchevoi A, Boosalis M, Ashfaq R, Ghebrehiwet B, Peerschke EI, Calvo F, Guillaume T. Receptor for the globular heads of C1q (gC1q-R, p33, hyaluronan-binding protein) is preferentially expressed by adenocarcinoma cells. Int J Cancer. 2004;110:741–750. doi: 10.1002/ijc.20105. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee HW, Chin L, Cordon-Cardos C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Sharpless NE. INK4a/ARF: A multifunctional tumor suppressor locus. Mutat Res. 2005;576:22–38. doi: 10.1016/j.mrfmmm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413:86–91. doi: 10.1038/35092592. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Tumor surveillance via the ARF-p53 pathway. Genes Dev. 1998;12:2984–2991. doi: 10.1101/gad.12.19.2984. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol. 2001;2:731–737. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Divorcing ARF and p53: An unsettled case. Nat Rev Cancer. 2006;6:663–673. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, Palmero I, Ryan K, Hara E, Vousden KH, Peters G. The alternative product from the human CDK2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998;17:5001–5014. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunayama J, Ando Y, Itoh N, Tomiyama A, Sakurada K, Sugiyama A, Kang D, Tashiro F, Gotoh Y, Kuchino Y, Kitanaka C. Physical and functional interaction between BH3-only protein Hrk and mitochondrial pore-forming protein p32. Cell Death Differ. 2004;11:771–781. doi: 10.1038/sj.cdd.4401418. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Wang Y, Finan JE, Middeldorp JM, Hayward SD. P32/TAP, a cellular protein that interacts with EBNA-1 of Epstein-Barr virus. Virology. 1997;236:18–29. doi: 10.1006/viro.1997.8739. [DOI] [PubMed] [Google Scholar]
- Weber JD, Kuo ML, Bothner B, Digiammarino EL, Kriwacki RW, Roussel MF, Sherr CJ. Cooperative signals governing ARF-MDM2 interaction and nucleolar localization of the complex. Mol Cell Biol. 2000;20:2517–2528. doi: 10.1128/mcb.20.7.2517-2528.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarbrough WG, Bessho M, Zanation A, Bisi JE, Xiong Y. Human tumor suppressor ARF impedes S-phase progression independent of p53. Cancer Res. 2002;62:1171–1177. [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y. Mutation in human ARF exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. Mol Cell. 1999;3:579–591. doi: 10.1016/s1097-2765(00)80351-2. [DOI] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.