

Abstract
HIV-2 and SIVMAC are AIDS-causing, zoonotic lentiviruses that jumped to humans and rhesus macaques, respectively, from SIVSM-bearing sooty mangabey monkeys. Cross-species transmission events such as these sometimes necessitate virus adaptation to species-specific, host restriction factors such as TRIM5. Here, a new human restriction activity is described that blocks viruses of the SIVSM/SIVMAC/HIV-2 lineage. Human T, B, and myeloid cell lines, peripheral blood mononuclear cells and dendritic cells were 4 to >100-fold less transducible by VSV G-pseudotyped SIVMAC, HIV-2, or SIVSM than by HIV-1. In contrast, transduction of six epithelial cell lines was equivalent to that by HIV-1. Substitution of HIV-1 CA with the SIVMAC or HIV-2 CA was sufficient to reduce HIV-1 transduction to the level of the respective vectors. Among such CA chimeras there was a general trend such that CAs from epidemic HIV-2 Group A and B isolates were the most infectious on human T cells, CA from a 1° sooty mangabey isolate was the least infectious, and non-epidemic HIV-2 Group D, E, F, and G CAs were in the middle. The CA-specific decrease in infectivity was observed with either HIV-1, HIV-2, ecotropic MLV, or ALV Env pseudotypes, indicating that it was independent of the virus entry pathway. As2O3, a drug that suppresses TRIM5-mediated restriction, increased human blood cell transduction by SIVMAC but not by HIV-1. Nonetheless, elimination of TRIM5 restriction activity did not rescue SIVMAC transduction. Also, in contrast to TRIM5-mediated restriction, the SIVMAC CA-specific block occurred after completion of reverse transcription and the formation of 2-LTR circles, but before establishment of the provirus. Transduction efficiency in heterokaryons generated by fusing epithelial cells with T cells resembled that in the T cells, indicative of a dominant-acting SIVMAC restriction activity in the latter. These results suggest that the nucleus of human blood cells possesses a restriction factor specific for the CA of HIV-2/SIVMAC/SIVSM and that cross-species transmission of SIVSM to human T cells necessitated adaptation of HIV-2 to this putative restriction factor.
Author Summary
HIV-1 and HIV-2, the two lentiviruses that cause AIDS in humans, are members of a family of such viruses that infect African primates. HIV-1 is a zoonosis that was transmitted to humans from chimpanzees. HIV-2 was transmitted to humans from sooty mangabey monkeys. In several documented cases of cross-species transmission of lentiviruses it has been shown that replication of the virus in the new host species necessitated that the virus adapt to species-specific antiviral factors in the host. Here we report that human blood cells possess an antiviral activity that exhibits specificity for viruses of the HIV-2/SIVMAC/SIVSM lineage, with restriction being greatest for SIVSM and the least for epidemic HIV-2. Here we show that this dominant-acting, antiviral activity is specific for the capsid and blocks the virus after it enters the nucleus. The evidence suggests that, in order to jump from sooty mangabey monkeys to humans, the capsid of these viruses changed in order to adapt to this antiviral activity. In keeping with the practice concerning anti-lentiviral activities we propose to call this new antiviral activity Lv4.
Introduction
Human immunodeficiency virus type 1 (HIV-1) is the major cause of the acquired immune deficiency syndrome (AIDS) pandemic. Among the immunodeficiency viruses that infect at least 40 of the primate species in sub-Saharan Africa, the simian immunodeficiency viruses (SIVs) found in central African chimpanzees and gorillas are monophyletic with HIV-1 [1,2]. Each of the four HIV-1 lineages (groups M, N, O, and P) is believed to have resulted from independent cross-species transmission of simian immunodeficiency viruses from chimpanzees (SIVCPZ), and perhaps from gorillas (SIVGOR) [3–6]. SIVCPZ itself is probably a recombinant virus that resulted from co-infection of a chimp with viruses transmitted from a red-capped mangabey (SIVRCM) and a greater spot-nosed monkey (SIVGSN) [7]. Until recently it was believed that SIVCPZ did not cause disease in chimpanzees but extensive observation of feral animals has demonstrated that this is not the case [8].
HIV-2, a second AIDS-causing virus that has highest prevalence in West Africa, was transmitted to people from sooty mangabey monkeys (Cercocebus atys) on multiple occasions [9–12]. There is no evidence for disease in sooty mangabey monkeys infected with SIVSM, but cross-species transmission to another non-native host, rhesus macaques (SIVMAC), resulted in AIDS [13,14].
Though transmission of primate lentiviruses to humans has occurred on multiple occasions and may still be occurring [15], these events are probably uncommon. Primate lentiviral sequences can be grouped into clades that are specific for a given host species [2]. Species crossovers are prevented in part by innate immune mechanisms, of which restriction by intracellular proteins is an important component. Proteins of the TRIM (Tripartite Motif) family can disrupt retroviral replication in a species-dependent manner [16–18]. TRIM proteins displaying anti-retroviral activity are present in all primates tested so far [19]. Moreover, phylogenetically and functionally related genes have been found in cattle [20,21] and in rabbits [22]. TRIM5α was the first member of this family to be identified as an anti-retroviral gene [23] and has been extensively studied. It targets incoming susceptible retroviruses, trapping them in cytoplasmic bodies that seem to form around the virus [24]. Inhibition of retroviral replication requires specific recognition of retroviral capsid motifs, and a TRIM5α-CA interaction can be detected in various settings [25–27]. Additionally, treatment with proteasome inhibitors partially relieves the restriction, suggesting that TRIM5α targets susceptible retroviruses to a proteasomal degradation pathway [28–30]. Finally, TRIM5α prevents nuclear transport of restricted retroviruses [28,30–32].
HIV-1 is inhibited by TRIM5α from a number of African and Asian monkey species, such as rhesus macaques, African green monkeys, and sooty mangabeys [19,33]. The human orthologue of TRIM5α restricts some non-primate lentiviruses such as the N-tropic strains of the murine leukemia virus (N-MLV) and the equine infectious anemia virus (EIAV) [34–36]. However, it has minimal activity against HIV-1 and various strains of SIVs such as SIVMAC and SIV from African green monkeys (SIVAGM) [32,36–39].
Thus, available data suggest that the early post-entry stages of SIVMAC replication are not inhibited by TRIM5α in human cells. These experiments, however, all used immortalized adherent cell lines such as TE671 (rhabdomyosarcoma) [32,40,41], HOS (osteosarcoma) [42] or HeLa cells (adenocarcinoma) [23,31,43]. Hofmann and colleagues compared the infectivity of vectors derived from SIVMAC or HIV-1 in a range of mammalian cell lines [44]. They found that HIV-1 vectors were up to 9-fold more infectious than SIVMAC vectors in several human cell lines, e.g. Raji (B lymphocyte) and in the T lymphocyte cell lines Jurkat, HuT78 and CEM. This raised the possibility that lentiviruses could be inhibited in a cell-type specific fashion in human cells. In the work presented here, we investigated restriction to SIVMAC replication in peripheral blood lymphocytes (PBLs) as well as in various cell lines. Our data reveal a TRIM5α-independent restriction activity targeting SIVMAC, and the related SIVSM and HIV-2, in human blood cells.
Results
Human blood cells are less permissive for SIVMAC, SIVSM, and HIV-2, than for HIV-1
Human cell lines were challenged with VSV G-pseudotyped, single-cycle vectors derived from HIV-1NL4-3 and SIVMAC239, as previously described [45]. In each case, nef was replaced with GFP coding sequence, such that the fluorescent reporter was expressed from the respective LTR. The two vectors were produced in parallel by collecting supernatant from transfected 293T cells. The vector-containing supernatants were checked for reverse transcriptase activity [46], normalized for titer on highly permissive CRFK feline kidney epithelial cells [47], and then used to infect a panel of human cell lines by serial dilution (Fig 1).
Fig 1. SIVMAC transduction of human blood-derived cell lines is less efficient than is transduction by HIV-1.

VSV G-pseudotyped HIV-1NL4-3GFP (black squares) and SIVMAC239GFP (white circles) were generated by plasmid transfection of 293T cells. In each plasmid, env was disrupted and nef replaced with GFP, such that the fluorescent reporter gene was expressed from the 5’ LTR. Vector stocks were normalized by titer on CRFK cells, and then used to challenge the indicated cell lines. 48 hrs post vector challenge, the percentage GFP-expressing cells was determined by FACS. Data is plotted as percent GFP+ (infected) cells (Y axis) versus CRFK infectious units (IU) x 1,000 (X axis).
SIVMAC transduction efficiency was 4 to 20-times less than that of HIV-1NL4-3 when the two vectors were used to challenge any of a panel of T cell lines, including Jurkat, SupT1, and CEM-SS cells, the Burkitt lymphoma-derived B cell line Raji, or the myelomonocytic cell lines U937 and THP-1 (Fig 1). The infectivity of SIVMAC was similar to that of HIV-1NL4-3 in adherent epithelial cell lines, including HeLa cells, HT1080 fibrosarcoma cells, TE671 rhabdomyosarcoma cells, U87 glioblastoma cells, and NP2 glioma cells (Fig 1).
Signal intensity by immunofluorescence microscopy of individual GFP-positive cells after SIVMAC transduction was at least as great as that after HIV-1NL4-3 transduction (Fig 2A). Mean fluorescence intensity by flow cytometry was 219.6 +/- 15.5 for SIVMAC and 170.3 +/- 11.3 for HIV-1NL4-3 (n = 6; p<0.01, Mann-Whitney). Based on these parameters, the decrease in apparent infectivity of SIVMAC did not appear to be explained by poor expression of the GFP reporter from the SIV LTR. The latter point was demonstrated more conclusively by using 3-part lentiviral vectors in which the GFP reporter was expressed from the HIV-1 and SIVMAC vectors using an identical spleen focus-forming virus (SFFV) promoter (Fig 2B); the relative decrease in CRFK-normalized, SIVMAC infectivity on Jurkat with the 3-part vector was at least as great in magnitude as it was with the 2-part vectors.
Fig 2. The decrease in T cell transduction efficiency by SIVMAC is not explained by differences in reporter gene expression.

