

Abstract
Stimulator of interferon genes (STING) is a cytosolic receptor that senses both exogenous and endogenous cytosolic cyclic dinucleotides (CDNs), activating TBK1/IRF3 (interferon regulatory factor 3), NF-κB (nuclear factor κB), and STAT6 (signal transducer and activator of transcription 6) signaling pathways to induce robust type I interferon and proinflammatory cytokine responses. CDN ligands were formulated with granulocyte-macrophage colony-stimulating factor (GM-CSF)–producing cellular cancer vaccines—termed STINGVAX—that demonstrated potent in vivo antitumor efficacy in multiple therapeutic models of established cancer. We found that rationally designed synthetic CDN derivative molecules, including one with an Rp,Rp dithio diastereomer and noncanonical c[A(2′,5′)pA(3′,5′)p] phosphate bridge structure, enhanced antitumor efficacy of STINGVAX in multiple aggressive therapeutic models of established cancer in mice. Antitumor activity was STING-dependent and correlated with increased activation of dendritic cells and tumor antigen–specific CD8+ T cells. Tumors from STINGVAX-treated mice demonstrated marked PD-L1 (programmed death ligand 1) up-regulation, which was associated with tumor-infiltrating CD8+IFNγ+ T cells. When combined with PD-1 (programmed death 1) blockade, STINGVAX induced regression of palpable, poorly immunogenic tumors that did not respond to PD-1 blockade alone.
INTRODUCTION
The efferent arm of the immune system has been a principal target in the development of cancer immunotherapies (1, 2), but the innate immune system plays a pivotal role in modulating the potency and specificity of these active vaccination strategies (3, 4). Clinical therapeutic cancer vaccines, therefore, have been formulated with antigen-presenting cell (APC)–activating adjuvants that typically stimulate the MyD88-TRIF signaling pathway (5). To date, these formulations with various forms of cancer vaccines have not yielded clinical efficacy. Recently, a prime-boost regimen combining an allogeneic granulocyte-macrophage colony-stimulating factor (GM-CSF)–secreting vaccine with a mesothelin-expressing Listeria monocytogenes vaccine demonstrated a survival advantage in pancreatic carcinoma patients (6). This vaccine trial illustrates the concept of combining tumor antigens (mesothelin), an APC-expanding cytokine (GM-CSF), and an APC-activating vector (Listeria).
Initially characterized as ubiquitous bacterial secondary messengers, cyclic dinucleotides (CDNs) [cyclic di-GMP (guanosine 5′-monophosphate) (CDG), cyclic di-AMP (adenosine 5′-monophosphate) (CDA), and cyclic GMP-AMP (cGAMP)] were shown to constitute a class of pathogen-associated molecular pattern molecules (PAMPs) that activate the TBK1/interferon regulatory factor 3 (IRF3)/type 1 interferon (IFN) signaling axis via the cytoplasmic pattern recognition receptor stimulator of interferon genes (STING) (7–15). Induction of IFN in Listeria-infected cells is STING-dependent, and it is lost in goldenticket mice harboring a mutant STING allele (13). Other observations, including the CDG-STING cocrystal structures, demonstrated that cytosolic microbial CDNs bind to STING to elicit an IFN and proinflammatory signaling cascade (16). The STING signaling pathway has emerged as a central Toll-like receptor (TLR)–independent mediator of host innate defense stimulated by cytosolic nucleic acids, either through direct binding of exogenous CDNs from bacteria or through binding of a structurally distinct CDN produced by a host cyclic GMP-AMP synthetase (cGAS) in response to cytosolic double-stranded DNA (dsDNA) (17–20). The STING pathway represents a central node linking cytosolic nucleic acids to a transcriptional response resulting in a MyD88-independent production of type I IFN.
Here, we demonstrate that CDNs are potent adjuvants in vaccine formulations that can provide therapeutic immunity against malignancies (21, 22). We have used CDNs to create a cell-based cancer vaccine—STINGVAX—that is ready for translation in clinical trials. We also describe the rational development of synthetic CDN derivatives modified to have increased stability in vivo and to enhance binding to human STING (hSTING). When coformulated with an irradiated GM-CSF–secreting whole-cell vaccine in the form of STINGVAX, the synthetic CDNs increased the antitumor efficacy in all the tumor models tested. STINGVAX-treated mice were characterized by enhanced tumor-infiltrating lymphocytes (TILs) along with up-regulation of programmed death ligand 1 (PD-L1), indicating that STINGVAX is ideally suited for combination with (programmed death 1) PD-1 blockade treatment (23–27). STINGVAX combined with PD-1 blockade did, indeed, induce regression of established tumors. These results support the rationale for clinical evaluation of STINGVAX, particularly in settings where immune checkpoint blockade alone is not effective.
RESULTS
STINGVAX is a potent cancer vaccine
We initially found that CDNs induced potent STING-dependent CD4+-, CD8+-, and T helper 1 (TH1)–biased humoral immunity that was specific for the coformulated ovalbumin (OVA) antigens (fig. S1). From these observations, we hypothesized that CDNs can expand antitumor cytotoxic T lymphocytes (CTL) elicited by cancer vaccines. We demonstrated that STINGVAX induced activation of dendritic cells (DCs) in vivo in the draining lymph nodes (DLNs), and observed that CDNs were comparable, if not superior, to lipopolysaccharide (LPS) in their capacity to activate DCs in the DLNs (Fig. 1A). GM-CSF–expressing vaccine cells alone did not induce a significant activation response in our in vivo assay (Fig. 1A, left panel). DC activation was associated with STING-dependent increase of phosphorylated IRF3 (Fig. 1A, right panel).
Fig. 1. STINGVAX is a potent cancer vaccine.

