

Abstract
Cochlear implantation is currently the treatment of choice for children with severe to profound hearing impairment. However, the outcomes with cochlear implants (CIs) vary significantly among recipients. The purpose of the present study is to identify the genetic determinants of poor CI outcomes. Twelve children with poor CI outcomes (the “cases”) and 30 “matched controls” with good CI outcomes were subjected to comprehensive genetic analyses using massively parallel sequencing, which targeted 129 known deafness genes. Audiological features, imaging findings, and auditory/speech performance with CIs were then correlated to the genetic diagnoses. We identified genetic variants which are associated with poor CI outcomes in 7 (58%) of the 12 cases; 4 cases had bi-allelic PCDH15 pathogenic mutations and 3 cases were homozygous for the DFNB59 p.G292R variant. Mutations in the WFS1, GJB3, ESRRB, LRTOMT, MYO3A, and POU3F4 genes were detected in 7 (23%) of the 30 matched controls. The allele frequencies of PCDH15 and DFNB59 variants were significantly higher in the cases than in the matched controls (both P < 0.001). In the 7 CI recipients with PCDH15 or DFNB59 variants, otoacoustic emissions were absent in both ears, and imaging findings were normal in all 7 implanted ears. PCDH15 or DFNB59 variants are associated with poor CI performance, yet children with PCDH15 or DFNB59 variants might show clinical features indistinguishable from those of other typical pediatric CI recipients. Accordingly, genetic examination is indicated in all CI candidates before operation.
INTRODUCTION
Approximately 1 in 1000 children suffer from severe to profound sensorineural hearing impairment (SNHI).1 For these children, cochlear implantation is currently regarded as the best-hearing rehabilitation strategy. Bypassing the deafened cochlea, a cochlear implant (CI) works by directly stimulating auditory nerve fibers, transmitting signals through the central auditory neural pathway, and ultimately yielding speech understanding in the auditory cortex. The benefits of cochlear implantation for spoken language, reading skills, and cognitive development have been well demonstrated.2 The outcomes after cochlear implantation, however, vary significantly among individuals. Many factors contribute to the outcomes, including age at implantation,3,4 residual hearing,5 presence of inner ear malformations,6 presence of cochlear nerve deficiency,7 parent–child interactions,2 and socioeconomic status.2
Genetic factors play an important role in pediatric SNHI, with more than 50% of cases having a genetic etiology.8 To date, more than 100 genes or loci with Mendelian inheritance have been related to deafness, and ∼50 genes have been identified to cause nonsyndromic hereditary hearing impairment (HHI) (The Hereditary Hearing Loss Homepage, http://hereditaryhearingloss.org/).9 It has been difficult to address the genetically heterogeneous HHI, as in practice only a limited number of genes could be screened using conventional Sanger sequencing. The advent of massively parallel sequencing (MPS), also known as next-generation sequencing, made comprehensive genetic analysis possible by enabling sequencing of an enormous volume of samples with faster turnaround and relatively low cost. The capability to test all candidate genes simultaneously has made MPS a powerful tool for genetic examination in many diseases with numerous possible causative genes, including mitochondrial diseases,10 familial hypercholesterolemia,11 lysosomal storage disorders,12 neuromuscular diseases,13 and hearing impairment.14
Theoretically, mutations in different deafness genes lead to different pathologies and might result in varied CI outcomes. Good outcomes have been documented in patients with certain common deafness-associated mutations, including GJB2 mutations,15SLC26A4 mutations,16 mitochondrial mutations,17 and OTOF mutations,18 because the effects of these mutations are confined to the inner ear and the function of the auditory nerve is spared.19 On the other hand, mutations in genes expressed in spiral ganglion neurons (SGNs), the neurons of auditory nerves, have been hypothesized to portend poor CI performance.20 It has been estimated that lifetime cost of cochlear implantation might exceed 1 million US dollars,20,21 but 3% to 7% pediatric recipients might become nonusers of their implants out of various reasons.22,23 It would be highly beneficial if these poor responders can be identified before the operation. To explore the genetic underpinnings of poor CI outcomes, we conducted comprehensive genetic analyses in children with poor CI outcomes and those with good CI outcomes, using MPS, and then correlated the genetic diagnoses to CI performance.
METHODS
Study Participants and Clinical Evaluations
Participants of the present study were selected from a cohort of ∼200 children with CIs.19 Inclusion criteria were as follows: CIs in use for more than 3 years and no previously detected mutations in common deafness genes. Exclusion criteria were as follows: syndromic hearing loss, acquired hearing loss, cochlear nerve deficiency in the implanted ears, and the presence of additional cognitive or psychological defects. All participants were Han Chinese and came from families whose native language was Mandarin. All children were unilaterally implanted with Nucleus 24 or Nucleus Freedom CIs at National Taiwan University Hospital or Chang Gung Memorial Hospital and received verbal education in mainstream schools or rehabilitation facilities after implantation. This study was approved by the Research Ethics Committees of both hospitals.
For each child, comprehensive family history, history, physical examination, audiological results, and imaging results were ascertained. The preoperative hearing level of the implanted ear was calculated as a 4-tone average (0.5, 1, 2, and 4 kHz).19 Otoacoustic emissions (OAEs) were recorded preoperatively to assess the physiology of hair cells. Preoperative imaging results were obtained using high-resolution computed tomography and magnetic resonance imaging, and abnormalities of the inner ear and cochlear nerve were determined according to criteria in the literature.24
Evaluation of Auditory and Speech Performance
Auditory performance in CI recipients was evaluated using the Categories of Auditory Performance (CAP) scale,19 which is an ordinal scale of auditory receptive ability composed of 8 categories ranging from “no awareness of environment” (CAP score = 0) to “use of telephone with known users” (CAP score = 7). Speech performance was assessed using the Speech Intelligibility Rating (SIR) scale,25 which classifies children's spontaneous speech intelligibility into 5 categories ranging from “unintelligible speech” (SIR score = 1) to “speech intelligible to all listeners” (SIR score = 5). The CAP and SIR scales have been confirmed as reliable instruments for measuring CI outcomes.26,27 Speech perception tests were also conducted to obtain objective recognition scores for 3 parameters: phonetically balanced word, easy sentence, and difficult sentence.25 The easy sentence test consisted of 15 sentences varying in length from 2 to 10 words, including 1 to 7 keywords familiar to the CI children in their daily conversations, such as “book” and “car”; whereas the difficult sentence test included 20 sentences varying in length from 2 to 12 words, containing 1 to 10 keywords of lower familiarity to the children, such as “examine” and “dormitory.”25
In our previous study, median CAP and SIR scores after 3 years of CI use were 6 and 4, respectively.25 We selected 12 children with CAP scores < 5 (unable to understand conversation without lip-reading) and SIR scores < 3 (speech intelligible only to listeners who concentrate and lip-read, or worse) despite more than 3 years of rehabilitation. We classified these 12 children as “cases” because of their poor CI outcomes. Thirty unrelated children with good CI performance (CAP score = 7 and SIR score = 5) were selected and matched to the cases in terms of implantation age, duration of rehabilitation, and preoperative hearing. These 30 children were the “matched controls” (Figure 1).