(A) CRFK cells (left panel) and Jurkat T cells (right panel) were transduced with VSV G-pseudotyped, single-cycle, two-part HIV-1NL4-3GFP or SIVMAC239-GFP vectors, as in Fig 1 . Virus stocks were normalized by reverse transcriptase activity prior to transduction. 48 hrs after transduction, cells were visualized by phase contrast and fluorescence microscopy. Shown are representative fields for each condition at 100x magnification. Fluorescence intensity of individual T cells transduced with SIVMAC239-GFP is at least as strong as that in cells transduced with HIV-1NL4-3GFP. (B) VSV G-pseudotyped, HIV-1NL4-3 (black squares) and SIVMAC239 (white circles) three-part vectors were generated by plasmid transfection of 293T cells. In each case, the viral genomic RNA was designed to transduce an identical SFFV-GFP reporter gene. Vector stocks were normalized by titer on CRFK cells, and then used to challenge Jurkat T cells. 48 hrs post vector challenge, the percentage GFP-expressing cells was determined by FACS. Data is plotted as percent GFP+ (infected) cells (Y axis) versus CRFK infectious units (IU) x 1,000 (X axis).
Next, the infectivity of SIVMAC was compared with that of HIV-1NL4-3 in primary human blood cells using the two-part vectors. Peripheral blood mononuclear cells (PBMCs) were prepared, stimulated with PHA for three days, and challenged with the single-cycle vectors. SIVMAC transduction was less efficient than for HIV-1NL4-3 (Fig 3A). The magnitude of this difference was ~20-fold. Similar magnitude differences were observed when three-part vectors were used to challenge human, monocyte-derived dendritic cells in the presence of Vpx+-VLPs (Fig 3B). The dendritic cell experiments were carried out as previously described by providing SIV Vpx in trans using SIV VLPs [48–50].
Fig 3. SIVMAC transduction of human peripheral blood mononuclear cells or of monocyte derived dendritic cells is less efficient than by HIV-1.

(A) VSV G-pseudotyped HIV-1NL4-3GFP (black squares) and SIVMAC239GFP (white circles) two-part vectors were generated by plasmid transfection of 293T cells. Vector stocks were normalized by titer on CRFK cells, and then used to challenge human peripheral blood mononuclear cells. (B) VSV G-pseudotyped, HIV-1NL4-3 (black squares) and SIVMAC239 (white circles) three-part vectors were generated by plasmid transfection of 293T cells. In each case, the viral genomic RNA was designed to transduce an identical SFFV-GFP reporter gene. Vector stocks were normalized by titer on CRFK cells, and then used to challenge monocyte derived dendritic cells (DCs). 2 days post-challenge, the percentage of GFP-expressing cells was determined by FACS. Data is plotted as percent GFP+ (infected) cells (Y axis) versus CRFK infectious units (IU) x 1,000 (X axis). Shown are representative data with cells from 4 independent blood donors.
SIVMAC and HIV-2 are believed to have arisen from cross-species transmission of SIVSM from sooty mangabey monkeys to rhesus macaques and humans, respectively [1,2]. We therefore investigated to what extent other members of the SIVSM lineage are capable of transducing Jurkat cells. An env-minus, VSV G-pseudotyped HIV-2ROD vector, in which nef was replaced with GFP, was normalized to the HIV-1NL4-3GFP and SIVMAC239GFP vectors by transduction titer on CRFK and used to transduce Jurkat T cells. The normalized titer for SIVMAC was roughly 20-fold lower than that for HIV-1NL4-3 on Jurkat cells (Fig 4A). HIV-2ROD transduction was nearly 10-fold lower than HIV-1NL4-3 on Jurkat cells (Fig 4A).
Fig 4. SIVMAC, HIV-2, and SIVSM transduction of human T cells is less efficient than transduction by HIV-1.
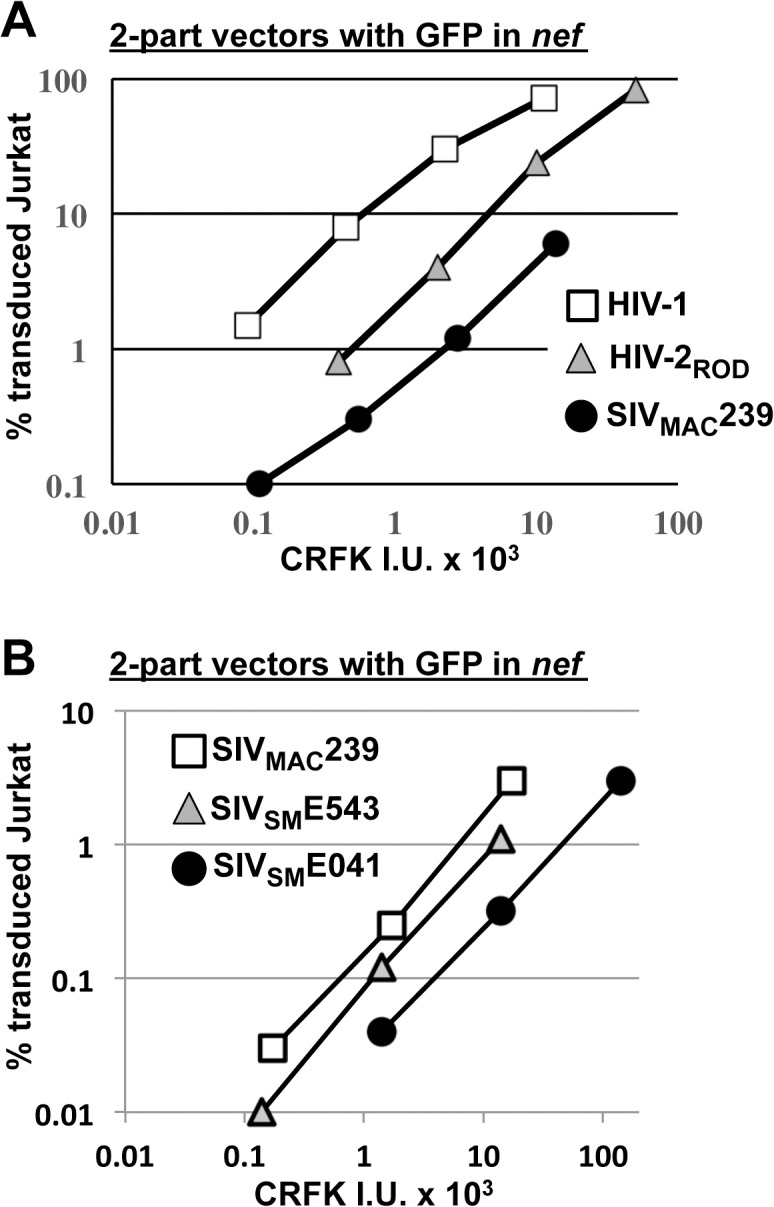
(A) Transduction efficiency of VSV G-pseudotyped two-part vectors for HIV-1NL4-3GFP (white squares), HIV-2RODGFP (grey triangles), or SIVMAC239GFP (black circles) on Jurkat T cells. (B) Chimeric vectors were generated in which gag-pol of SIVMAC239GFP (white squares) was replaced with gag-pol from SIVSME543 (grey triangles) or SIVSM041 (black circles). In each case (A and B), VSV G-pseudotyped vectors were generated by plasmid transfection of 293T cells. Vector stocks were normalized by titer on CRFK cells, and then used to challenge Jurkat T cells. 48 hrs post-challenge, the percentage of GFP-expressing cells was determined by FACS. Data is plotted as percent GFP+ (infected) cells (Y axis) versus CRFK infectious units (IU) x 1000 (X axis).
SIVMAC239, the virus utilized in the experiments above, is highly adapted to rhesus macaques, having been passaged many times in these animals since the 1960s [13,51]. SIVSME041 is a virus that was isolated directly from sooty mangabey monkeys [52]. SIVSME543 was passed twice through rhesus macaques [53] and would therefore be expected to have a modest level of adaptation to the new host. As compared with SIVMAC239, a three-part vector generated from the non-adapted SIVSME041 [54] had decreased Jurkat-specific transduction efficiency (Fig 4B). A three-part vector generated from SIVSME543 [54], the SIVSM virus that had been serially replicated in a non-native host (macaques), had transduction activity more similar to that of SIVMAC239 (Fig 4B). These results suggest that, in order to efficiently infect humans or rhesus macaques, SIVSM must acquire resistance to a putative restriction activity present in blood cells.
The capsid of SIVMAC, HIV-2, or SIVSM is sufficient to decrease HIV-1 transduction efficiency in a T cell-specific manner
The experiments described above suggest that SIVMAC, SIVSM, and HIV-2 transduction is sensitive to a restriction activity that is elaborated by human blood cells. Since capsid (CA) is the retroviral determinant that confers sensitivity to several restriction factors, including Fv1 [17], TRIM5 [23,45], and Mx2 [55–57], the transduction efficiency of the 2-part HIV-1 vector described above was compared with that of an isogenic vector in which CA coding sequence was replaced with that from SIVMAC239 or HIV-2ROD. Neither of the two chimeras had transduction activity on CRFK cells or on HeLa cells.
Since restriction factor sensitivity determinants are often located within the N-terminal two-thirds of CA [58], we then trimmed the C-terminal coding sequences of HIV-2ROD and SIVMAC239 CA back to amino acid 202, using HIV-1 CA sequences to encode amino acids 203 to 230 (Fig 5A). When normalized by RT activity [46] the two chimeras exhibited transduction efficiency on CRFK and HeLa cells very similar to the parental vector (Fig 5B and 5C, respectively). In contrast, the chimeric vectors bearing SIVMAC239 or HIV-2ROD CA transduced Jurkat T cells less efficiently, with a magnitude reduction that correlated with the respective parental vectors (Fig 5D).
Fig 5. The capsid of SIVMAC, HIV-2, or SIVSM is sufficient to decrease HIV-1 transduction efficiency in a T cell-specific manner.