(A) STINGVAX can activate DLN DC via IRF3 phosphorylation. CDNs, STINGVAX, GM-vaccine (GM-vac), LPS with GM-vaccine, or phosphate-buffered saline (PBS) control was inoculated into naïve C57BL/6 mice, and DLNs from the site of injection were harvested after 3 days. DCs gated with CD11c and MHCII were examined for CD86 expression [% mean fluorescence intensity (MFI)]. Bone marrow–derived DCs were treated with C-di-AMP [CDNs (20 μg/ml) and LPS (10 μg/ml) for 24 hours], and the Western blot was probed with antibodies to pIRF3 and β-actin. STING−/− labeled samples were from goldenticket mice that have a functionally defective STING gene. (B) STINGVAX treatment of established B16 melanoma demonstrated a significant antitumor effect in vivo, and this effect was dose-dependent. Mice bearing palpable B16 melanoma were treated with a single injection of STINGVAX [using 2 to 200 μg of CDNs (CDA) per vaccine], with STINGVAX showing a better anti-tumor effect than GM-vaccine or CDNs at doses greater than 20 μg. (C) STINGVAX increased tumor-infiltrating CD8+ T lymphocytes. B16 tumors treated with either GM-vac or STINGVAX were stained with αCD8-FITC (fluorescein isothiocyanate) and counterstained with DAPI (4′,6-diamidino-2-phenylindole). Scale bar, 100 μm. CD8+IFNγ+ TILs from these treated mice were harvested and quantitated relative to tumor mass. Data are means ±SEM from five tumor samples. (D) STINGVAX increased the number of tumor-specific CTL in treated mice. In vivo CTL assay was performed on day 20. Splenocytes were isolated from each of the treated groups, and p15E peptide–specific in vivo T cell activity (y axis labeled as “% specific killing”) was measured. Data are means ±SEM, n = 3. (E) The in vivo efficacy of STINGVAX was STING-dependent. Palpable B16 tumors were treated in wild-type (WT) and goldenticket (STING−/−) mice. The animals were treated with STINGVAX (20 μg of CDA), and the tumors were compared as in (B). (F) STINGVAX (CDA) was used to treat tumor-bearing mice subjected to CD4 (GK1.5) or CD8 (c2.43) depletion (left panel). STINGVAX (CDA) antitumor efficacy was abrogated in IFNαR mice (right panel). For (B), (E), and (F), each group had 10 mice, and data represent means ± SEM. Each graph is representative of five experiments.
To test antitumor efficacy in vivo, we treated mice bearing palpable B16 melanoma tumors with a single dose of STINGVAX injected into the contralateral side from the site of established tumor. Compared to the unformulated GM-CSF–secreting tumor cell vaccine (GM-vaccine; see Materials and Methods) (28) and its combination with other control adjuvants including monophosphoryl lipid A (MPL), poly(I:C) (polyinosinic-polycytidylic acid), and R848, STINGVAX (formulated with CDA as the CDN) demonstrated an increased antitumor response (Fig. 1B and fig. S2). Dose-response experiments revealed that the antitumor effect of STINGVAX was diminished when formulated with less than 20 μg CDNs per 106 vaccine cells per mouse (Fig. 1B). Direct tumor cell killing was not observed with CDA alone (fig. S3), suggesting the requirement for vaccine cell–induced CD8+ T cell priming. When the tumor tissue was analyzed, STINGVAX-treated tumors had quantitatively increased CD8+ TILs (Fig. 1C). This increase in TILs in STINGVAX-treated mice correlated with increased tumor antigen–specific priming as demonstrated by in vivo CTL assay (Fig. 1D). STINGVAX efficacy was indistinguishable regardless of whether it was formulated with equimolar CDA or CDG (fig. S4).
To determine whether the increased antitumor efficacy of STINGVAX required STING signaling, we performed the B16 treatment assay in STING-mutant goldenticket mice and found that the antitumor efficacy of STINGVAX required functional STING (Fig. 1E). In addition, STINGVAX efficacy was diminished in CD8+ T cell–depleted mice (Fig. 1F, left panel), demonstrating that CD8+ T cells were essential for inhibition of tumor outgrowth. Depletion of CD4+ T cells only partially abrogated the antitumor effect, probably due to the depletion of both regulatory and nonregulatory CD4+ T cells (Fig. 1F, left panel). When we tested STINGVAX in IFNαR−/− mice, the antitumor effect was largely lost, demonstrating the importance of type I IFN in its efficacy (Fig. 1F, right panel).
We also tested STINGVAX in colon carcinoma (CT26), upper aero-digestive squamous cell carcinoma (SCCFVII), and pancreatic carcinoma models (Panc02), and found that the in vivo antitumor responses for STINGVAX were better than those for GM-vaccine in all the tumor models tested (Fig. 2). Once again, subcutaneous injection of CDN alone had no effect on the in vivo growth rate of these tumors. In a pancreatic cancer model, Panc02 tumor cells inoculated into the hemispleen metastasize to the liver via splenic vessels, and the mouse survival is directly dependent on liver metastatic burden (29, 30). As shown in Fig. 2C, STINGVAX increased the survival in this model of metastatic pancreatic cancer (31).
Fig. 2. STINGVAX induced antitumor responses in all the tumor models tested.

(A) Balb/c mice bearing palpable CT26 colon carcinoma were treated with STINGVAX (CDA), GM-vac, CDNs (CDA), and PBS controls. STINGVAX significantly decreased CT26 tumor growth compared to GM-vaccine or CDNs (CDA) alone in vivo. (B) C3H/HeOUJ mice bearing palpable SCCFVII tongue carcinomas were treated with STINGVAX, GM-vac (from SCCFVII–GM-vac), CDNs, and PBS controls. (C) STINGVAX was used in a liver metastasis model of Panc02 pancreatic carcinoma inoculated into a hemispleen. Cyclophosphamide (Cy) was injected to reduce regulatory T cells for all the groups. Survival was monitored for 90 days. STINGVAX-treated mice demonstrated significantly improved overall survival compared to control groups (log-rank test). (A) to (C) were replicated five times with 10 mice per group. Data are means ± SEM for (A) and (B).
Synthetic CDN derivatives have potent human immunostimulatory capacity
We then sought to develop synthetic CDN compounds with increased activity compared to the natural canonical STING ligands produced by bacteria or by host cGAS. We also wanted to address the polymorphism of the hSTING alleles that may not be responsive to bacterial CDNs (32). To reduce degradation by phosphodiesterases, dithio analogs of CDG, in which the nonbridging oxygens were replaced with sulfur atoms, were previously synthesized (33). Recognizing that the phosphorus atoms in the internucleotide phosphate bridge constitute a chiral center, we tested the immunostimulatory properties of purified [RP,RP] and [RP,SP] dithio CDA diastereomers, separated by preparatory high-performance liquid chromatography (Fig. 3A and fig. S5). The phosphodiesterase-resistant [RP,RP] dithio CDA diastereomer (RR-S2 CDA) induced higher levels of IFNβ in murine DC2.4 cells than the [RP,SP] dithio CDA diastereomer (RS-S2 CDA), which was comparable to the unmodified parent molecule (Fig. 3, B and C).