FIGURE 1.
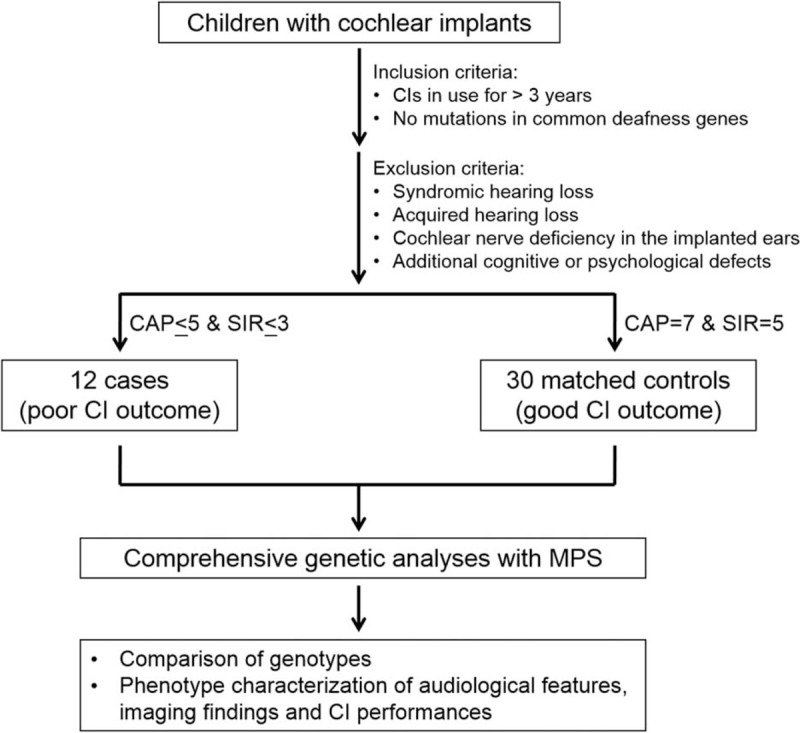
Study design. From a cohort of children with cochlear implants (CIs), 12 with poor CI outcomes were selected as “cases” and 30 with good CI outcomes were selected as “matched controls.” These 42 children were subjected to comprehensive genetic analyses using massively parallel sequencing, which targeted 129 known deafness genes. Genotypes were then compared between “cases” and “matched controls,” and phenotypes were correlated to the genetic diagnoses. CAP, Categories of Auditory Performance; MPS, massively parallel sequencing; SIR, Speech Intelligibility Rating.
Massively Parallel Sequencing and Data Analyses
Genomic DNA was extracted from peripheral blood or saliva samples from the CI patients and their family members. DNA libraries were generated from fragmented genomic DNA and subjected to enrichment with custom probes targeting 129 known human deafness genes.28 The enriched DNA was sequenced using an Illumina HiSeq 2000 system, and sequence analyses were then performed as previously described.28 In brief, the paired-end sequence reads were aligned, sorted, and converted by BWA and Picard (http://picard.sourceforge.net) software. Variant calling was performed using GATK, and variants were annotated using ANNOVAR. Integrative Genomics Viewer was used to view the mapping and annotation of sequences. Pindel was used for detecting structural variants such as large deletions/insertions, inversions, and duplications, and ExomeDepth for detecting copy number variants.
Variants with allele frequencies above 5% in both the 1000 Genomes Project29 and the NHLBI-ESP 6500 exome project (http://evs.gs.washington.edu/EVS/) were excluded because they are unlikely to be the genetic cause of severe single-gene diseases such as SNHI. We retained all highly possible disease-causing variants for further analyses, including frameshift and nonframeshift insertion/deletion (indel) variants, nonsense variants, and splice site variants. PolyPhen-230 and SIFT31 were used to predict the deleterious effects of amino acid substitutions, and we excluded missense variants with PolyPhen-2 scores < 0.95 or SIFT scores > 0.05, except for variants that had been reported in the Deafness Variation Database (http://deafnessvariationdatabase.org/). Sanger sequencing was performed to confirm the filtered variants and examine cosegregation of the genotype and SNHI phenotype among the family members. Allele frequencies of variants segregating with the phenotype were also verified in a panel of 100 normal-hearing Han Chinese.
Analyses of Genotypes and Phenotypes
To investigate the genetic determinants of CI outcomes, the frequencies of variant alleles of specific genes were compared between the cases and matched controls. Audiological features, imaging findings, and auditory and speech performance with CIs were further analyzed in children with genetic variants associated with poor CI outcomes.
RESULTS
Demographic Characteristic
The demographic characteristics of the 12 cases and 30 matched controls are summarized in Table 1. There was no significant difference in implantation age (Student t test, P = 0.84), residual hearing before operation (Student t test, P = 0.48), rehabilitation duration (Student t test, P = 0.25), and sex distribution (Fisher exact test, P = 0.31) between the 2 groups.
TABLE 1.