(A) Chimeric vectors were generated in which the coding sequence for HIV-1 CA amino acid residues 1 to 202 of the two-part HIV-1NL4-3GFP vector (white squares) was replaced with sequence encoding the corresponding amino acid residues from HIV-2ROD (grey triangles) or SIVMAC239 (black circles). VSV G-pseudotyped vector was generated for each by transfection of 293T cells. Stocks were normalized by RT and used to challenge CRFK cells (B) or HeLa cells (C). Stocks were then normalized for CRFK transduction activity and used to challenge Jurkat T cells (D). 48 hrs post-challenge, the percentage of GFP-expressing cells was determined by FACS. Data is plotted as percent GFP+ (infected) cells (Y axis) versus RT activity (B and C), or versus CRFK infectious units (IU) x 1000 (X axis). (E) Chimeric vectors were generated in which the coding sequence for HIV-1 CA amino acid residues 1 to 202 of the HIV-1 gag-pol expression vector (white) was replaced with sequence encoding the corresponding amino acid residues from various HIV-2 isolates (grey) or SIVSME041 (black). Three-part, VSV G-pseudotyped, SFFV-GFP bearing vectors were generated for each CA chimera by transfection of 293T cells. Stocks were then normalized for CRFK transduction activity and used to challenge Jurkat T cells. 48 hrs post-challenge, the percentage of GFP-expressing cells was determined by FACS. Data is plotted as CRFK normalized transduction of Jurkat cells, relative to the parental HIV-1 vector (F). Accession numbers for the different CA coding sequences are as follows: HIV-2(AB), 731744; HIV-2(A), GH123; HIV-2(D), L33083; HIV-2(E), L33087; HIV-2(F), U75441; HIV-2(H), AY5308; SIVSME041, HM059825.
Having established that CA from either SIVMAC239 or HIV-2ROD is sufficient to reduce Jurkat T cell transduction efficiency by a 2-part HIV-1 vector (Fig 5D), fifteen additional chimeras were generated in the context of a 3-part HIV-1 vector using CA coding sequences from nine different HIV-2 Groups (Fig 5E). Many of the non-epidemic HIV-2 Groups in the database consist of single isolates, for which only partial HIV-2 CA sequences (encoding amino acids 1 to 162) are available [11]. In the case of these partial CAs, HIV-2ROD sequence was substituted for the missing HIV-2 sequences (amino acids 163 to 202). As with the 2-part vectors, no infectivity was observed unless CA amino acids 203–230 were provided by HIV-1. Among the chimeras generated, representatives from Groups AB, A, D, E, F, and H, and from a primary SIVSM, were sufficiently infectious to evaluate CRFK-normalized transduction efficiency on Jurkat T cells. As a general trend, chimeras generated with CA from the epidemic Groups (A and B) were the most infectious on Jurkat T cells, those from the non-epidemic Groups (D, E, F, and H) were less infectious, and that from SIVSM was the least infectious (Fig 5F). These results suggest that SIVSM must acquire resistance to the putative CA-specific restriction activity present in human blood lymphoid cells in order to efficiently infect human blood cells.
The defect in Jurkat transduction associated with SIVMAC CA is independent of the viral entry pathway
All of the experiments above were conducted with vectors pseudotyped with VSV G. To determine if the decreased transduction efficiency associated with the SIVMAC CA is observed with other glycoproteins, a two-part, env-minus HIV-1 vector with GFP in place of nef, or an isogenic vector in which CA1-202 coding sequences were replaced with those from SIVMAC239, were pseudotyped with Env glycoproteins from either HIV-1HXB2, HIV-2MCN, ecotropic MLV, or ALV-A (Fig 6). The transduction titer of each pseudotyped vector was first measured on HeLa cells bearing either human CD4, murine mCAT1 ecotropic receptor, or avian TVA receptor. Each was then used to challenge Jurkat T cells that had been stably transduced to bear the cognate receptors, as appropriate. 48 hrs post-challenge, the percentage of GFP-expressing cells was determined by FACS. In each case, the chimeric vector bearing SIVMAC CA1-202 was as defective as the VSV G-pseudotyped vector (Fig 6A–6D). These results demonstrate that the Jurkat transduction defect associated with the SIVMAC CA is independent of the pathway of viral entry.
Fig 6. The transduction defect associated with SIVMAC CA is independent of the virus entry pathway.

A two-part, env-minus HIV-1 vector with GFP in place of nef (black squares), or an isogenic vector in which CA1-202 coding sequences were replaced with those from SIVMAC239 (white circles), were produced by 293T transfection. Each vector was pseudotyped with Env glycoprotein from either HIV-1HXB2(A), HIV-2MCN (B), ecotropic MLV (C), or ALV-A (D) and transduction efficiency was measured on HeLa cells bearing human CD4 (A and B), the mCAT1 ecotropic receptor (C), or the avian TVA receptor (D), and then used to challenge Jurkat cells bearing the same receptors. 48 hrs post-challenge, the percentage of GFP-expressing cells was determined by FACS.
As2O3 increases SIVMAC transduction of human blood cells
Given the results described above, evidence was sought that the cell type-specific defect in SIVMAC transduction efficiency might be due to a dominant-acting, human blood-specific, restriction factor. Restriction activity of the capsid-specific restriction factors Fv1 and TRIM5 is saturated by large quantities of virus-like particles (VLPs) bearing restriction-sensitive cores [17]. Flat, epithelial cells work well as viral targets in TRIM5 saturation experiments; in contrast, saturation experiments have not been possible in T cell lines [59,60]. Attempts here to saturate putative SIVMAC-specific restriction activity in Jurkat T cells with SIV VLPs were also unsuccessful.
As2O3 rescues retroviruses from CA-specific restriction by TRIM5 but has no effect on retrovirus transduction efficiency in the absence of TRIM5-mediated restriction [31,35,47,61,62]. The exact mechanism by which As2O3 blocks TRIM5-mediated restriction is not known, though the effect results in increased reverse transcription and correlates with disruption of mitochondrial membrane potential [31,61].
To test the hypothesis that SIVMAC transduction of human blood cells might be restricted by TRIM5, or by a cellular factor with similar properties, the effect of As2O3 on SIVMAC transduction was assessed. As2O3 had no effect on the transduction efficiency of VSV G-pseudotyped, 2-part vectors for either SIVMAC239 or HIV-1NL4-3 in TE671 (Fig 7A), an adherent rhabdomyosarcoma cell line in which SIVMAC infectivity was equivalent to that of HIV-1NL4-3 (Fig 1). In contrast, As2O3 increased SIVMAC transduction of Jurkat T cells 3-fold, and transduction of PBMCs or primary CD4+ T cells 7-fold (Fig 7B–7D). HIV-1NL4-3 T cell transduction of any of these cells was increased less than 2-fold by As2O3 (Fig 7B–7D). Thus, As2O3 enhanced SIVMAC transduction of human blood cells in which relative transduction efficiency of SIVMAC was compromised. These results are consistent with the presence of a TRIM5-like, SIVMAC-specific, restriction factor in human blood cells.
Fig 7. As2O3 specifically increases SIVMAC infectivity in human blood cells.

TE671 cells (A), Jurkat T cells (B), human PBMC (C), or human CD4+ T cells (D) were transduced with two-part, VSV G-pseudotyped HIV-1NL4-3-GFP or SIVMACGFP vectors using a predetermined quantity of virus such that 1% of cells were infected. As2O3 was added 1 hr prior to vector challenge and maintained for 12 hrs post-infection, at the concentrations indicated on the X axis. 48 hrs post-challenge the percentage of GFP-expressing cells was determined. The Y axis shows the fold increase relative to infection without As2O3.
SIVMAC transduction efficiency in human CD4+ T cells does not increase with disruption of endogenous TRIM5α or CypA
TRIM5 is a well-characterized host cell restriction factor that decreases retroviral transduction in a capsid-specific fashion [23,45]. Though ectopic expression of human TRIM5α in adherent cell lines shows minimal restriction activity against SIVMAC [23,34] it was important to determine whether endogenous human TRIM5α contributes to the SIVMAC transduction block in human blood cells. To investigate this possibility, a miR30-based TRIM5 knockdown cassette was delivered to Jurkat T cells using a lentiviral vector as previously described [48,63,64] (Fig 8). The vector also expresses a puromycin-resistance gene that was exploited to select pools of transduced cells. Cyclophilin A (CypA), an HIV-1 capsid binding protein [65] that promotes TRIM5-mediated restriction in some cell types [66], and appears to protect against an unknown restriction activity in other cells [17], was also targeted for knockdown with a lentiviral vector. As a control for miR30 lentiviral vector transduction and puromycin selection, Jurkat T cells were transduced with an otherwise isogenic lentiviral vector targeting luciferase (Luc), a gene that is not present in these cells.
Fig 8. Knockdown of TRIM5 or of cyclophilin A has no effect on SIVMAC transduction of Jurkat CD4+ T cells.

Jurkat T cells (A) or primary human CD4+ T cells (B) were transduced with lentiviral vectors bearing a puromycin resistance cassette and miR30-based knockdown cassettes targeting either luciferase (black squares), CypA (gray diamonds), or TRIM5 (white triangles). Puromycin-resistant pools of transduced cells were challenged with VSV G-pseudotyped N-MLVGFP, B-MLVGFP, HIV-1NL-GFP, SIVmac-GFP, or EIAVGFP, as indicated. The percentage of GFP+ (infected) cells at 48 hrs is reported. HIV-1NL-GFP and SIVmac-GFP vectors were two-part vectors, with GFP in place of nef. N-MLVGFP, B-MLVGFP, and EIAVGFP were three-part vectors.
TRIM5 knockdown efficiency in Jurkat T cells cannot be assessed by western blot since endogenous human TRIM5 is not detectable in these cells using available antibodies. Instead, knockdown efficiency can be deduced by comparing the infectivity of a pair of viruses, one of which is restricted by human TRIM5 (N-MLV), and the other which is not restricted (B-MLV) [59]. The three pools of puromycin-resistant Jurkat T cells–either knocked down for TRIM5, CypA, or Luc—were therefore challenged with N-tropic or B-tropic MLV-GFP reporter viruses. As shown previously [59], N-tropic MLV was much less infectious than B-tropic MLV in the control (luciferase) knockdown cells (Fig 8A). TRIM5 knockdown increased N-MLV transduction efficiency up to the level achieved by the non-restricted B-tropic MLV (Fig 8A) but no effect on the transduction efficiency of HIV-1NL4-3 or SIVMAC was observed (Fig 8A). Also, as shown previously (3, 4), knockdown of CypA had no effect on N-tropic MLV, B-tropic MLV, or SIVMAC (Fig 8A), though CypA knockdown decreased HIV-1NL4-3 transduction efficiency by 3 to 4-fold (Fig 8A). Thus, the low relative transduction of Jurkat T cells by SIVMAC was not increased by knockdown of TRIM5 or CypA.