Fig. 3. RR-S2 CDNs have increased activity compared to canonical CDNs.

(A) Structure of RR-S2 and RS-S2 CDA diastereomers showing alternative positions of sulfur (S) atom substitutions for nonbridging oxygens. (B) Induction of IFNβ in mouse DC2.4 cells. IFNβ was measured using L929-ISRE IFN reporter cells. Data are means ± SD of five samples. HBSS, Hanks’ balanced salt solution. (C) The indicated CDN (100 μM) was incubated overnight with snake venom phosphodiesterase (SVPD; 0.1 μg/μl). After boiling and centrifugation, the capacity for the samples to induce IFNβ in DC2.4 cells was tested as in (B). (D) CDNs mixed with Effectene reagent (Qiagen) were added to BMMs isolated from WT or goldenticket (STING−/−) mice at 5 μM. After 6 hours of incubation, induced downstream expression of IRF3-dependent (IFNB1), NF-κB–dependent (TNF, IL-6), and STAT6-dependent (CCL2/MCP-1) proinflammatory cytokines was assessed by qRT-PCR, and relative expression was determined by comparison with unstimulated BMMs and Gapdh and Yhwaz reference genes.
Specific polymorphisms of hSTING have been shown to be refractory to bacterial CDNs with canonical 3′,5′ phosphate bridge linkages, but retain responsiveness to c[G(2′,5′pA(3′,5′)p] (2′-3′cGAMP) produced by host cell cGAS (16–18, 32). To advance STINGVAX for clinical trials, we synthesized CDN compounds that contained the same 2′-5′, 3′-5′ non-canonical (or mixed) linkage phosphate bridge as 2′-3′cGAMP (fig. S5). The noncanonical mixed linkage (ML) has been reported to increase the binding affinity for hSTING by at least 10-fold (34), resulting in a 20-Å ligand-induced conformational shift of the STING homodimer (35).
To determine the activity of these synthetic CDNs in vitro, we initially measured induction of TBK1-dependent (IFNβ), nuclear factor κB (NF-κB)–dependent [tumor necrosis factor–α (TNFα), interleukin-6 (IL-6)], and signal transducer and activator of transcription 6 (STAT6)–dependent [monocyte chemoattractant protein 1 (MCP-1)] cytokines in murine bone marrow macrophages (BMMs) by quantitative reverse transcription polymerase chain reaction (qRT-PCR). Dithio modification, but not the structure of the phosphate linkage, conferred the most robust induction of cytokines, whereas no cytokine expression was noted in STING-null mice (Fig. 3D).
In contrast to murine cells, synthetic CDNs containing a 2′-3′ ML phosphate bridge were more potent activators of human APCs compared to canonical CDNs (Fig. 4). In THP1 cells, which contain the HAQ STING allele (H71, A230, R232, 293Q), the ML phosphate bridge conferred increased signaling of both adenosine and guanosine nucleotide–based molecules compared to canonical CDNs (Fig. 4A). The [RP,RP] dithio modification of both canonical and noncanonical ML CDNs also increased IFN levels (Fig. 4A). When monocytes and DCs isolated from a donor homozygous for wild-type STING (hSTINGWT) (36) were tested, 2′-3′ CDNs potently stimulated the production of IFNα in human monocytes and DCs (Fig. 4B). Additionally, all of the CDNs tested enhanced major histocompatibility complex class I (MHCI) and costimulatory marker (CD80, CD83, and CD86) expression to a similar extent as did LPS (Fig. 4C).
Fig. 4. ML–RR-S2–CDA potently activates human APCs.

(A) Human THP1-Blue cells with IRF3 reporter gene were stimulated with 50 μM of CDNs, and the concentration of secreted embryonic alkaline phosphatase (SEAP) reporter was measured by spectrophotometry. Histogram represents mean ± SD of triplicate samples. (B) PBMCs from a WT STING human donor were stimulated with 50 μM of the indicated CDNs. After 24 hours, intracellular IFNα on gated CD14+ monocytes or on CD11c+HLA-DR+ DCs (mDCs) was measured by fluorescence-activated cell sorting (FACS). (C) Cultured human DCs were stimulated with 50 μM synthetic CDNs or LPS (1 μg/ml) as a positive control. After 48 hours, MHCI (HLA-ABC), CD80, CD83, and CD86 were measured on the gated CD11c+ DCs. Bar graphs indicate the average MFI (top panel); representative histograms show CD80, CD86, CD83, and MHCI expression in human DCs. Filled histograms correspond to unstimulated cells, the dotted line represents LPS stimulation, and the solid line represents stimulation with ML–RR-S2–CDA (bottom). (D) Synthetic noncanonical CDNs can stimulate all the hSTING alleles with increased potency. Human PBMCs from four donors, each with a different STING genotype (STINGWT, STINGHAQ, STINGWT/REF, or STINGHAQ/REF), were stimulated with 10 μM of the indicated CDNs. After 6 hours of incubation, the supernatants were harvested for analysis of TNFα protein (top), and the cells were harvested for analysis of IFNβ induction by qRT-PCR (bottom). (E) CDN-stimulated DCs can activate TH1 response in human T cells. Human DCs (CD11c+ cells) were probed for IL-12 expression after treatment with GM-CSF, LPS, or ML–RR-S2–CDA. Human DCs treated with GM-CSF, LPS, or ML–RR-S2–CDA were used in MLRs to stimulate human T cells (gated for CD8+ cells). Intracellular IFNγ was measured in CD8+-gated cells. Left panels display mean MFI (IL-12, left; IFNγ, right), and right panels show representative histograms from gated flow analysis. Data are means ± SD of five samples.
We tested these synthetic CDNs on peripheral blood mononuclear cells (PBMCs) from other donors with different STING genotypes (STINGWT, STINGHAQ, STINGWT/REF, and STINGHAQ/REF), and we found that they can stimulate human monocytes with greater potency than canonical CDNs, regardless of the STING genotypes (Fig. 4D) (37). CDG and CDA containing both the ML and dithio configuration robustly induced type I IFN transcript in all the genotypes tested, whereas both canonical and noncanonical CDA containing the dithio modification appeared superior at inducing secretion of TNFα.
Because these synthetic CDNs activated human APC, we sought to determine whether DCs activated by CDNs could stimulate TH1 responses in vitro. Consistent with the human APC activation data shown above, our synthetic ML RR-CDN also induced a potent enhancement of IL-12 on human DCs (Fig. 4E). When these activated DCs were used in mixed lymphocyte reaction (MLR), CDN-treated DCs induced an enhanced IFNγ response that was stronger than the response induced with GM-CSF–treated DCs (Fig. 4E).