Demographic Characteristics of the 12 Cases (Poor CI Outcomes) and 30 Matched Controls (Good CI Outcomes)

Identification of Genetic Variants
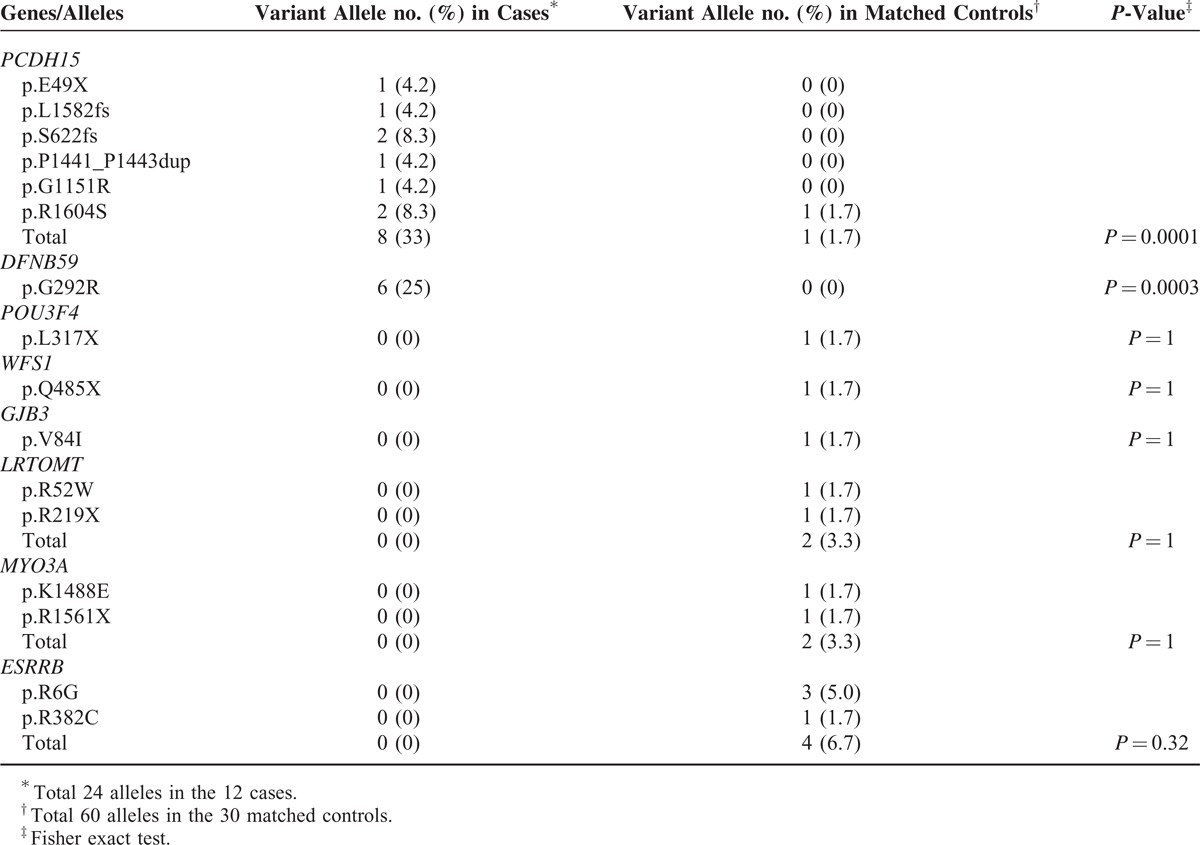
Variants associated with CI outcomes were detected in 7 of the 12 cases (58%) and 7 of the 30 matched controls (23%) (Table 2). In 4 cases, a total of 6 novel PCDH15 variants were identified (Figure 2A); and each case had 2 variant PCDH15 alleles which cosegregated with the SNHI phenotype in the pedigrees (Figure 2B). Of the 2 missense variants (p.G1151R and p.R1604S) which predictably resulted in amino acid changes, the pathogenicity of p.G1151R was supported by its low SIFT (0.01) and high PolyPhen-2 (1) scores, its low frequency (0.00279553) in 1000 Genomes, its absence in the 6500 NHLBI exomes and the 100 normal-hearing Han Chinese (Table 2), and the evolutionary conservation of the p.G1151 amino acid residue (Figure 2C). The pathogenicity of p.R1604S was less confirmable given its low PolyPhen-2 score (0.129) and the lower evolutionary conservation of the p.R1604 amino acid residue (Figure 2C). Nonetheless, the frequency of this variant was very low in the general population (<0.005 in both the 6500 NHLBI exomes and 1000 Genomes), so it was extremely unlikely that the hearing-impaired proband of a nonconsanguineous family segregated 2 variant alleles by chance. Therefore, we inferred that p.R1604S was causally related to SNHI in family DE3321. The 4 CI recipients with PCDH15 mutations revealed similar audiographic features, having bilateral symmetric flat-type audiograms of profound severity (Figure 2D). The MPS results visualized by Integrative Genomics Viewer are shown in Figure 2E.
TABLE 2.
Genetic Variants Identified in the 7 Cases and 7 Matched Controls
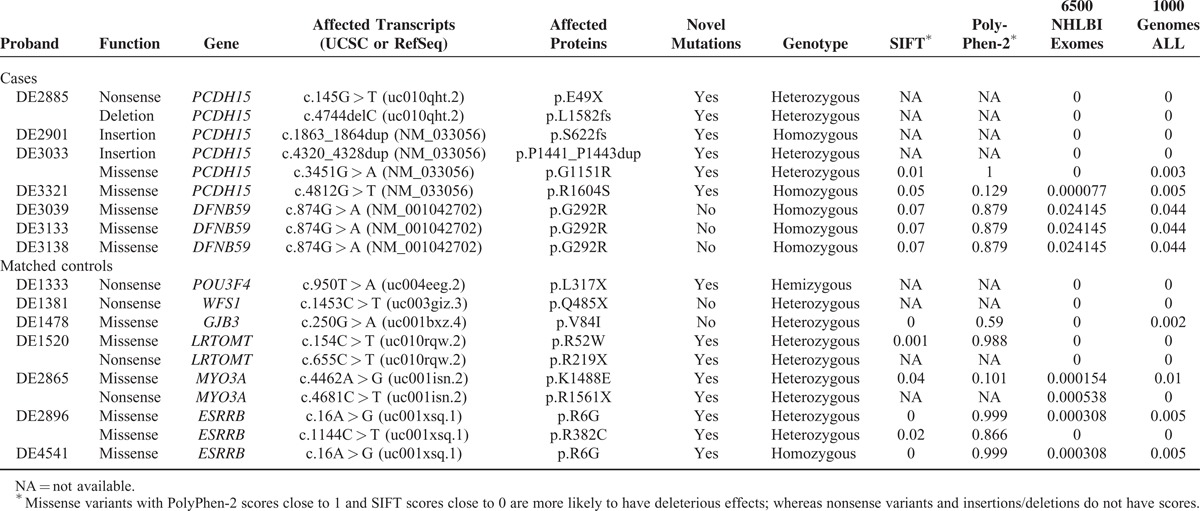
FIGURE 2.