To extend these findings to primary cells, human CD4+ T cells were enriched from peripheral blood by positive-selection with magnetic beads, stimulated with plate-bound anti-CD3 and anti-CD28 antibodies, and transduced with the same lentiviral vectors for stable knockdown of TRIM5, CypA, or luciferase, as previously described [67] (Fig 8B). Transduced cells were propagated in puromycin-resistant pools. Transduction with a control vector in which the puromycin resistance cassette was replaced with GFP demonstrated that primary transduction efficiency, in the absence of drug selection, was greater than 90%. Growth of transduced CD4+ T cells in tissue culture was maintained in an ongoing fashion by TCR re-stimulation every two weeks [67].
CD4+ T cells from one of two representative blood donors are shown in Fig 8B. The titer of the N-tropic and B-tropic MLV vectors on the stably-transduced, primary human CD4+ T cells, was not sufficient to assess the efficiency of TRIM5 knockdown. Instead, a lentiviral vector derived from the equine infectious anemia virus (EIAV-GFP) was utilized [68]. As previously shown in human HeLa cells [34], knockdown of TRIM5 increased EIAV-GFP transduction efficiency (Fig 8B). Though the absolute infectivity of EIAV-GFP in the luciferase and CypA knockdown cells was at the limit of detection, it was possible to document an increase in EIAV-GFP transduction efficiency of at least 50-fold in the TRIM5 knockdown cells, confirming that the TRIM5 knockdown was robust. As expected, CypA knockdown caused a modest reduction in HIV-1NL4-3 infectivity (Fig 8B). SIVMAC was 50- to 100-times less infectious than HIV-1NL4-3 in all of the CD4+ T cell knockdown lines tested (Fig 8B). Thus, neither TRIM5 knockdown nor CypA knockdown increased SIVMAC transduction efficiency, in Jurkat T cells or in primary CD4+ T cells.
The block to SIVMAC transduction in Jurkat T cells occurs prior to establishment of the provirus, but after entry into the target cell nucleus
To determine where in the retroviral replication cycle the relative block to SIVMAC transduction occurs, CRFK cells and Jurkat T cells were challenged with the single-cycle, 2-part, HIV-1NL4-3 GFP reporter vector, or the isogenic vector bearing the SIVMAC239 CA, that were diagramed schematically in Fig 5A. Full-length linear viral cDNA, 2-LTR circle viral cDNA, and proviral DNA as assessed by Alu-PCR were quantitated by real-time PCR, using previously described protocols [69,70]. The relative level of PCR product obtained with the vector bearing SIVMAC239 CA was expressed as a percentage of that obtained with the vector bearing HIV-1NL4-3 CA, with the latter set at 100%. In CRFK cells, infection with the two vectors resulted in comparable amounts of full-length linear and 2-LTR circles (Fig 9A). As compared with the vector bearing HIV-1NL4-3 CA, transduction of Jurkat T cells with the vector bearing SIVMAC239 CA resulted in the same amount of full-length linear cDNA and 2-LTR circles, but 10-fold less product for Alu-PCR (Fig 9A).
Fig 9. The block to SIVMAC infection of Jurkat T cells occurs after formation of 2-LTR circles.

CRFK and Jurkat (A), or Hela and Jurkat (B), or PBMCs (C) were infected with VSV G-pseudotyped HIV-1NL4-3-GFP, or with isogenic vector bearing the SIVMAC239 CA residues 1 to 202. 24 hrs post-infection, DNA was collected from the cells and subjected to qPCR using primers specific for full-length linear viral cDNA, 2-LTR circles, or proviral DNA, as indicated. Shown is the abundance of signal from vector bearing the SIVMAC239 CA1-202, relative to the amount of signal from HIV-1NL4-3-GFP. In each case, infection was performed in the presence of an RT inhibitor to control for background levels of signal.
Since Alu repeats are primate-specific [71], Alu-PCR could not be performed using the feline CRFK cells as transduction targets. Therefore, similar experiments were performed with HeLa cells (Fig 9B). In addition, a PCR protocol for 2-LTR circles was used in which one of the PCR primers spans the circle junction; this distinguishes bona fide 2-LTR circles from auto-integrants [72]. No defect in full-length linear cDNA or 2-LTR circles was detected when transduction of Jurkat cells with the vector bearing SIVMAC239 CA was compared with the vector bearing HIV-1NL4-3 CA (Fig 9B). As compared with HeLa cells, a specific defect in provirus establishment in Jurkat T cells by the vector bearing SIVMAC239 CA was observed (Fig 9B). Similar results were obtained using human PBMCs as target cells, though the signal from Alu-PCR was insufficient to quantitate the magnitude difference between HIV-1 and SIVMAC (Fig 9C). These results indicate that reverse transcription and nuclear transport by particles bearing SIVMAC CA is equivalent to that of particles bearing HIV-1NL4-3 CA, and that the relative block to SIVMAC transduction likely occurs after entry into the nucleus, prior to integration.
Poor relative infectivity of SIVMAC239 in human blood cells results from a dominant-acting restriction activity
Human blood cells such as Jurkat T cells might be less permissive for SIVMAC transduction because they lack a factor, which is present in epithelial cell lines such as HeLa, that promotes SIVMAC transduction. Alternatively, human blood cells might possess an inhibitor of SIVMAC transduction that is absent from the adherent cell lines. To determine which of these two possibilities is correct, Jurkat T cells were fused with HeLa cells using polyethylene glycol. The resulting heterokaryons were then challenged with the single-cycle, HIV-1NL4-3 GFP reporter vector (hCA-GFP), or the isogenic vector bearing the SIVMAC239 CA (sCA-GFP), that were shown schematically in Fig 5A.
A flow cytometry-based assay was established that discriminates infected heterokaryons from those cells that fail to form heterokaryons (Fig 10). Primary flow cytometry data for a single representative experiment is shown in Fig 10A; Fig 10B shows a bar plot of the results for three independent experiments. The HeLa cells that were used in the fusion stably synthesized TagRFP-657, a far-red fluorescent protein [73]. The Jurkat T cells that were used in the fusion stably bore the avian leukosis virus TvA receptor on their surface. The HIV-1 CA-GFP and SIV CA-GFP vectors were pseudotyped with avian leukosis virus subtype A (ALV-A) Env so that the vectors were able to enter Jurkat-TvA cells but not the HeLa-RFP cells. Heterokaryons formed by fusion of the two cell types would bear the cognate receptor for ALV-A Env and would also be positive for RFP. Infected heterokaryons, then, would be positive for GFP and RFP.
Fig 10. Evidence for a dominant-acting, capsid-specific, restriction activity in Jurkat T cells.

(A) Jurkat and HeLa cells stably expressing the ALV-A receptor (TvA) or TagRFP-657, as indicated, were fused by treatment with PEG and transduced with ALV-A Env-pseudotyped HIV-1NL4-3-GFP, or with isogenic vector bearing the SIVMAC239 CA1-202. Shown are flow cytometry dot plots obtained 48 hrs post-transduction. HeLa-TagRFP-657 cells are only permissive to infection with ALV-A Env-pseudotyped vectors after fusion with Jurkat-TvA. Infected heterokaryons were visualized as GFP and TagRFP-657 double-positive cells. As a positive transduction control, TagRFP-657 and TvA were also co-expressed in HeLa cells, as indicated. The percentage of transduced cells are indicated. (B) Bar graph showing the infectivity of the SIVMAC239 CA1-202-bearing vector relative to the isogenic vector bearing HIV-1 CA, for the HeLa, Jurkat and heterokaryons. Data from the flow cytometry data shown in A, and two repeat experiments, is shown with the standard deviation.
As a control, HeLa-RFP cells were engineered to express TvA (HeLa-RFP-TvA); in these cells, transduction with SIV CA-GFP was 1.6-fold higher than with HIV-1 CA-GFP (Fig 10A). Challenge of the Jurkat-TvA cells with HIV-1 CA-GFP and SIV CA-GFP recapitulated the phenotype of the parental Jurkat cells. That is, transduction of Jurkat-TvA cells with SIV CA-GFP was 5.6-fold less efficient than with HIV-1 CA-GFP, though, when these values are corrected for the transduction efficiency on HeLa-TVA, the difference is 9-fold (Fig 10A). When Jurkat-TvA cells were mixed with HeLa-RFP cells in the absence of polyethylene glycol, no GFP/RFP double-positive cells were detected, and transduction with SIV CA-GFP was 6-fold (corrected) less efficient than with HIV-1 CA-GFP (Fig 10A). When Jurkat-TvA cells were mixed with HeLa-RFP cells in the presence of polyethylene glycol, GFP/RFP double positive cells were detected, and transduction of this population with SIV CA-GFP was 7.3-fold (corrected) less efficient than with HIV-1 CA-GFP (Fig 10A). The bar graph in Fig 10B shows the results for three experiments with the standard deviation. The results of this heterokaryon assay indicate that Jurkat T cells possess a dominant-acting restriction activity specific for SIV CA.
Discussion
The characteristics of a previously unreported retroviral restriction activity in human blood cells are described here. The first clue to the existence of this restriction activity was that SIVMAC239 transduced human blood cells less efficiently than did HIV-1. Lower SIVMAC239 transduction efficiency relative to HIV-1 was observed with all human blood-derived cells tested here, including cell lines of lymphoid and myeloid lineage, human PBMCs and primary CD4+ T cells, and, as previously described [49,50,74], monocyte-derived dendritic cells and macrophages. In contrast to blood-derived cells, fibroblasts, fibrosarcoma, epithelial carcinoma, and glioblastoma cell lines were transduced as efficiently by SIVMAC239 as by HIV-1.