STINGVAX with modified CDNs has enhanced antitumor efficacy
Given these in vitro findings showing increased STING signaling with our rationally designed CDNs, we compared STINGVAX potency when formulated with RR-S2 CDA or with canonical CDA. STINGVAX formulated with RR-S2 CDA conferred higher efficacy in both the B16 and TRAMP treatment models as compared to the parental CDA molecule (Fig. 5A). Because murine STING lacks the polymorphisms of hSTING alleles, we compared two formulations of STINGVAX (RR-S2 CDA versus ML–RR-S2–CDA) and, as expected, found only modest differences in their antitumor activity (fig. S6). To test whether the enhanced in vivo responses seen in Fig. 5A were activating the STING pathway, we performed Western blots to test whether RR-S2 CDA enhanced IRF3 phosphorylation in vivo (Fig. 5B). Additionally, after incubating murine DCs with CDA and RR-S2 CDA, we detected enhanced IFNα expression with RR-S2 CDA in comparison to the canonical CDA (Fig. 5C). We did observe some increased IFNα response in goldenticket mice with RR-CDA that may be potentially STING-independent. Cumulatively, these findings demonstrated that CDNs with RR dithio linkage would provide improved STING-dependent IRF3 and type I IFN responses as a vaccine adjuvant in pre-clinical settings, whereas the ML and RR-S2 modifications should be appropriate in clinical trials.
Fig. 5. Synthetic RR-S2 CDA increased STINGVAX’s potency.

(A) STINGVAX was formulated with either canonical CDA or RR-S2 CDA, with equimolar CDN amounts per mouse (20 μg per vaccine), and used to treat palpable B16 and TRAMP tumors. To improve the sensitivity of the in vivo treatment assay between the different STINGVAX formulations, we increased the initial tumor burden for the B16 tumor to 105 cells per inoculation for these experiments. Each group had 10 mice, and data represent means ± SEM. Each graph is representative of three to five experiments. (B) WT and goldenticket (STING−/−) bone marrow–derived murine DCs were incubated with CDA, and the cell lysates were probed with pIRF3 antibody. (C) DCs harvested from lymph nodes of WT and goldenticket (GT) mice were incubated with CDNs overnight, and their IFNα levels were quantitated by flow cytometry. Shown are the data for CD11c+B220+-gated DCs and the MFI change relative to untreated WT DC controls (WT). Bottom panel illustrates the IFNα MFI change relative to WT untreated DC (WT), and the top panel is a representative histogram. Three replicates were performed, and data represent means ± SEM.
STINGVAX with PD-1 blockade cures mice bearing established tumors
Although we showed that STINGVAX is a potent, antigen-specific CTL-generating vaccine, we were concerned that the induction of immune checkpoint molecules within the tumor microenvironment (TME) might blunt antigen-specific T cell responses generated against the tumor. When we probed the TME for the up-regulation of PD-L1 on tumor cells in vaccine-treated mice, we found that CDN-dependent CD8+IFNγ+ T cell infiltration correlated with an increased expression of PD-L1 (Fig. 6A). Mice treated with IFNγ neutralizing antibody showed no up-regulation of PD-L1, consistent with an adaptive immune resistance mechanism for PD-L1 induction on tumors (fig. S7). We reasoned, therefore, that blocking the PD-1–PD-L1 interaction would unleash the tumor-specific CTL response. PD-1–blocking antibody and GM-vac alone modestly enhanced the antitumor response compared to the HBSS control group (Fig. 6B, upper panel). However, when PD-1–blocking antibody was combined with STINGVAX formulated with RR-S2 CDA, much improved antitumor responses were observed, with some of the established B16 tumors showing regression (Fig. 6B, lower panels). When we tested this combinatorial therapy in the CT26 model, all established tumors regressed (Fig. 7). In these mice that showed regression of the primary tumor, a second inoculation of CT26 cells resulted in no tumor growth, demonstrating that this combined therapy resulted in long-term tumor-specific memory (fig. S8). Notably, neither B16 nor CT26 regressed in response to anti–PD-1 treatment alone when treatment was started in mice with established tumors (Figs. 6 and 7).
Fig. 6. STINGVAX can up-regulate PD-L1.

(A) STINGVAX treatment induces up-regulation of PD-L1 on tumors, correlating with increased infiltration of IFNγ+CD8+ T cells. Scale bars, 100 μm. Harvested tumor tissues from treated mice were probed with CD8, IFNγ, and PD-L1 conjugates, followed by DAPI counter-stain. Colocalized CD8 and IFNγ costaining positive cells are shown in yellow; CD8, green (FITC); IFNγ, red [phycoerythrin (PE)]. PD-L1–PE conjugate (red) was used for the right panel. DAPI (blue) was used as a counterstain. The absolute number of CD8+IFNγ+ and PD-L1+ cells per mass of tumor tissue is shown in the right panels. Data are means ±SEM from five samples. (B) Combination of STINGVAX (with CDA) and PD-1–blocking antibody slightly improved the antitumor response of established B16 tumors compared to the STINGVAX, but did not reach statistical significance (top panel). However, the combination of STINGVAX (RR-S2 CDA) + anti–PD-1 significantly improved the antitumor response when compared against the combination of STINGVAX (CDA) + anti–PD-1. The smaller panels on the bottom right are volume plots of individual mice (overlapping curves represent multiple mice with regression). Each group had 10 mice, and data represent means ± SEM. The graph is representative of five experiments. n.s., not significant.
Fig. 7. STINGVAX with PD-1 blockade can induce tumor regression.
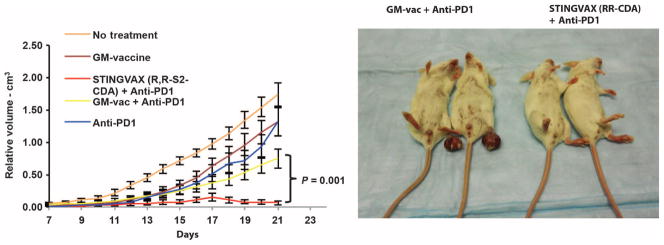
Combination of STINGVAX (RR-S2 CDA) and PD-1–blocking antibody can cure all of the mice with established CT26 tumors. The right panel depicts the mice and tumors at day 20. Each group had 10 mice, and data represent means ± SEM. The graph is representative of five experiments.