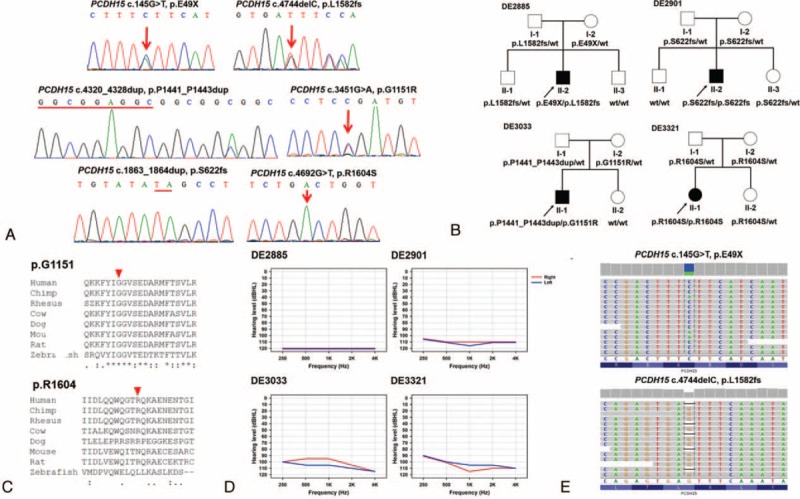
PCDH15 mutations in 4 families with poor CI outcomes. (A) The 6 PCDH15 mutations identified in the present study, including p.E49X, p.L1582fs, p.P1441_P1443dup, p.G1151R, p.S622fs, and p.R1604S. Sequencing data are shown on either the forward or the reverse strand, while PCDH15 is a reverse-stranded gene. (B) Pedigrees of the 4 families, showing that bi-allelic PCDH15 mutations cosegregated with the phenotype of hearing impairment in the family members. (C) Evolutionary conservation of the PCDH15 p.G1151 and p.R1604 amino acid residues. Arrowhead: variant site. (D) Audiograms of the 4 CI recipients with PCDH15 mutations. All 4 recipients had bilateral symmetric flat-type audiograms of profound severity. Hearing levels of the right ear and left ear are marked with red and blue lines, respectively. (E) Representative plots of the massively parallel sequencing results. Integrative Genomics Viewer showed a single nucleotide substitution (c.145G > T) and a single nucleotide deletion (c.4744delC) in the same patient (DE2885). Sequencing data are shown on the forward strand, while PCDH15 is a reverse-stranded gene.
Three cases were homozygous for the DFNB59 p.G292R (c.874G > A) variant. This variant was previously reported as a rare nonpathogenic polymorphism.32,33 However, the allele frequency of DFNB59 p.G292R in the 12 cases, which were recruited from 12 unrelated families, was significantly higher than that in the 1000 Genomes (0.25 vs. 0.044, Fisher exact test, P = 0.0005). In addition, except the 3 cases with poor CI outcomes in the present study, homozygosity for DFNB59 p.G292R has never been found in any of the >70 hearing-impaired Taiwanese families we performed MPS previously.28,34 Accordingly, from a statistical perspective, DFNB59 p.G292R might be associated with poor CI performance in the Taiwanese population.
Genetic causes of SNHI were identified in 7 matched controls. The mutations were scattered in 6 genes completely different from the 2 genes in the cases (Table 2). Two known autosomal dominant mutations, WFS1 p.Q485X35 and GJB3 p.V84I,36,37 were detected in 2 matched controls. Novel mutations in 4 other genes were detected in the remaining 5 matched controls: 2 had bi-allelic mutations in ESRRB, 1 had bi-allelic mutations in LRTOMT, 1 had bi-allelic mutations in MYO3A, and 1 had a hemizygous mutation in POU3F4.
Comparison of Variant Frequencies Between Cases and Matched Controls
The allele frequencies of the detected variants in the 12 cases and 30 matched controls are shown in Table 3. The allele frequency of PCDH15 mutations was 33% and 1.7% in cases and matched controls, respectively, and the difference between the 2 groups was significant (Fisher exact test, P = 0.0001). Similarly, the allele frequency of the DFNB59 p.G292R variant was significantly higher in cases than in matched controls (25% vs. 0%, Fisher exact test, P = 0.0003). In other words, variants in PCDH15 and DFNB59 were significantly associated with poor CI outcomes. In contrast, although mutations in certain genes occurred more frequently in matched controls than in cases, the difference in mutation frequency between the 2 groups was not statistically significant.
TABLE 3.