The presence of a dominant-acting, SIVMAC239-specific, restriction factor in human blood cells—as opposed to the lack of a cofactor for SIVMAC239 replication in these cells—was supported by the finding of a block to SIVMAC239 replication in Jurkat/HeLa-heterokaryons of equal magnitude to the block in Jurkat T cells. Similar heterokaryon experiments demonstrated the presence of a dominant restriction activity prior to the cloning of the retroviral restriction factors APOBEC3G, TRIM5, and TETHERIN [42,43,75,76]. Several methods for quantitating transduction of heterokaryon target cells were tried here, all of which gave similar qualitative results. Of these assays, the heterokaryon assay presented in Fig 10 gave us the clearest assessment of heterokaryon transduction efficiency; it exploits the specificity of the ALV/TVA interaction [77], and the clean spectral separation of GFP from the far-red fluorescent protein TagRFP-657 [73].
As is the custom for naming dominant-acting, lentiviral restriction activities of unknown identity [40,43,78,79], the SIVMAC239-specific restriction activity described here will be called Lv4. Whether this activity is due to a single factor, or due to a multi-factor complex, remains to be determined. Knockdown experiments presented here showed that Lv4 is distinct from TRIM5 (Fig 8), the protein responsible for Lv1 activity [23,45]. Lv2 is an HIV-2 env-specific restriction activity [80]. Lv4 restricts vectors that are pseudotyped with VSV G or with Env from Lv2-resistant HIV-2 clone MCN (Fig 6) so it must be distinct from Lv2. For that matter it also restricts vectors pseudotyped with HIV-1 Env, MLV ecotropic Env, or ALV-A Env (Fig 6) so it acts independent of the viral entry pathway. Lv3 restricts HIV-1 in an env-specific fashion [79] and so it must also be distinct from Lv4.
SIVMAC239 CA was sufficient to transfer Lv4-sensitivity when it was substituted for HIV-1 CA (Fig 5). This observation puts Lv4 in good company with a growing family of restriction factors that target the retroviral CA. The CA-specific restriction factors Fv1 and TRIM5 can be saturated by virus-like particles (VLPs) bearing restriction-sensitive CA [40,43,81,82]. Attempts to saturate Lv4 with SIVMAC239 virus-like particles were unsuccessful, though this result was not unexpected since Lv4 was only observed in blood cells, and saturation of CA-specific restriction activities in non-adherent cells that grow in suspension has not been reported [59].
Others have shown that SIV transduces human T cell lines less efficiently than HIV-1 and they provided suggestive evidence that this difference was independent of TRIM5 and CypA [83]. Here, after demonstrating that Lv4 activity does not require TRIM5 through knockdown experiments in either Jurkat or PBMC (Fig 8), our attention was directed to other potential CA-specific candidates. Disruption of TNPO3 results in accumulation of the CA-binding protein CPSF6 in the cytoplasm and an associated block to HIV-1 nuclear entry [72]. Though inhibition of SIVMAC by CPSF6 was slightly greater than that of HIV-1 [84], this differential effect was much smaller than was observed with Lv4. Additionally, Lv4 blocks SIVMAC at a later stage in the lentiviral life cycle than does CPSF6 (Fig 9), as demonstrated using the same assay for bona fide HIV-1 2-LTR circles [72]. In response to the identification of MX2 in a targeted screen for HIV-1 inhibitors among interferon stimulated genes (ISGs) [85], and prior to identification of MxB as a lentivirus CA-specific inhibitor [55–57], MX2 was found to inhibit HIV-1 and SIV equally well when ectopically expressed in either HT1080 or HeLa cells, and thus ruled out as Lv4.
Like the restriction activity conferred by TRIM5 [47,61,67], Lv4 was suppressed by arsenic (Fig 5). Efficient knockdown of TRIM5 in Jurkat T cells or in primary CD4+ T cells, though, had no effect on SIVMAC titer (Fig 8), indicating that Lv4 is distinct from TRIM5. How arsenic works to suppress restriction activity is not known. Among its many effects, arsenic inhibits NFκB signaling by oxidizing a critical cysteine in IKKα/β [86]. This suggests that arsenic might inhibit TRIM5 restriction activity by oxidizing critical cysteines. The fact that Lv4 is inhibited by arsenic suggests that, like TRIM5, it too might be a cysteine-containing protein. Attempts to identify the host factor responsible for Lv4 activity by ectopically expressing a panel of 36 TRIM family members [87], each of which possess cysteine-rich zinc-fingers and b boxes, has so far failed to identify an SIVMAC-specific inhibitor. That being said, the cell type-specific suppression of TRIM5 restriction activity by arsenic [47] suggests that arsenic targets a common cellular co-factor required for TRIM5 and Lv4 restriction activity. Such a co-factor might be an innate immune signaling molecule like those shown to be required for TRIM5-mediated restriction [48].
TRIM5 blocks retroviruses soon after entry into the cell cytoplasm [88]. This is evident as a block to the accumulation of viral cDNA [23]. If this block to reverse transcription is removed by arsenic or by proteasome inhibitors, a downstream block is encountered at the level of nuclear transport, with a decrease in viral cDNA circles [30,31]. The capsid binding proteins MX2 and, conditionally, CPSF6, both appear to block infection prior to entry in the nucleus [56,57,72,89]. The block due to Lv4 occurred before integration, but after completion of reverse transcription and nuclear entry, as indicated by levels of nascent viral cDNA, viral cDNA circles, and Alu-PCR (Fig 9). Thus, any putative factor underlying Lv4 activity likely interacts with CA within the nucleus and acts to block integration. These results are consistent with the steadily increasing evidence, acquired over many years, that CA plays an essential role within the nucleus of newly infected cells [90–93].
Finally, sensitivity to Lv4 was not unique to SIVMAC but shared by other viruses in the same family, including HIV-2 and SIVSM (Fig 4). Most studies here were performed with SIVMAC because the restriction activity was more robust than for HIV-2, but it was not so severe as for SIVSM, which precluded quantitation of restriction activity against the latter virus. The relative restriction activity targeting these viruses is consistent with a model in which replication of HIV-2 necessitated adaption of the SIVSM CA, such that it became relatively resistant to Lv4. There was indeed a trend such that HIV-2 isolates from non-epidemic Groups were generally more sensitive to Lv4 than were epidemic HIV-2 strains (Fig 5F). Though HIV-2 infects humans, relative to HIV-1 this virus is still restricted by Lv4. Thus, Lv4 may contribute to the fact that HIV-2-infected individuals are less likely to progress to AIDS than are those people infected with HIV-1 [94].
Materials and Methods
Plasmid DNAs
HIV-1NL4-3GFP, SIVMAC239GFP, HIV-2RODGFP, SIVSME041GFP, and SIVSME543GFP encode modified proviral clones for the respective viruses [31,45,54,95]; each of these plasmids lacks functional env and encodes GFP instead of Nef. For some experiments, coding sequences for residues 1 to 202 of HIV-1NL4-3GFP were replaced by overlapping PCR with the corresponding CA coding sequences from HIV-2ROD, SIVMAC239, SIVSME041 or SIVSME543 [54,96,97]. CA1-202 chimeras were also generated within the context of p8.9NdSB [31,45,54,95]; the restriction sites BlpI and BstEII were introduced flanking CA coding sequences and the following sequences, synthesized by GenScript, were inserted at these restrictions sites:
>HIV-2(AB), 731744