DISCUSSION
The potent IFN responses elicited from cytosolic dsDNA as danger signals, as well as pathogen-derived CDNs, have evolved to demarcate “affected” cells that may require closer immunosurveillance (36). With the emergence of STING as a central regulator of this innate immune response, we have engineered STINGVAX, a combinatorial cancer vaccine that targets the STING pathway and colocalizes CDNs with tumor antigens and GM-CSF to amplify tumor-specific CTL in the context of established tumor. A critical element of our vaccine design revolves around the structural specificity of CDNs binding to STING. [RP,RP] configuration (RR-S2 CDA) in combination with the noncanonical 2′,3′ phosphate bridge linkage alterations can enhance hSTING activation. Our report illustrates the tight link between the structural binding mechanisms of CDNs to STING and the activation of innate immunity to generate clinically efficacious adjuvants for cancer vaccines.
Our study is limited by using non-autochthonous models that may not directly address toleragenic TME. However, all our transplanted tumor treatments were initiated once the tumors were palpable, by which point tumor tolerance should be already established. Future preclinical studies will need to be directed toward OVA or hemagglutinin transgenic models amenable to a more robust inquiry into how CDNs affect antigen-specific tolerance. This report is also limited in mechanistic details on how CDNs confer increased immunologic potency when coformulated with GM-vaccine cells. Ween-vision several possibilities that include (i) activation of the STING in the tumor cell vaccine to activate recruited DCs, (ii) MHC up-regulation in STINGVAX to activate pre-existing tumor-primed T cells, and (iii) the tumor cell vaccine simply serving as a CDN depot. These possible mechanisms of action are not mutually exclusive and will need to be investigated in future studies.
The ease of STINGVAX formulation, the proven safety profile of GM-CSF–secreting vaccines in multiple clinical trials, and the recent efficacy of these cellular vaccine platforms combined with Listeria vaccines in pancreatic cancer patients all provide rationale for translation of STINGVAX (6). Although a recent report showed that CDG can provide therapeutic benefit to 4T1 tumors, these canonical CDNs have limited translational value given the presence of hSTING alleles that are refractory to canonical CDNs (38). Furthermore, although this study suggested direct tumor cell toxicity as the mechanism, we did not observe this in our in vitro or in vivo studies. We had initial concerns about potential STINGVAX toxicity because it is a potent inducer of IFN. However, none of the mice injected with STINGVAX showed gross evidence of cytokine storm, even when the dose of CDNs reached 200 μg per mouse.
The up-regulation of PD-L1 on the tumor and the associated ability to induce regression of tumors treated with the combination of STINGVAX and anti–PD-1 illustrate the adaptive immune resistance mechanism (25, 27). As a potent inducer of IFNγ-secreting T cells and PD-L1 in the TME, STINGVAX (ML–RR-S2–CDA) is well suited for clinical combination with immune checkpoint–blocking antibodies, particularly anti–PD-1. The recent clinical trials with nivolumab and ipilimumab demonstrated durable responses in advanced cancer patients, but the overall response rates ranged from 20 to 40% of the patients treated (24). Because preclinical studies have shown improved in vivo responses when vaccines are combined with both PD-1 and CTLA-4 blockade (39), combining multiple immune checkpoint inhibitors with STINGVAX is also a valid clinical strategy. Recent reports that cumulatively demonstrated colocalization of CD3, IFNγ, and PD-L1 in multiple human tumor types complement our mechanistic studies on the induction of PD-L1 on tumor cells (40). Cumulatively, our findings justify the need for combining STINGVAX with immune checkpoint inhibition in clinical trials.
MATERIALS AND METHODS
Study design
This was a preclinical study of CDNs as adjuvants for cancer vaccines. We hypothesized that CDNs formulated with an APC-mobilizing vaccine in the form of STINGVAX can induce a potent IFN response in the TME to render a tumor more responsive to checkpoint inhibition. We used multiple murine models to test if STINGVAX can induce an antitumor response in vivo in established tumors. Each treatment and control group had 10 mice per group, and the experiments were replicated at least three to five times. To ensure translatability of CDNs, rationally synthesized CDNs were screened for optimal IFN responses in human myeloid cells. We also modeled the human TME to ensure that these myeloid-activating CDNs can induce IFNγ responses in human lymphocytes. Finally, we formulated these synthetic CDNs into STINGVAX and combined them with PD-1 blockade in murine models that were refractory to PD-1 blockade alone.
Mice, cell lines, and antibodies
Six- to 10-week-old female C57BL/6, Balb/c, C3H/HeOUJ, C57BL/6-Tg 8247Ng/J (TRAMP), and C57BL/6J-Tmem173gt/J (goldenticket) mice (The Jackson Laboratory) were housed according to the rules of the Johns Hopkins Hospital Animal Care and Use Committee. B16, CT26, SCCFVII, Panc02, B16 GM-vaccine, SCCFVII GM-vaccine, and Panc02 GM-vaccine were cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS). GM-vaccine cells were prepared from GM-CSF–expressing autologous tumor cells that were lethally irradiated with 150 Gy before injection. CT26 GM-vaccine consisted of lethally irradiated CT26 and allogeneic B78H1 secreting GM-CSF. For all the vaccines, GM-CSF expression ranged from 50 to 500 ng per 106 cells per 24 hours. Antibodies against CD3, CD4, CD8, CD11c, CD14, HLA-DR, MHCII, IFNα, IFNγ, CD80, CD83, and CD86 were purchased from BD Biosciences. CD11c+ cells were isolated by anti-mouse CD11c MicroBeads (Miltenyi). GK1.5 and clone 2.43 (InVivoMAb) were used to deplete CD4 and CD8, respectively. Hybridoma expressing PD-1–blocking antibody (clone G4) was obtained from C. Drake.
Vaccine formulations
STINGVAX was prepared by incubating 2 to 200 μg of CDNs with lethally irradiated (150 Gy) GM-vaccine cells (106) at 4°C for 30 min. STINGVAX for each tumor type was formulated from each of the GM-vaccine derived from B16, CT26, SCCFVII, TRAMP, and Panc02 tumor lines. CDNs used for STINGVAX formulation consisted of CDA, CDG, RR-S2 CDA, or ML–RR-S2–CDA. The standard CDN amount used was 20 μg per vaccine. STINGVAX was injected subcutaneously into the contralateral limb of tumor-bearing mice. For the Panc02 model, the vaccines were injected subcutaneously into the flanks after cyclophosphamide injection.