Comparison of Variant Frequencies Between Cases and Matched Controls
Clinical Features and CI Outcomes in Patients With PCDH15 or DFNB59 Variants
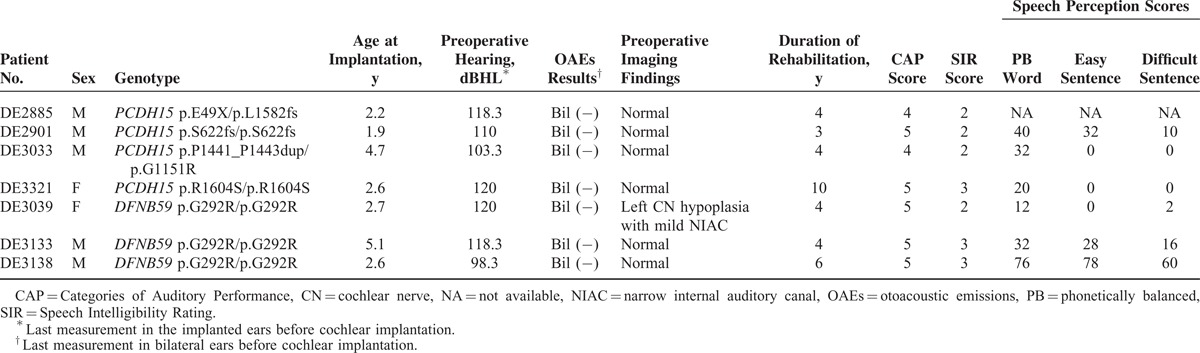
The phenotypes of the 7 CI recipients with PCDH15 or DFNB59 variants are summarized in Table 4. None of the 7 recipients was diagnosed as having auditory neuropathy (AN) before cochlear implantation, as OAEs were absent in both ears of these patients. Cochlear nerve hypoplasia was identified in the nonimplanted ear of 1 patient (DE3039) homozygous for the DFNB59 p.G292R variant, whereas imaging findings were normal in the other 6 patients. Speech perception scores were documented in 6 patients. One patient (DE3138) homozygous for DFNB59 p.G292R showed moderate speech perception scores, while the other 5 patients revealed poor scores in all 3 parameters despite more than 3 years of rehabilitation.
TABLE 4.
Clinical Features and Outcomes in the CI Recipients With PCDH15 or DFNB59 Variants
DISCUSSION
In the present study, we identified genetic determinants in 7 of the 12 cases with poor CI outcomes: 4 cases had bi-allelic PCDH15 mutations and 3 were homozygous for the DFNB59 p.G292R variant. The allele frequencies of PCDH15 and DFNB59 variants were significantly higher in the 12 cases than in the 30 matched controls with good CI outcomes, indicating that variants in these 2 genes were specifically associated with poor CI performance. Moreover, children with PCDH15 or DFNB59 variants revealed audiological and imaging results indistinguishable from those of other typical CI recipients with cochlear SNHI. These findings have important clinical implications. Genetic testing to identify SNHI patients likely to have poor CI outcomes could help in counseling patients preoperatively. For these patients, the genetic information is critical for determining appropriate rehabilitation programs and setting the expectations of physicians, audiologists, schools, and families, as these patients might need more aggressive aural rehabilitation postimplantation.
Prior to the present study, CI outcomes had never been reported in patients with bi-allelic PCDH15 or DFNB59 mutations. PCDH15 encodes protocadherin-15, and PCDH15 mutations have been related to Usher syndrome type 1F and nonsyndromic deafness DFNB23.38 Besides being a structural protein at the tip links of stereocilia,39 protocadherin-15 is also expressed in hair cell synapses and SGNs, suggesting its role in synaptic maturation.40 Mice with defective Pcdh15 gene showed a reduction in the number of SGNs in addition to disordered arrangement of stereocilia in hair cells.41 It has been documented that CI performance is satisfactory in most patients with type 1 Usher syndrome,42 but in a patient with digenic CDH23 and PCDH15 mutations, there was no development of open-set word recognition at 2 years after implantation.43 Therefore, pathogenetic mechanisms of PCDH15 mutations might differ from other Usher syndrome-related genetic mutations in involving auditory nerves additionally, thus contributing to poor CI performance.
The DFNB59 gene encodes pejvakin, which is expressed in hair cells, SGNs, and brainstem auditory nuclei in mammals.44,45 Corresponding to the histological distributions, mutations in DFNB59 have been described in families with either AN or cochlear SNHI.32,33,44–46 Because SGNs and brainstem auditory nuclei are affected, it is conceivable that patients with DFNB59 variants may demonstrate poor performance with CIs. The DNFB59 p.G292R variant is of particular interest. Although this variant was reported in deafness individuals of several pedigrees, it was also found in control individuals with a relatively high allele frequency in 1000 genomes (0.044). Therefore, it was considered to be a benign polymorphism rather than a pathogenic mutation.32,33 However, to exclude the pathogenecity/susceptibility of an autosomal recessive locus simply by allele frequency might be too arbitrary. Current genetic data from the literature could be consistent with the hypothesis that DNFB59 p.G292R is a pathogenic allele with incomplete penetrance. Another possible explanation is that the DFNB59 p.G292R variant is not a causative mutation for deafness, but it is in linkage disequilibrium with an unknown pathogenic mutation which is associated with poor CI outcomes in the Taiwanese population. Under this assumption, although we did not identify the true causative mutation in these 3 patients, the DFNB59 p.G292R variant still might serve as a predictor for CI performance. Further studies are warranted to confirm the association between DFNB59 variants and poor CI outcomes.
In the present study, deleterious variants of the WFS1, GJB3, ESRRB, LRTOMT, MYO3A, and POU3F4 genes were detected in children with good CI outcomes. Good CI performance has also been reported in children with mutations in a number of nonsyndromic HHI genes, including GJB2,15SLC26A4,16OTOF,18TMC1,47COCH,48 and LOXHD1.49 By performing MPS of 58 deafness genes in 8 patients with CI or electrical acoustic stimulation, Miyagawa et al50 determined that patients with mutations in the MYO15A, TECTA, and ACTG1 genes also showed relatively good auditory performance after operation. Good CI performance has been attributed to the fact that the expression and pathology of these genes are confined to the cochlea.19,20,50 Among the 6 genes related to good CI outcomes in the present study, LRTOMT,51MYO3A,52 and POU3F453 are expressed only in the cochlea; GJB3 is expressed in the cochlea and auditory nerves54; and WFS155 and ESRRB56 are expressed in the cochlea and SGNs. Favorable CI outcomes have been documented in patients with GJB337 and WFS157 mutations, whereas mutations in POU3F4 have been associated with varied CI outcomes, possibly confounded by additional cognitive or behavioral issues.58,59
Taken together, our results indicate that mutations in genes confined to the cochlea are associated with good CI outcomes in the absence of other cognitive or psychological problems, whereas mutations in genes expressed in SGNs and/or brainstem auditory nuclei might be associated with poor CI outcomes. Eppsteiner et al20 conducted comprehensive genetic screening of 29 adult CI recipients with idiopathic adult-onset severe-to-profound hearing loss and identified bi-allelic TMPRSS3 mutations in 2 of 6 patients with poor performance. Because TMPRSS3 is expressed in SGNs, the authors proposed that mutations in genes expressed in SGNs might portend poor CI performance. Of note, benefits from cochlear implantation were reported in patients with TMPRSS3 mutations in 3 other studies.50,60,61 As shown in the present study, although most children with PCDH15 or DFNB59 variants had poor speech perception scores, all 7 children achieved CAP scores of 4 to 5 and SIR scores of 2 to 3. Moreover, good CI outcomes were observed in children with mutations in GJB3, WFS1, and ESRRB. In other words, among the genes expressed in SGNs and/or brainstem auditory nuclei, mutations in certain genes, such as PCDH15, DFNB59, and TMPRSS3, appear to result in more severe pathologies in the auditory neural pathway, thus compromising the utility of CIs. However, the physiology of the auditory neural pathway is not completely abolished, as patients with mutations in PCDH15, DFNB59, and TMPRSS3 still gained some benefits from CIs.