GCTCAGCAAGCAGCAGCTGACACAGGAAACAACAGCCAGGTCAGCCAAAATTACCCAGTGCAACAAGTAGCTGGCAATTATGTCCATGTGCCGTTAAGTCCCCGAACCTTAAATGCCTGGGTAAAATTAGTGGAGGAAAAGAAGTTCGGGGCAGAAATAGTACCAGGATTTCAGGCACTATCAGAGGGATGTACCCCTTATGATATCAATCAAATGCTAAATTGTGTGGGAGAACACCAGGCAGCCATGCAAGTCATTAGAGAAATAATCAATGAAGAGGCGGCAGACTGGGACCAGCAACACCCGATACCAGGTCCACTGCCAGCAGGACAACTTAGAGACCCCAGAGGATCAGATATAGCGGGAACCACCAGCACAGTAGAGGAACAAATACAGTGGATGTACAGGGGTCAAAATTCCGTCCCAGTGGGGAACATTTATAGAAGATGGATTCAATTAGGATTGCAGAAATGTGTCAGGATGTACAATCCTACTAATATACTAGATGTAAAACAAGGGCCAAAAGAACCCTTCCAAAGCTATGTAGATAGATTCTACAAAAGCCTACGGGCAGAACAAGCAGACACAGCCGTGAGAGCATGGATGACAGAAACACTACTGGTCCAGAATGCTAACCCAGATTGCAAGCTAGTACTC
>HIV-2(A), GH123
TGTACAACAGACAGGCGGTGGCAACTATATCCACGTGCCACTGAGCCCCCGAACTCTAAATGCTTGGGTAAAATTAGTAGAGGACAAGAAGTTCGGGGCAGAAGTAGTGCCAGGATTTCAAGCACTCTCAGAAGGCTGCACGCCCTATGATATCAACCAAATGCTTAATTGTGTGGGCGATCACCAAGCAGCTATGCAAATAATCAGAGAGATTATCAATGACGAAGCAGCAGATTGGGATGCACAGCACCCAATACCAGGCCCCTTACCAGCAGGGCAGCTTAGAGACCCAAGGGGGTCTGACATAGCAGGAACAACTAGCACAGTAGAAGAACAGATCCAGTGGATGTATAGGCCACAAAATCCCGTGCCGGTAGGGAACATCTACAGAAGATGGATCCAGATAGGGCTACAGAAGTGTGTCAGGATGTACAACCCAACTAACATCTTAGACGTAAAGCAGGGACCAAAGGAACCGTTCCAGAGCTATGTGGACAGGTTCTATAAAAGCTTGAGGGCAGAACAAACAGATCCGGCAGTAAAGAACTGGATGACCCAAACGCTGCTAATACAGAATGCCAACCCAGACTGCAAGTTAGTACTA
>HIV-2(D), L33083
AGTGCAGCAAGTCGGCGGAAATTATGTCCACCTACCGCTGAGTCCCAGAACATTAAATGCATGGGTTAAGTTAGTGGAGGACAAAAAATTCGGGGCAGAGGTAGTGCCAGGGTTTCAGGCACTATCGGAAGGCTGCACTCCGTATGACATCAATCAGATGCTAAATTGTGTAGGAGAACATCAGGCAGCCATGCAGATCATAAGGGAAATAATCAATGATGAGGCAGCAGATTGGGATCAGCAGCATCCACAACCAGGCCCACTACCAGCAGGACAGCTCAGAGATCCACGAGGATCTGATATAGCAGGAACCACTAGCACAGTGGAGGAACAAATACAGTGGATGTACAGGCAGCAGAATCCCATACCAGTTGGAAATATCTATAGGAGATGGATCCAGCTAGGGTTACAGAAATGTGTCAGAATGTACAACCCAACTAACATTCTGGATATAAAACAAGGGCCAAAAGAGACGTTCCAGAGCTATGTAGATAGATTCTACAA
AAGCTTGAGGGCAGAACAAACAGACCCAGCAGTGAAAAATTGGATGACACAAACACTGCTGATTCAGAATGCTAACCCAGATTGCAAGTTAGTACTA
>HIV-2(E), L33087
AGTGCAACAGATAGGAAATAACTATGTGCACTCTCCACTGTCCCCAAGAACATTGAATGCATGGGTCAAATTAGTAGAAGAAAAGAAATTTGGAGCAGAGGTAGTGCCAGGCTTCCAGGCATTATCAGAAGGATGCACCCCGTATGACATCAACCAGATGCTTAATTGCGTGGGGGAACATCAGGCAGCCATGCAAATTATCAGAGAGATAATCAATGAAGAAGCAGCAGATTGGGACGTACAGCATCCAAGAGGGCAACCGCCAGCACAGGGCCTAAGAGACCCATCAGGATCAGACATAGCAGGGACAACCAGTACCCCCGCAGAACAAATAGAGTGGATGTACAGGAATCCAAATCCAATCCCTGTGGGAGACATCTATAGAAGATGGATCCAGCTAGGGCTCCAGAAATGTGTCAGAATGTATAATCCAACAAACATTCTGGACGTCAAACAGGGGCCCAAAGAATCTTTTCAGAGCTATGTAGATAGATTCTACAAAAG
CTTGAGGGCAGAACAAACAGACCCAGCAGTGAAAAATTGGATGACACAAACACTGCTGATTCAGAATGCTAACCCAGATTGCAAGTTAGTACTA
>HIV-2(F), U75441
AGTGCAGCAGGTAGGAGGAAATTACACCCATATTCCTCTGAGTCCGAGGACATTAAATGCTTGGGTTAAATTAGTAGAGGAAAAGAAATTTGGGGCAGAAATAGTGCCAGGCTTCCAAGCATTGTCAGAAGGCTGCACCCCTTATGATATTAATCAAATGTTAAATTGTGTAGGGGAACATCAGGCAGCCATGCAAATAATCAGGGAAATAATCAATGAAGAAGCAGCCGACTGGGATCAGAATCATCCAAGGCAGCTGCCAGCGCCACCAGGGCTGCGTGATCCGTCAGGATCTGACATTGCAGGAACAACTAGTACAGTACAAGAACAGATAGAATGGATGTACAGACAGGGTAACTCAATCCCAGTAGGGGACATTTACAGAAGATGGATCCAAATAGGCCTTCAAAAATGTGTAAGAATGTACAATCCTACTAATATCCTAGATGTAAAACAGGGACCAAAAGAACCATTTCAAAGCTATGTAGATAGATTCTACAAAAG
CTTGAGGGCAGAACAAACAGACCCAGCAGTGAAAAATTGGATGACACAAACACTGCTGATTCAGAATGCTAACCCAGATTGCAAGTTAGTACTA
>HIV-2(H), AY5308
GGTGCAGCAGATAGGTGGCAATTATGCCCACCTACCTCTAAGTCCTAGAACACTCAATGCCTGGGTAAAACTGGTAGAGGAGAAAAAATTTGGAGCAGAAGTAGTGCCAGGATTTCAGGCACTCTCAGAGGGCTGCACGCCCTATGATATTAATCAAATGTTAAATTGCGTGGGAGAACATCAAGCTGCTATGCAAATTATCAGGGAAATAATTAATGATGAAGCAGCAGATTGGGACACACAGCACCCAAACCAAGGCCCACCACCAGCAGGGCAACTTAGAGAGCCAAGAGGTTCTGATATTGCAGGAACAACTAGCACAGTGGAAGAGCAGATACAGTGGATGTACAGGCCGCAAAATCCAATACCGGTGGGTAACATCTATCGGAGATGGATCCAATTGGGCCTACAAAAATGTGTTAGAATGTACAATCCAACTAATATCTTAGATATAAAGCAAGGGCCAAAGGAGCCATTTCAAAGTTATGTAGATAGATTCTACAA
AAGTTTGAGAGCAGAACAAACAGATCCAGCAGTGAAAAATTGGATGACTCAGACGCTGCTGATTCAGAATGCTAACCCAGACTGCAAACTCGTGTTA
>SIVSME041, HM059825
AGTGCAGCAAGTAGGTGGCAATTATACCCACCTACCCTTAAGTCCAAGAACATTAAATGCTTGGGTAAAATTGATAGAAGAGAAAAAATTTGGGGCAGAAGTAGTGCCAGGATTCCAAGCACTATCAGAAGGCTGCACTCCCTATGACATCAATCAGATGCTAAATTGTGTAGGGGAGCATCAATCAGCCATGCAAATTATTAGAGAAATTATAAATGAAGAAGCTGCTGATTGGGATTTACAACACCCACAGCCAGGTCCAATACCAGCAGGACAACTTAGAGACCCGAGAGGATCAGACATTGCAGGAACTACTAGCACAGTAGAAGAACAAATTCAATGGATGTATAGGCAGCAAAACCCTATACCAGTAGGTAACATTTACAGAAGGTGGATCCAATTAGGGCTGCAAAAATGTGTAAGGATGTATAATCCAACAAACATTTTAGATGTGAAACAAGGACCAAAAGAGCCATTTCAAAGCTATGTAGATAGATTCTACAA
GAGTCTAAGAGCAGAACAAACAGACCCAGCAGTGAAAAATTGGATGACTCAAACACTGCTGATTCAAAATGCTAACCCAGATTGCAAATTGGTGCTC
pMD2.G encodes the vesicular stomatitis virus glycoprotein (VSV G) and psPAX2 encodes HIV-1 Gag and Gag-Pol [98]. pCIG3N and pCIG3B encode N-tropic and B-tropic versions of murine leukemia virus (MLV) Gag-Pol and pCNCG is an MLV-derived vector expressing GFP [61,99]. pONY3.1 is an equine infectious anemia virus (EIAV) gag-pol plasmid and pONY8.0 is an EIAV GFP-packaging vector [68].
pAPM is a lentiviral vector expressing puromycin-resistance and a miR30-based knockdown cassette from the spleen focus forming virus LTR [48,63,64]. The knockdown targeting sequences used here were as follows: luciferase: 5’-tacaaacgctctcatcgacaag-3’, cyclophilin A (CypA): 5’-ctggattgcagagttaagttta-3’, TRIM5: 5’-tgccaagcatgcctcactgcaa-3’. pAIP and pAIB are lentiviral vectors expressing puromycin and blasticidin resistance respectively. The HIV-1 Env glycoprotein expression plasmid was based on HXB2 [46] and the HIV-2 Env was from the MCN clone [100]. MLV ecotropic Env was expressed from pFBMOSALF [101] and its cognate receptor, mCAT1, was stably expressed with the pBABE-puro MLV-based vector. Codon optimized TvA with a triple HA tag derived from pKZ261 [102] was cloned into pAIP (pAIP-TvA). ALV-A env glycoprotein for virion pseudotyping was expressed from pAB6 [103]. Far red fluorescence protein TagRFP-657 [73] was cloned into pAIB for stable expression (pAIB-RFP).
Cells
Cell lines were either grown in DMEM (293T, TE671, HeLa, NP2, U87, HT1080, and Crandall feline kidney fibroblasts, CRFK cells) or RPMI (Jurkat, SupT1, CEM-SS, Raji, U937, and THP-1), supplemented with 10% fetal calf serum as described before [61,104,105].
PBMC were separated by Ficoll density centrifugation, stimulated with PHA for 3 days, and cultured in RPMI supplemented with antibiotics, 10% fetal bovine serum, and 20 IU/ml hIL-2 [67,106].
CD4+ T lymphocytes were enriched from PBMC by positive selection using magnetic beads (Miltenyi Biotec). Typically the resulting population was >99% CD4+. Cells were stimulated for 24 hrs on NUNC maxisorp plates that had been coated with 2 μg/ml anti-CD3 antibody and 2 μg/ml anti-CD28 antibody (BD Biosciences) in RPMI with 10% FBS, glutamax (Invitrogen), and 20 IU/ml hIL-2. Two wks after primary stimulation, cells were re-stimulated using plate-bound anti-CD3 and anti-CD28 antibodies.
Production of viral stocks
VSV G-pseudotyped viral stocks of HIV-1, SIVMAC239, and the CA chimera vectors described above, were prepared by co-transfection of the indicated plasmids with pMD2.G in 293T cells, as described [31]. Virion stocks were normalized by reverse transcriptase assay [31] and by titer on non-restrictive CRFK cells or HeLa cells [107]. For production of the shRNA-expressing APM vectors, 8 x 106 cells were plated per 10-cm plate. The next day, cells were transfected using Lipofectamine 2000 (Invitrogen) and 20 μg of pAPM, 15 μg of psPAX2 and 5 μg of pMD2.G. Supernatant was collected and passed through a 0.45 μM filter at 48 hrs and at 72 hrs post-transfection, and used immediately to transduce target cells.
Challenge with GFP reporter virus
Reporter virus-containing supernatant was titrated onto 4 x 104 of the indicated target cells, in 0.4 ml media per well, in 24-well plates. As2O3 (Sigma) was prepared as described [31] and, where indicated, added to the cell culture 15 mins prior to virus addition. Cell supernatant was replaced with fresh medium without drug, 12 hrs after addition of virus. Cells were trypsinized when necessary and analyzed by flow cytometry 48 hrs after infection, as described [61].