In vivo tumor models
C57BL/6 mice were injected with 1 × 104 to 5 × 104 B16 cells in the footpads. In some cases, 105 B16 were injected to increase the tumor burden at the time of treatment initiation. Once palpable tumor developed (7 to 10 days), 100 μl of 106 B16 GM-vaccine or STINGVAX was injected subcutaneously into the contralateral limb. Palpable tumor measurements were initiated once all three dimensions reached between 2 and 4 mm, and the tumor volume was calculated by the following formula: length (mm) × width (mm) × height (mm) × 0.5326 × 0.01 (41). C3H/HeOUJ mice and Balb/c mice were used with SCCFVII/SF and CT26 cells, respectively, with comparable methods (42). Tumors were measured daily. For the Panc02 hemispleen model, C57BL/6 mice were inoculated with 2 × 106 Panc02 tumor cells into a single hemispleen, thereby seeding the liver via splenic vessels (43). A single dose of cyclophosphamide (100 mg/kg) was administered intraperitoneally on day 3 after tumor injection for all the groups. Panc02 STINGVAX was administered daily on days 4, 7, and 14. For the PD-1 blockade experiments, anti–PD-1 (G4) (100 μg per mouse per injection) was injected intraperitoneally twice a week once the tumor was palpable. Neutralizing IFNγ (Bio-XCell) antibody was given intraperitoneally twice a week during the duration of the vaccine treatments in some experiments.
Western blot
Murine DCs (BMM or DLN) were treated with CDNs (20 μg/3 ml) for 24 hours, rinsed, lysed [radioimmunoprecipitation assay (RIPA), Sigma, 20 min on ice], and fractionated (20 μg of protein per lane) with 10% SDS–polyacrylamide gel electrophoresis gel. After transferring onto nitrocellulose membrane and blocking [TBST—5% nonfat milk, 50 mM tris-HCl (pH 7.5), 150 mM NaCl, 0.1% Tween 20], the membrane was probed with phospho-IRF3 (Millipore) and β-actin (Cell Signaling) antibodies. The membranes were washed three times with TBST and incubated with the appropriate horseradish peroxidase–conjugated secondary antibody (1 hour, room temperature), and the peroxidase activity was detected with enhanced chemiluminescence reagent (Pierce).
In vitro leukocyte activation assays
CD3- and CD19-depleted (Dynabeads) DCs were cultured with GolgiStop (BD) and LPS (0.1 μg/ml) or CDNs (2 to 20 μg/ml) for 5 hours. DCs were stained for anti-mouse CD11c, CD86, CD80, and MHCII. Gated DC population (CD11c+MHCII+) was probed for intracellular cytokine analysis (IL-12, IFNα/β, and TNFα) after permeabilization with Cytokit (BD). For the lymphocyte analysis, harvested splenocytes or lymph node lymphocytes were stimulated with phorbol 12-myristate 13-acetate (PMA; 1 μM/ml) and ionomycin (1 μg/ml) and GolgiStop. Suspended cells were stained for mouse CD3, CD4, CD8, and intracellular IFNγ. Isotype controls were used to delineate positive staining.
IFN induction assay
THP1-Blue cells (4 × 105), a human monocyte cell line stably transfected with an IRF-inducible SEAP reporter gene (InvivoGen), were incubated with 50 μM CDNs or controls for 30 min at 37°C. After 30 min, cells were washed, plated in RPMI containing 10% FBS, and incubated at 37°C with 5% CO2. Twenty μl of the supernatant was collected after overnight incubation and added to 180 μl of QUANTI-Blue reagent (InvivoGen). After 45 min of incubation, absorbance (655 nm) measurements were taken. Data are representative of at least two independent experiments. For the luciferase assay, mouse DC2.4 cells (1× 105) were incubated with CDNs or controls for 30 min at 37°C. Cells were washed with RPMI containing 10% FBS, and incubated at 37°C with 5% CO2 for 4 hours. Ten μl of supernatants were incubated with L929 cells (5 ×104). After 4 hours, lucerifase activity was quantified by addition of luciferin reagent (Promega) and reading on a luminometer.
Stimulation of human monocytes and DCs
Human PBMCs obtained from healthy donors were stimulated with 50 μM CDNs for 6 hours, followed by brefeldin A for an additional 16 hours. Gated monocytes (CD14+) or mDCs (CD11c+, HLA-DR+) were stained with intracellular anti-IFNα. Human CD11c+ cells were cultured with GM-CSF (50 ng/ml) and IL-4 (25 ng/ml) (R&D) for 7 days and then stimulated for 48 hours with LPS (1 μg/ml) or 50 μM CDNs. Surface expression of MHCI, CD80, CD83, and CD86 was determined by FACS. In some cases, human PBMCs obtained from donors with all the different STING alleles (HemaCare) were sequenced as follows: genomic DNA isolated from 104 PBMCs (Epicentre) was used to amplify regions of exons 3, 6, and 7 of hSTING. Primers for amplification were as follows: hSTING exon 3, GCTGAGACAGGAGCTTTGG (forward) and AGCCAGAGAGGTTCAAGGA (reverse); hSTING exon 6, GGCCAATGACCTGGGTCTCA (forward) and CACCCAGAATAGCATCCAGC (reverse); hSTING exon 7, TCAGAGTTGGGTATCAGAGGC (forward) and ATCTGGTGTGCTGGGAAGAGG (reverse). The same primers used for amplification were used for sequencing. Genotypes were determined as outlined in (32). For qRT-PCR assay for IFNβ1 expression, 106 cells were stimulated with 10 μM CDNs. Relative normalized expression was determined with unstimulated cells serving as a control for each donor, and GUSB and PGK1 were used as reference genes. TNFα protein in the cell supernatants was measured using CBA Flex Sets according to the manufacturer’s instructions.