Pathologies in SGNs and/or brainstem auditory nuclei clinically might be featured by AN.62 Cochlear implantation in patients with AN requires special consideration because the outcome is more unpredictable than in patients with cochlear SNHI.63–65 Despite the expression of PCDH15 and DFNB59 in SGNs and/or brainstem auditory nuclei, none of the 7 cases with mutations was diagnosed as having AN before cochlear implantation. In other words, it might be difficult to distinguish patients with PCDH15 or DFNB59 mutations from typical pediatric CI candidates based on preoperative audiological evaluations. From this standpoint, genetic diagnosis can be invaluable in elucidating the etiologies and predicting CI outcomes before implantation.
The interpretation and extrapolation of the results in the present study, however, should be done with caution. First, although this study represents the largest series to date of complete MPS analyses in children with long-term CI use, the participants are of a single ethnic background; hence, multicenter studies across populations might be necessary to validate our observations. Second, all 129 genes screened by our MPS panel are known deafness genes; thus, in families without detected mutations, particularly those with multiple affected members, SNHI might be attributed to mutations in unknown deafness genes. For these individuals, whole-exome sequencing might help elucidate the underlying pathophysiology. Third, it has been demonstrated that the optimal time to implant a young deaf child is within age 3.5 years in childhood, and is best by the first 2 years of life.66 The average implantation age in the 12 cases with poor outcomes was 3.2 years, which is close to the upper limit of the optimal time period and already beyond the best time to implant a child. A possible explanation for the late implantation is that the coverage rate of newborn hearing screening in Taiwan had not increased to 90% until 2012,67 resulting in delayed diagnosis in certain hearing-impaired children. Although we used a “matched controls” study design to control for the confounding effects of the implantation age on the CI outcomes, we could not exclude the possibility that poor performers in the present study might have done better if they were implanted earlier in life.
CONCLUSION
We demonstrated PCDH15 and DFNB59 variants were associated with poor CI performance in hearing-impaired children, probably attributable to pathology in SGNs and/or brainstem auditory nuclei. Because children with PCDH15 or DFNB59 variants might have clinical features indistinguishable from those of other typical pediatric CI candidates, comprehensive genetic examination might be indicated in all CI candidates before operation.
Acknowledgments
The authors would like to thank National Research Program for Biopharmaceuticals (NRPB, NSC 1022325-B-492-001) and National Center for High-performance Computing (NCHC) of National Applied Research Laboratories (NARLabs) of Taiwan for providing computational resources and storage resources. We thank Min-Hua Liao for technical support and the Shih-Mien Tu Foundation for patient recruitment. We thank Ming-Tai Chang, Chung-Tsai Su, Yun-Chian Cheng, and Chien-Yu Chen for using Anome GAP software package to independently confirm the results of our bioinformatic analysis results. We also wish to thank all subjects and their parents for participating in the present study.
Footnotes
Abbreviations: AN = auditory neuropathy, CAP = Categories of Auditory Performance, CI = cochlear implant, HHI = hereditary hearing impairment, MPS = massively parallel sequencing, OAE = otoacoustic emission, SGN = spiral ganglion neurons, SIR = Speech Intelligibility Rating, SNHI = sensorineural hearing impairment.
This study was supported by research grants from the National Health Research Institute (NHRI-EX103-10147PI), the National Science Council of the Executive Yuan of the Republic of China (NSC 100-2314-B-303-015-MY3), National Taiwan University (UN103-032) and Chang-Gung Memorial Hospital Research Program (CMRPG3B0181-3).