RNA interference using lentivirus vectors
Jurkat cells or primary CD4+ T cells were spinfected with shRNA-encoding APM vectors twice, at 24 hr and 48 hr after stimulation with plate-bound anti-CD3 and anti-CD28 antibodies. Spinfection was done at 1,130 rcf for 90 mins, using 2 ml of freshly produced virus supernatant for each well of a 6-well plate containing 5 x 105 stimulated lymphocytes. Cells were put in 5 μg/ml of puromycin for 72 hrs, 2 days after the first spinfection.
Reverse transcriptase assay
Virus-containing supernatant was harvested 48 hr post-transfection, clarified by low-speed centrifugation, and filtered through 0.45 μm pore filters (Sarstedt). Reverse transcriptase (RT) activity in the supernatant was quantified using a modified Sybr green I-based, real-time PCR, enhanced RT assay [108,109]. Virions in cell-free supernatant were disrupted by adding an equal volume of a solution containing 0.25% Triton X-100, 50 mM KCl, 100 mM Tris-HCl pH 7.4, and 0.4 U/μl RNase inhibitor (RiboLock, MBI Fermentas). Virion lysate was then added to a single-step, RT PCR assay with 35 nM MS2 RNA (Roche) as template, 500 nM of each primer (5’-TCCTGCTCAACTTCCTGTCGAG-3’ and 5’-CACAGGTCAAACCTCCTAGGAATG-3’), and hot-start Taq (Promega), all in 20 mM Tris-Cl pH 8.3, 5 mM (NH4)2SO4, 20 mM KCl, 5 mM MgCl2, 0.1 mg/ml BSA, 1/20,000 SYBR Green I (Sigma), and 200 μM dNTPs. All reactions and quantitation of product were carried out with a Biorad CFX96 cycler. The RT step was 42°C for 20 min, and the PCR was programmed for 40 cycles of denaturation at 95°C for 5 s, annealing 55°C for 5 s, extension at 72°C for 20 s and acquisition at 80°C for 5 s. A standard curve was obtained using known concentrations of recombinant HIV-1 RT (Ambion).
Quantitation of viral cDNA
Cell-free virions were normalized by RT-activity and incubated with CRFK, Hela or Jurkat cells in 6-well plates for 12 hrs, for full-length linear cDNA and 2-LTR circles, or 48 hrs, for Alu PCR. For each virus and cell type, infections were also performed in the presence of 40 μM AZT, to control for contamination of plasmid DNA in the PCR reaction. Cells were harvested and washed extensively with PBS. Total DNA was extracted (Qiagen, Qiamp DNA mini kit), quantified, and subjected to real-time PCR with a Biorad CFX96 cycler.
Full-length linear retroviral cDNA and 2-LTR circles were detected with SYBR-Green I based reactions using 100 ng template DNA and 320 nM of each primer pair (5’-ACAAGCTAGTACCAGTTGAGCCAGATAAG-3’ and 5’-gccgtgcgcgcttcagcaagc-3’ for full length linear; 5’-AACTAGGGAACCCACTGCTTAAG-3’ and 5’-TCCACAGATCAAGGATATCTTGTC-5’ or 5’- CAGTGTGGAAAATCTCTAGCAGTAC-3’ for 2-LTR circles) in 20 mM Tris-Cl pH 8.3, 5 mM (NH4)2SO4, 20 mM KCl, 5 mM MgCl2, 0.1 mg/ml BSA, 1/20,000 SYBR Green I (Sigma), and 200 μM dNTPs. The PCR was programmed for 40 cycles of denaturation at 95°C for 5 s, annealing 55°C for 5 s, extension at 72°C for 20 s and acquisition at 80°C for 5 s. Provirus was quantified by Taqman-based ALU-PCR according to the protocol described by Butler et al. [69] using 200 ng of template DNA, primers 5’-AACTAGGGAACCCACTGCTTAAG-3′ and 5′-TGCTGGGATTACAGGCGTGAG-3′ and probe 5′-(FAM)-ACACTACTTGAAGCACTCAAGGCAAGCTTT-(TAMRA)-3′. PCR was performed with a CFX96 cycler (Biorad): 95°C for 15 seconds and 60°C for 90 seconds, for 50 cycles. Relative quantification of retroviral cDNA sequences and ALU PCR was with respect to standard curves prepared from serial dilutions of DNA derived from the cell culture with the highest infection, diluted in DNA extracted from non-infected cells.
Microscopy
CRFK and Jurkat cells transduced with VSV G-pseudotyped HIV-1NL4-3GFP or SIVMAC239-GFP vectors were visualized by phase contrast and fluorescence microscopy 4 days after vector challenge. Pictures of live cell cultures were taken at 100x magnification using a Nikon Eclipse Ti microscope equipped with a DS-QiMC digital camera and NIS elements software.
Heterokaryon assay
2 x 107 Hela-RFP and 2 x 107 Jurkat-TvA were washed with serum-free DMEM and slowly resuspended over 1 min in 500 μl of Polyethylene Glycol 1500 (PEG-1500, GE Healthcare), at 37°C. Cells were incubated for another 2 mins and then 2 ml of serum-free DMEM was added slowly over a period of 4 minutes at 37°C with constant, gentle agitation. An additional 5 ml of serum-free DMEM was added and cells were incubated for 5 min at 37°C. Cells were then pelleted and resuspended in complete medium before seeding in 24-well plates. 6 hours later, cells were challenged with ALV-A Env-pseudotyped vectors. A negative fusion control sample was also produced with no PEG addition. Infected cell cultures were analyzed using a FACS-Canto (BD) 48 hrs after vector challenge. Fluorescence acquisition was performed using blue (488 nm) and red (633 nm) lasers. Dead cells were excluded from the analysis based on propidium iodide staining.
Ethics statement
Human peripheral blood mononuclear cells (PBMC) were obtained from anonymous, untraceable blood donors. This research is therefore considered non-human subjects research by our Institutional Review Board, based on NIH guidelines (45 CFR 46.102(f)): http://grants.nih.gov/grants/policy/hs/faqs_aps_definitions.htm.
Acknowledgments
We are grateful to Aine McKnight, Welkin Johnson, Andrea Kirmaier, Walther Mothes, and Pradeep Uchil for plasmids.
Data Availability
All relevant data are within the paper.
Funding Statement
This work was supported by the National Institute of Health (http://www.nih.gov/) grant RO1AI59159 to JL, National Institute of Health (http://www.nih.gov/) Grant DP1DA034990 to JL, and Swiss National Science Foundation (http://www.snf.ch) grant 3100A0-128655 to JL; European Union Marie Curie (http://ec.europa.eu/research/mariecurieactions/index_en.htm) Grant 237265 to MP and JL; European Union Career Integration (http://ec.europa.eu/research/mariecurieactions/index_en.htm) Grant 322130 to MP; and Canadian Institutes for Health Research (http://www.cihr-irsc.gc.ca/e/193.html) grant 102712 to LB. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Sharp PM, Hahn BH (2011) Origins of HIV and the AIDS pandemic. Cold Spring Harb Perspect Med 1: a006841 10.1101/cshperspect.a006841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sharp PM, Hahn BH (2010) The evolution of HIV-1 and the origin of AIDS. Philosophical Transactions of the Royal Society B: Biological Sciences 365: 2487–2494. 10.1098/rstb.2010.0031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gao F, Bailes E, Robertson DL, Chen Y, Rodenburg CM, et al. (1999) Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature 397: 436–441. 10.1038/17130 [DOI] [PubMed] [Google Scholar]
- 4. Keele BF, Van Heuverswyn F, Li Y, Bailes E, Takehisa J, et al. (2006) Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science 313: 523–526. 10.1126/science.1126531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Plantier J-C, Leoz M, Dickerson JE, De Oliveira F, Cordonnier F, et al. (2009) A new human immunodeficiency virus derived from gorillas. Nat Med 15: 871–872. 10.1038/nm.2016 [DOI] [PubMed] [Google Scholar]
- 6. Van Heuverswyn F, Li Y, Neel C, Bailes E, Keele BF, et al. (2006) Human immunodeficiency viruses: SIV infection in wild gorillas. Nature 444: 164 10.1038/444164a [DOI] [PubMed] [Google Scholar]
- 7. Bailes E, Gao F, Bibollet-Ruche F, Courgnaud V, Peeters M, et al. (2003) Hybrid origin of SIV in chimpanzees. Science 300: 1713 10.1126/science.1080657 [DOI] [PubMed] [Google Scholar]
- 8. Keele BF, Jones JH, Terio KA, Estes JD, Rudicell RS, et al. (2009) Increased mortality and AIDS-like immunopathology in wild chimpanzees infected with SIVcpz. Nature 460: 515–519. 10.1038/nature08200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clavel F, Guyader M, Guetard D, Sallé M, Montagnier L, et al. (1986) Molecular cloning and polymorphism of the human immune deficiency virus type 2. Nature 324: 691–695. 10.1038/324691a0 [DOI] [PubMed] [Google Scholar]
- 10. Damond F, Worobey M, Campa P, Farfara I, Colin G, et al. (2004) Identification of a highly divergent HIV type 2 and proposal for a change in HIV type 2 classification. AIDS Res Hum Retroviruses 20: 666–672. 10.1089/0889222041217392 [DOI] [PubMed] [Google Scholar]
- 11. Gao F, Yue L, Robertson DL, Hill SC, Hui H, et al. (1994) Genetic diversity of human immunodeficiency virus type 2: evidence for distinct sequence subtypes with differences in virus biology. J Virol 68: 7433–7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hirsch VM, Olmsted RA, Murphey-Corb M, Purcell RH, Johnson PR (1989) An African primate lentivirus (SIVsm) closely related to HIV-2. Nature 339: 389–392. 10.1038/339389a0 [DOI] [PubMed] [Google Scholar]
- 13. Apetrei C, Kaur A, Lerche NW, Metzger M, Pandrea I, et al. (2005) Molecular epidemiology of simian immunodeficiency virus SIVsm in U.S. primate centers unravels the origin of SIVmac and SIVstm. J Virol 79: 8991–9005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Letvin NL, Daniel MD, Sehgal PK, Desrosiers RC, Hunt RD, et al. (1985) Induction of AIDS-like disease in macaque monkeys with T-cell tropic retrovirus STLV-III. Science 230: 71–73. [DOI] [PubMed] [Google Scholar]
- 15. Ayouba A, Akoua-Koffi C, Calvignac-Spencer S, Esteban A, Locatelli S, et al. (2013) Evidence for continuing cross-species transmission of SIVsmm to humans: characterization of a new HIV-2 lineage in rural Côte d'Ivoire. AIDS 27: 2488–2491. 10.1097/01.aids.0000432443.22684.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goff SP (2004) Retrovirus restriction factors. Mol Cell 16: 849–859. 10.1016/j.molcel.2004.12.001 [DOI] [PubMed] [Google Scholar]
- 17. Luban J (2007) Cyclophilin A, TRIM5, and resistance to human immunodeficiency virus type 1 infection. J Virol 81: 1054–1061. 10.1128/JVI.01519-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nisole S, Stoye JP, Saïb A (2005) TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol 3: 799–808. 10.1038/nrmicro1248 [DOI] [PubMed] [Google Scholar]