Real-time RT-PCR assay on BMMs
BMMs were isolated from tibias and femurs of C57BL/6 and goldenticket mice and cultured in RPMI medium with 5% colony stimulating factor 1, 5% FBS, 1× L-glutamine, and 1× penicillin/streptomycin for 7 days before use. BMMs (106) were stimulated with CDNs at 5 μM in HBSS with the addition of Effectene (Qiagen) transfection reagent (per kit protocol) for 6 hours at 37°C, 5% CO2. After 6 hours of incubation, the gene expression of type I IFNβ (Ifnβ1), proinflammatory cytokines MCP-1 (Ccl2), Tnfα, and Il-6 was assessed by RT-PCR using the PrimePCR RNA purification and cDNA analysis system, run on a CFX96 gene cycler (Bio-Rad). Relative normalized expression was determined for each gene (with wild-type unstimulated BMMs as a control) to account for the different efficiencies of PCR amplification for the target (Etarget) and the reference (Ereference) and transform the logarithmic scaled raw data unit of cycle threshold (CT) into the linear unit of normalized expression. The reference genes used were Gapdh and Ywhaz, confirmed to have a coefficient variable (CV) below 0.5 and an M value below 1, and thus not vary with different treatment conditions. Data are representative of two independent experiments.
In vivo CTL assay
Splenocytes were labeled with 0.5 and 5 μM carboxyfluorescein succinimidyl ester (CFSE; Molecular Probes). The 5 μM CFSE–labeled cells were pulsed with P15E (10 μg/ml) (KSPWFTTL) peptide. The 0.5 μM CFSE–labeled cells were pulsed with β-galactosidase (β-gal) peptide (TPHPARIGL). Mice were injected intravenously with a 1:1 mixture of these cells, and the splenocytes were isolated after 24 hours and analyzed by flow cytometry. Antigen-specific killing was calculated using the following formula: .
Immunohistofluorescence
Frozen sections (10 μm thick) were fixed with acetone and blocked with 1% bovine serum albumin (BSA) for 30 min at room temperature. For paraffin-embedded tissue, the sections were fixed in 4% paraformaldehyde before blocking with 1% BSA. αCD4 and αCD8 FITC conjugates and αIFNγ and αPD-L1 primary antibodies were incubated for 1 hour at 4°C. Cy3 conjugate antibody was used as secondary antibody in some cases. DAPI was used as the nuclear counterstain. Positive cells in 10 randomly selected fields at ×40 magnification were quantitated. Images were taken on a Nikon Eclipse E800 microscope with a Nikon DS-Qi1Mc camera and NIS-Elements Advanced Research 3.0 software.
Statistical analysis
We used paired t test to calculate two-tailed P values to estimate the statistical significance of differences between two treatment groups using Excel software. Error bars are SEM. P values are labeled in the figures. Kaplan-Meier curves were generated using GraphPad Prism software and analyzed with log-rank test.
Supplementary Material
Acknowledgments
We thank C. Drake and B. Francisca for reagents (clone G4) and for useful discussions.
Funding: NIH R01 CA178613 (Y.K.); Melanoma Research Alliance (Y.K. and D.M.P.); 1PO1 AI63302 and 1R01 AI27655 (D.A.P.); NIH K23 CA148964-01, Viragh Foundation and the Skip Viragh Pancreatic Cancer Center at Johns Hopkins, and the National Cancer Institute Specialized Programs of Research Excellence (SPORE) in Gastrointestinal Cancers P50 CA062924 (L.Z.). Y.K. has received a sponsored research grant from Aduro Biotech Inc.
Footnotes
www.sciencetranslationalmedicine.org/cgi/content/full/7/283/283ra52/DC1
Materials and Methods
Fig. S1. CDNs can induce cellular and humoral immunity.
Fig. S2. CDNs are more potent adjuvants than TLR agonists.
Fig. S3. CDNs do not kill tumor cells directly.
Fig. S4. CDA and CDG are equally effective when formulated into STINGVAX.
Fig. S5. CDNs with dithiophosphate backbone can be synthesized.
Fig. S6. 2′-3′ linkage did not have improved antitumor efficacy in the murine system.
Fig. S7. PD-L1 induction with STINGVAX treatment is IFNγ-dependent.
Fig. S8. STINGVAX-treated mice become immune to repeat tumor challenge.
Reference (44)
Author contributions: J.F. designed and performed the experiments. D.B.K., M.L., L.H.G., S.M.M., E.L., K.M., J.J.L., P.L., W.L., K.E.S., Q.Z., K.C.S., and L.Z. performed the experiments. D.A.P., J.J.W., and D.M.P. designed the experiments. D.M.P., T.W.D.J., and Y.K. designed and wrote the manuscript.
Competing Interests: D.M.P. is an uncompensated consultant for Amplimmune, Aduro Biotech, and ImmuneXcite. D.A.P. has a consulting relationship with and a financial interest in Aduro Biotech, and Y.K., D.A.P., and the company stand to benefit from the commercialization of the results of this research.
Data and materials availability: Synthetic CDNs and GM-vaccines are available upon request. Cells, mice, antibodies, and cited reagents are all commercially available.