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Nadol JB., Jr Hearing loss. N Engl J Med 1993; 329:1092–1102. [DOI] [PubMed] [Google Scholar]
- 2.Niparko JK, Tobey EA, Thal DJ, et al. Spoken language development in children following cochlear implantation. JAMA 2010; 303:1498–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nikolopoulos TP, O’Donoghue GM, Archbold S. Age at implantation: its importance in pediatric cochlear implantation. Laryngoscope 1999; 109:595–599. [DOI] [PubMed] [Google Scholar]
- 4.Waltzman SB, Roland JT., Jr Cochlear implantation in children younger than 12 months. Pediatrics 2005; 116:e487–e493. [DOI] [PubMed] [Google Scholar]
- 5.Cullen RD, Higgins C, Buss E, et al. Cochlear implantation in patients with substantial residual hearing. Laryngoscope 2004; 114:2218–2223. [DOI] [PubMed] [Google Scholar]
- 6.Papsin BC. Cochlear implantation in children with anomalous cochleovestibular anatomy. Laryngoscope 2005; 115 (1 Pt 2 Suppl. 106):1–26. [DOI] [PubMed] [Google Scholar]
- 7.Walton J, Gibson WP, Sanli H, et al. Predicting cochlear implant outcomes in children with auditory neuropathy. Otol Neurotol 2008; 29:302–309. [DOI] [PubMed] [Google Scholar]
- 8.Smith RJ, Bale JF, Jr, White KR. Sensorineural hearing loss in children. Lancet 2005; 365:879–890. [DOI] [PubMed] [Google Scholar]
- 9.Hilgert N, Smith RJ, Van Camp G. Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mutat Res 2009; 681:189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calvo SE, Compton AG, Hershman SG, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med 2012; 4:118ra110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maglio C, Mancina RM, Motta BM, et al. Genetic diagnosis of familial hypercholesterolaemia by targeted next-generation sequencing. J Intern Med 2014; 276:396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernandez-Marmiesse A, Morey M, Pineda M, et al. Assessment of a targeted resequencing assay as a support tool in the diagnosis of lysosomal storage disorders. Orphanet J Rare Dis 2014; 9:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vasli N, Bohm J, Le Gras S, et al. Next generation sequencing for molecular diagnosis of neuromuscular diseases. Acta Neuropathol 2012; 124:273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shearer AE, Black-Ziegelbein EA, Hildebrand MS, et al. Advancing genetic testing for deafness with genomic technology. J Med Genet 2013; 50:627–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dahl HH, Wake M, Sarant J, et al. Language and speech perception outcomes in hearing-impaired children with and without connexin 26 mutations. Audiol Neurootol 2003; 8:263–268. [DOI] [PubMed] [Google Scholar]
- 16.Wu CC, Lee YC, Chen PJ, et al. Predominance of genetic diagnosis and imaging results as predictors in determining the speech perception performance outcome after cochlear implantation in children. Arch Pediatr Adolesc Med 2008; 162:269–276. [DOI] [PubMed] [Google Scholar]
- 17.Tono T, Ushisako Y, Kiyomizu K, et al. Cochlear implantation in a patient with profound hearing loss with the A1555G mitochondrial mutation. Am J Otol 1998; 19:754–757. [PubMed] [Google Scholar]
- 18.Rouillon I, Marcolla A, Roux I, et al. Results of cochlear implantation in two children with mutations in the OTOF gene. Int J Pediatr Otorhinolaryngol 2006; 70:689–696. [DOI] [PubMed] [Google Scholar]
- 19.Wu CC, Liu TC, Wang SH, et al. Genetic characteristics in children with cochlear implants and the corresponding auditory performance. Laryngoscope 2011; 121:1287–1293. [DOI] [PubMed] [Google Scholar]
- 20.Eppsteiner RW, Shearer AE, Hildebrand MS, et al. Prediction of cochlear implant performance by genetic mutation: the spiral ganglion hypothesis. Hear Res 2012; 292:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohr PE, Feldman JJ, Dunbar JL, et al. The societal costs of severe to profound hearing loss in the United States. Int J Technol Assess Health Care 2000; 16:1120–1135. [DOI] [PubMed] [Google Scholar]
- 22.Archbold SM, Nikolopoulos TP, Lloyd-Richmond H. Long-term use of cochlear implant systems in paediatric recipients and factors contributing to non-use. Cochlear Implants Int 2009; 10:25–40. [DOI] [PubMed] [Google Scholar]
- 23.Raine CH, Summerfield Q, Strachan DR, et al. The cost and analysis of nonuse of cochlear implants. Otol Neurotol 2008; 29:221–224. [DOI] [PubMed] [Google Scholar]
- 24.Wu CC, Chen PJ, Chiu YH, et al. Prospective mutation screening of three common deafness genes in a large Taiwanese Cohort with idiopathic bilateral sensorineural hearing impairment reveals a difference in the results between families from hospitals and those from rehabilitation facilities. Audiol Neurootol 2008; 13:172–181. [DOI] [PubMed] [Google Scholar]
- 25.Fang HY, Ko HC, Wang NM, et al. Auditory performance and speech intelligibility of Mandarin-speaking children implanted before age 5. Int J Pediatr Otorhinolaryngol 2014; 78:799–803. [DOI] [PubMed] [Google Scholar]
- 26.Archbold S, Lutman ME, Nikolopoulos T. Categories of auditory performance: inter-user reliability. Br J Audiol 1998; 32:7–12. [DOI] [PubMed] [Google Scholar]
- 27.Allen C, Nikolopoulos TP, Dyar D, et al. Reliability of a rating scale for measuring speech intelligibility after pediatric cochlear implantation. Otol Neurotol 2001; 22:631–633. [DOI] [PubMed] [Google Scholar]
- 28.Wu CC, Lin YH, Lu YC, et al. Application of massively parallel sequencing to genetic diagnosis in multiplex families with idiopathic sensorineural hearing impairment. PLoS One 2013; 8:e57369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abecasis GR, Auton A, Brooks LD, et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 49:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 32.Hashemzadeh Chaleshtori M, Simpson MA, Farrokhi E, et al. Novel mutations in the pejvakin gene are associated with autosomal recessive non-syndromic hearing loss in Iranian families. Clin Genet 2007; 72:261–263. [DOI] [PubMed] [Google Scholar]
- 33.Collin RW, Kalay E, Oostrik J, et al. Involvement of DFNB59 mutations in autosomal recessive nonsyndromic hearing impairment. Hum Mutat 2007; 28:718–723. [DOI] [PubMed] [Google Scholar]
- 34.Lin YH, Wu CC, Hsu TY, et al. Identification of a novel GATA3 mutation in a deaf Taiwanese family by massively parallel sequencing. Mutat Res 2015; 771:1–5. [DOI] [PubMed] [Google Scholar]
- 35.Hardy C, Khanim F, Torres R, et al. Clinical and molecular genetic analysis of 19 Wolfram syndrome kindreds demonstrating a wide spectrum of mutations in WFS1. Am J Hum Genet 1999; 65:1279–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oh SK, Choi SY, Yu SH, et al. Evaluation of the pathogenicity of GJB3 and GJB6 variants associated with nonsyndromic hearing loss. Biochim Biophys Acta 2013; 1832:285–291. [DOI] [PubMed] [Google Scholar]
- 37.Chu EA, Mhatre AN, Lustig LR, et al. Implication of mutations in Connexin 31 in cochlear implant outcome. Gene Funct Dis 2001; 2:214–220. [Google Scholar]
- 38.Ahmed ZM, Riazuddin S, Ahmad J, et al. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet 2003; 12:3215–3223. [DOI] [PubMed] [Google Scholar]
- 39.Kazmierczak P, Sakaguchi H, Tokita J, et al. Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature 2007; 449:87–91. [DOI] [PubMed] [Google Scholar]
- 40.Zallocchi M, Meehan DT, Delimont D, et al. Role for a novel Usher protein complex in hair cell synaptic maturation. PLoS One 2012; 7:e30573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Washington JL, III, Pitts D, Wright CG, et al. Characterization of a new allele of Ames waltzer generated by ENU mutagenesis. Hear Res 2005; 202:161–169. [DOI] [PubMed] [Google Scholar]
- 42.Pennings RJ, Damen GW, Snik AF, et al. Audiologic performance and benefit of cochlear implantation in Usher syndrome type I. Laryngoscope 2006; 116:717–722. [DOI] [PubMed] [Google Scholar]
- 43.Liu XZ, Angeli SI, Rajput K, et al. Cochlear implantation in individuals with Usher type 1 syndrome. Int J Pediatr Otorhinolaryngol 2008; 72:841–847. [DOI] [PubMed] [Google Scholar]
- 44.Delmaghani S, del Castillo FJ, Michel V, et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat Genet 2006; 38:770–778. [DOI] [PubMed] [Google Scholar]
- 45.Schwander M, Sczaniecka A, Grillet N, et al. A forward genetics screen in mice identifies recessive deafness traits and reveals that pejvakin is essential for outer hair cell function. J Neurosci 2007; 27:2163–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ebermann I, Walger M, Scholl HP, et al. Truncating mutation of the DFNB59 gene causes cochlear hearing impairment and central vestibular dysfunction. Hum Mutat 2007; 28:571–577. [DOI] [PubMed] [Google Scholar]
- 47.Makishima T, Kurima K, Brewer CC, et al. Early onset and rapid progression of dominant nonsyndromic DFNA36 hearing loss. Otol Neurotol 2004; 25:714–719. [DOI] [PubMed] [Google Scholar]
- 48.Vermeire K, Brokx JP, Wuyts FL, et al. Good speech recognition and quality-of-life scores after cochlear implantation in patients with DFNA9. Otol Neurotol 2006; 27:44–49. [DOI] [PubMed] [Google Scholar]
- 49.Grillet N, Schwander M, Hildebrand MS, et al. Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am J Hum Genet 2009; 85:328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyagawa M, Nishio SY, Ikeda T, et al. Massively parallel DNA sequencing successfully identifies new causative mutations in deafness genes in patients with cochlear implantation and EAS. PLoS One 2013; 8:e75793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahmed ZM, Masmoudi S, Kalay E, et al. Mutations of LRTOMT, a fusion gene with alternative reading frames, cause nonsyndromic deafness in humans. Nat Genet 2008; 40:1335–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walsh T, Walsh V, Vreugde S, et al. From flies’ eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc Natl Acad Sci U S A 2002; 99:7518–7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Minowa O, Ikeda K, Sugitani Y, et al. Altered cochlear fibrocytes in a mouse model of DFN3 nonsyndromic deafness. Science 1999; 285:1408–1411. [DOI] [PubMed] [Google Scholar]
- 54.Lopez-Bigas N, Olive M, Rabionet R, et al. Connexin 31 (GJB3) is expressed in the peripheral and auditory nerves and causes neuropathy and hearing impairment. Hum Mol Genet 2001; 10:947–952. [DOI] [PubMed] [Google Scholar]
- 55.Cryns K, Thys S, Van Laer L, et al. The WFS1 gene, responsible for low frequency sensorineural hearing loss and Wolfram syndrome, is expressed in a variety of inner ear cells. Histochem Cell Biol 2003; 119:247–256. [DOI] [PubMed] [Google Scholar]
- 56.Collin RW, Kalay E, Tariq M, et al. Mutations of ESRRB encoding estrogen-related receptor beta cause autosomal-recessive nonsyndromic hearing impairment DFNB35. Am J Hum Genet 2008; 82:125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hogewind BF, Pennings RJ, Hol FA, et al. Autosomal dominant optic neuropathy and sensorineual hearing loss associated with a novel mutation of WFS1. Mol Vis 2010; 16:26–35. [PMC free article] [PubMed] [Google Scholar]
- 58.Lee HK, Lee SH, Lee KY, et al. Novel POU3F4 mutations and clinical features of DFN3 patients with cochlear implants. Clin Genet 2009; 75:572–575. [DOI] [PubMed] [Google Scholar]
- 59.Stankovic KM, Hennessey AM, Herrmann B, et al. Cochlear implantation in children with congenital X-linked deafness due to novel mutations in POU3F4 gene. Ann Otol Rhinol Laryngol 2010; 119:815–822. [DOI] [PubMed] [Google Scholar]
- 60.Elbracht M, Senderek J, Eggermann T, et al. Autosomal recessive postlingual hearing loss (DFNB8): compound heterozygosity for two novel TMPRSS3 mutations in German siblings. J Med Genet 2007; 44:e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weegerink NJ, Schraders M, Oostrik J, et al. Genotype-phenotype correlation in DFNB8/10 families with TMPRSS3 mutations. J Assoc Res Otolaryngol 2011; 12:753–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Starr A, Sininger YS, Pratt H. The varieties of auditory neuropathy. J Basic Clin Physiol Pharmacol 2000; 11:215–230. [DOI] [PubMed] [Google Scholar]
- 63.Rance G, Beer DE, Cone-Wesson B, et al. Clinical findings for a group of infants and young children with auditory neuropathy. Ear Hear 1999; 20:238–252. [DOI] [PubMed] [Google Scholar]
- 64.Miyamoto RT, Kirk KI, Renshaw J, et al. Cochlear implantation in auditory neuropathy. Laryngoscope 1999; 109 (2 Pt 1):181–185. [DOI] [PubMed] [Google Scholar]
- 65.Nikolopoulos TP. Auditory dyssynchrony or auditory neuropathy: understanding the pathophysiology and exploring methods of treatment. Int J Pediatr Otorhinolaryngol 2014; 78:171–173. [DOI] [PubMed] [Google Scholar]
- 66.Sharma A, Campbell J. A sensitive period for cochlear implantation in deaf children. J Matern Fetal Neonatal Med 2011; 24 (Suppl. 1):151–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang CM, Yang IY, Ma YC, et al. The effectiveness of the promotion of newborn hearing screening in Taiwan. Int J Pediatr Otorhinolaryngol 2014; 78:14–18. [DOI] [PubMed] [Google Scholar]