REFERENCES AND NOTES
- 1.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, Vogel AN, Kalos M, Riley JL, Deeks SG, Mitsuyasu RT, Bernstein WB, Aronson NE, Levine BL, Bushman FD, June CH. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4:132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steinman RM. Decisions about dendritic cells: Past, present, and future. Annu Rev Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 4.Mellman I. Dendritic cells: Master regulators of the immune response. Cancer Immunol Res. 2013;1:145–149. doi: 10.1158/2326-6066.CIR-13-0102. [DOI] [PubMed] [Google Scholar]
- 5.Dubensky TW, Jr, Reed SG. Adjuvants for cancer vaccines. Semin Immunol. 2010;22:155–161. doi: 10.1016/j.smim.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 6.Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, Springett G, Morse M, Zeh H, Cohen D, Fine RL, Onners B, Uram JN, Laheru DA, Lutz ER, Solt S, Murphy AL, Skoble J, Lemmens E, Grous J, Dubensky T, Jr, Brockstedt DG, Jaffee EM. Safety and survival With GVAX pancreas prime and Listeria monocytogenes–expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol. 2015 doi: 10.1200/JCO.2014.57.4244. JCO.2014.57.4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karaolis DK, Means TK, Yang D, Takahashi M, Yoshimura T, Muraille E, Philpott D, Schroeder JT, Hyodo M, Hayakawa Y, Talbot BG, Brouillette E, Malouin F. Bacterial c-di-GMP is an immunostimulatory molecule. J Immunol. 2007;178:2171–2181. doi: 10.4049/jimmunol.178.4.2171. [DOI] [PubMed] [Google Scholar]
- 8.McWhirter SM, Barbalat R, Monroe KM, Fontana MF, Hyodo M, Joncker NT, Ishii KJ, Akira S, Colonna M, Chen ZJ, Fitzgerald KA, Hayakawa Y, Vance RE. A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J Exp Med. 2009;206:1899–1911. doi: 10.1084/jem.20082874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barker JR, Koestler BJ, Carpenter VK, Burdette DL, Waters CM, Vance RE, Valdivia RH. STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. MBio. 2013;4:e00018–13. doi: 10.1128/mBio.00018-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science. 2010;328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamayo R, Pratt JT, Camilli A. Roles of cyclic diguanylate in the regulation of bacterial pathogenesis. Annu Rev Microbiol. 2007;61:131–148. doi: 10.1146/annurev.micro.61.080706.093426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, Vance RE. The N-ethyl-N-nitrosourea-induced goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun. 2011;79:688–694. doi: 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barber GN. Cytoplasmic DNA innate immune pathways. Immunol Rev. 2011;243:99–108. doi: 10.1111/j.1600-065X.2011.01051.x. [DOI] [PubMed] [Google Scholar]
- 15.Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 2012;5:ra20. doi: 10.1126/scisignal.2002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol. 2013;14:19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- 17.Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 2013;3:1355–1361. doi: 10.1016/j.celrep.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, Hopfner KP, Ludwig J, Hornung V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380–384. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, Tuschl T, Patel DJ. Cyclic [G(2′,5′)pA(3′,5′)p] Is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 2013;153:1094–1107. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type-I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ebensen T, Libanova R, Schulze K, Yevsa T, Morr M, Guzmán CA. Bis-(3′,5′)-cyclic dimeric adenosine monophosphate: Strong Th1/Th2/Th17 promoting mucosal adjuvant. Vaccine. 2011;29:5210–5220. doi: 10.1016/j.vaccine.2011.05.026. [DOI] [PubMed] [Google Scholar]
- 22.Madhun AS, Haaheim LR, Nøstbakken JK, Ebensen T, Chichester J, Yusibov V, Guzman CA, Cox RJ. Intranasal c-di-GMP-adjuvanted plant-derived H5 influenza vaccine induces multi-functional Th1 CD4+ cells and strong mucosal and systemic antibody responses in mice. Vaccine. 2011;29:4973–4982. doi: 10.1016/j.vaccine.2011.04.094. [DOI] [PubMed] [Google Scholar]
- 23.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, Chen L. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, Activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu J, Malm IJ, Kadayakkara DK, Levitsky H, Pardoll D, Kim YJ. Preclinical evidence that PD1 blockade cooperates with cancer vaccine TEGVAX to elicit regression of established tumors. Cancer Res. 2014;74:4042–4052. doi: 10.1158/0008-5472.CAN-13-2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, Jackson V, Hamada H, Pardoll D, Mulligan RC. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte–macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshimura K, Jain A, Allen HE, Laird LS, Chia CY, Ravi S, Brockstedt DG, Giedlin MA, Bahjat KS, Leong ML, Slansky JE, Cook DN, Dubensky TW, Pardoll DM, Schulick RD. Selective targeting of antitumor immune responses with engineered live-attenuated Listeria monocytogenes. Cancer Res. 2006;66:1096–1104. doi: 10.1158/0008-5472.CAN-05-2307. [DOI] [PubMed] [Google Scholar]
- 30.Soares KC, Foley K, Olino K, Leubner A, Mayo SC, Jain A, Jaffee E, Schulick RD, Yoshimura K, Edil B, Zheng L. A preclinical murine model of hepatic metastases. J Vis Exp. 2014;51677 doi: 10.3791/51677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leao IC, Ganesan P, Armstrong TD, Jaffee EM. Effective depletion of regulatory T cells allows the recruitment of mesothelin-specific CD8+ T cells to the antitumor immune response against a mesothelin-expressing mouse pancreatic adenocarcinoma. Clin Transl Sci. 2008;1:228–239. doi: 10.1111/j.1752-8062.2008.00070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yi G, Brendel VP, Shu C, Li P, Palanathan S, Cheng Kao C. Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PLOS One. 2013;8:e77846. doi: 10.1371/journal.pone.0077846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan H, Wang X, KuoLee R, Chen W. Synthesis and immunostimulatory properties of the phosphorothioate analogues of cdiGMP. Bioorg Med Chem Lett. 2008;18:5631–5634. doi: 10.1016/j.bmcl.2008.08.088. [DOI] [PubMed] [Google Scholar]
- 34.Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, Hartmann G, Barchet W, Tuschl T, Patel DJ. Structure-function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell. 2013;154:748–762. doi: 10.1016/j.cell.2013.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell. 2013;51:226–235. doi: 10.1016/j.molcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vance RE, Isberg RR, Portnoy DA. Patterns of pathogenesis: Discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe. 2009;6:10–21. doi: 10.1016/j.chom.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dubensky TW, Jr, Kanne DB, Leong ML. Rationale, progress and development of vaccines utilizing STING-activating cyclic dinucleotide adjuvants. Ther Adv Vaccines. 2013;1:131–143. doi: 10.1177/2051013613501988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO, Zhou J, Hayakawa Y, Karaolis DK, Gravekamp C. STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol Res. 2014;2:901–910. doi: 10.1158/2326-6066.CIR-13-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T cell rejection function in tumors. Cancer Res. 2013;73:3591–3603. doi: 10.1158/0008-5472.CAN-12-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti–PD-1 therapy. Clin Cancer Res. 2014;20:5064–5074. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24:148–154. doi: 10.1007/BF00300234. [DOI] [PubMed] [Google Scholar]
- 42.Davis MB, Vasquez-Dunddel D, Fu J, Albesiano E, Pardoll D, Kim YJ. Intratumoral administration of TLR4 agonist absorbed into a cellular vector improves antitumor responses. Clin Cancer Res. 2011;17:3984–3992. doi: 10.1158/1078-0432.CCR-10-3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng L, Foley K, Huang L, Leubner A, Mo G, Olino K, Edil BH, Mizuma M, Sharma R, Le DT, Anders RA, Illei PB, Van Eyk JE, Maitra A, Laheru D, Jaffee EM. Tyrosine 23 phosphorylation-dependent cell-surface localization of annexin A2 is required for invasion and metastases of pancreatic cancer. PLOS One. 2011;6:e19390. doi: 10.1371/journal.pone.0019390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaffney BL, Veliath E, Zhao J, Jones RA. One-flask syntheses of c-di-GMP and the [Rp,Rp] and [Rp,Sp] thiophosphate analogues. Org Lett. 2010;12:3269–3271. doi: 10.1021/ol101236b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.