- 19. Song B, Javanbakht H, Perron M, Park DH, Stremlau M, et al. (2005) Retrovirus restriction by TRIM5alpha variants from Old World and New World primates. J Virol 79: 3930–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Si Z, Vandegraaff N, O'Huigin C, Song B, Yuan W, et al. (2006) Evolution of a cytoplasmic tripartite motif (TRIM) protein in cows that restricts retroviral infection. Proc Natl Acad Sci USA 103: 7454–7459. 10.1073/pnas.0600771103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ylinen LMJ, Keckesova Z, Webb BLJ, Gifford RJM, Smith TPL, et al. (2006) Isolation of an active Lv1 gene from cattle indicates that tripartite motif protein-mediated innate immunity to retroviral infection is widespread among mammals. J Virol 80: 7332–7338. 10.1128/JVI.00516-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schaller T, Hué S, Towers GJ (2007) An active TRIM5 protein in rabbits indicates a common antiviral ancestor for mammalian TRIM5 proteins. J Virol 81: 11713–11721. 10.1128/JVI.01468-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, et al. (2004) The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427: 848–853. 10.1038/nature02343 [DOI] [PubMed] [Google Scholar]
- 24. Campbell EM, Perez O, Anderson JL, Hope TJ (2008) Visualization of a proteasome-independent intermediate during restriction of HIV-1 by rhesus TRIM5alpha. J Cell Biol 180: 549–561. 10.1083/jcb.200706154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li X, Li Y, Stremlau M, Yuan W, Song B, et al. (2006) Functional replacement of the RING, B-box 2, and coiled-coil domains of tripartite motif 5alpha (TRIM5alpha) by heterologous TRIM domains. J Virol 80: 6198–6206. 10.1128/JVI.00283-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sebastian S, Luban J (2005) TRIM5alpha selectively binds a restriction-sensitive retroviral capsid. Retrovirology 2: 40 10.1186/1742-4690-2-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stremlau M, Perron M, Lee M, Li Y, Song B, et al. (2006) Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc Natl Acad Sci USA 103: 5514–5519. 10.1073/pnas.0509996103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anderson JL, Campbell EM, Wu X, Vandegraaff N, Engelman A, et al. (2006) Proteasome inhibition reveals that a functional preintegration complex intermediate can be generated during restriction by diverse TRIM5 proteins. J Virol 80: 9754–9760. 10.1128/JVI.01052-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rold CJ, Aiken C (2008) Proteasomal degradation of TRIM5alpha during retrovirus restriction. PLoS Pathog 4: e1000074 10.1371/journal.ppat.1000074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu X, Anderson JL, Campbell EM, Joseph AM, Hope TJ (2006) Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc Natl Acad Sci USA 103: 7465–7470. 10.1073/pnas.0510483103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Berthoux L, Sebastian S, Sokolskaja E, Luban J (2004) Lv1 inhibition of human immunodeficiency virus type 1 is counteracted by factors that stimulate synthesis or nuclear translocation of viral cDNA. J Virol 78: 11739–11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ylinen LMJ, Keckesova Z, Wilson SJ, Ranasinghe S, Towers GJ (2005) Differential restriction of human immunodeficiency virus type 2 and simian immunodeficiency virus SIVmac by TRIM5alpha alleles. J Virol 79: 11580–11587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ohkura S, Yap MW, Sheldon T, Stoye JP (2006) All three variable regions of the TRIM5alpha B30.2 domain can contribute to the specificity of retrovirus restriction. J Virol 80: 8554–8565. 10.1128/JVI.00688-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hatziioannou T, Perez-Caballero D, Yang A, Cowan S, Bieniasz PD (2004) Retrovirus resistance factors Ref1 and Lv1 are species-specific variants of TRIM5alpha. Proc Natl Acad Sci USA 101: 10774–10779. 10.1073/pnas.0402361101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Keckesova Z, Ylinen LMJ, Towers GJ (2004) The human and African green monkey TRIM5alpha genes encode Ref1 and Lv1 retroviral restriction factor activities. Proc Natl Acad Sci USA 101: 10780–10785. 10.1073/pnas.0402474101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yap MW, Nisole S, Lynch C, Stoye JP (2004) Trim5alpha protein restricts both HIV-1 and murine leukemia virus. Proc Natl Acad Sci USA 101: 10786–10791. 10.1073/pnas.0402876101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hatziioannou T, Cowan S, Schwedler von UK, Sundquist WI, Bieniasz PD (2004) Species-specific tropism determinants in the human immunodeficiency virus type 1 capsid. J Virol 78: 6005–6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stremlau M, Perron M, Welikala S, Sodroski J (2005) Species-specific variation in the B30.2(SPRY) domain of TRIM5alpha determines the potency of human immunodeficiency virus restriction. J Virol 79: 3139–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yap MW, Nisole S, Stoye JP (2005) A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr Biol 15: 73–78. 10.1016/j.cub.2004.12.042 [DOI] [PubMed] [Google Scholar]
- 40. Besnier C, Takeuchi Y, Towers G (2002) Restriction of lentivirus in monkeys. Proc Natl Acad Sci USA 99: 11920–11925. 10.1073/pnas.172384599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hatziioannou T, Cowan S, Goff SP, Bieniasz PD, Towers GJ (2003) Restriction of multiple divergent retroviruses by Lv1 and Ref1. EMBO J 22: 385–394. 10.1093/emboj/cdg042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Münk C, Brandt SM, Lucero G, Landau NR (2002) A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc Natl Acad Sci USA 99: 13843–13848. 10.1073/pnas.212400099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cowan S, Hatziioannou T, Cunningham T, Muesing MA, Göttlinger HG, et al. (2002) Cellular inhibitors with Fv1-like activity restrict human and simian immunodeficiency virus tropism. Proc Natl Acad Sci USA 99: 11914–11919. 10.1073/pnas.162299499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hofmann W, Schubert D, LaBonte J, Munson L, Gibson S, et al. (1999) Species-specific, postentry barriers to primate immunodeficiency virus infection. J Virol 73: 10020–10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sayah DM, Sokolskaja E, Berthoux L, Luban J (2004) Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430: 569–573. 10.1038/nature02777 [DOI] [PubMed] [Google Scholar]
- 46. Lai RPJ, Yan J, Heeney J, McClure MO, Göttlinger H, et al. (2011) Nef decreases HIV-1 sensitivity to neutralizing antibodies that target the membrane-proximal external region of TMgp41. PLoS Pathog 7: e1002442 10.1371/journal.ppat.1002442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sebastian S, Sokolskaja E, Luban J (2006) Arsenic counteracts human immunodeficiency virus type 1 restriction by various TRIM5 orthologues in a cell type-dependent manner. J Virol 80: 2051–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pertel T, Hausmann S, Morger D, Züger S, Guerra J, et al. (2011) TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472: 361–365. 10.1038/nature09976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pertel T, Reinhard C, Luban J (2011) Vpx rescues HIV-1 transduction of dendritic cells from the antiviral state established by type 1 interferon. Retrovirology 8: 49 10.1186/1742-4690-8-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Reinhard C, Bottinelli D, Kim B, Luban J (2014) Vpx rescue of HIV-1 from the antiviral state in mature dendritic cells is independent of the intracellular deoxynucleotide concentration. Retrovirology 11: 12 10.1186/1742-4690-11-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Apetrei C, Lerche NW, Pandrea I, Gormus B, Silvestri G, et al. (2006) Kuru experiments triggered the emergence of pathogenic SIVmac. AIDS 20: 317–321. 10.1097/01.aids.0000206498.71041.0e [DOI] [PubMed] [Google Scholar]
- 52. Ling B, Apetrei C, Pandrea I, Veazey RS, Lackner AA, et al. (2004) Classic AIDS in a sooty mangabey after an 18-year natural infection. J Virol 78: 8902–8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hirsch V, Adger-Johnson D, Campbell B, Goldstein S, Brown C, et al. (1997) A molecularly cloned, pathogenic, neutralization-resistant simian immunodeficiency virus, SIVsmE543-3. J Virol 71: 1608–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kirmaier A, Wu F, Newman RM, Hall LR, Morgan JS, et al. (2010) TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol 8 10.1371/journal.pbio.1000462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu Z, Pan Q, Ding S, Qian J, Xu F, et al. (2013) The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 14: 398–410. 10.1016/j.chom.2013.08.015 [DOI] [PubMed] [Google Scholar]
- 56. Goujon C, Moncorgé O, Bauby H, Doyle T, Ward CC, et al. (2013) Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502: 559–562. 10.1038/nature12542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, et al. (2013) MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature. 10.1038/nature12653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Grütter MG, Luban J (2012) TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Curr Opin Virol 2: 142–150. 10.1016/j.coviro.2012.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sokolskaja E, Berthoux L, Luban J (2006) Cyclophilin A and TRIM5alpha independently regulate human immunodeficiency virus type 1 infectivity in human cells. J Virol 80: 2855–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Towers GJ, Hatziioannou T, Cowan S, Goff SP, Luban J, et al. (2003) Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat Med 9: 1138–1143. 10.1038/nm910 [DOI] [PubMed] [Google Scholar]
- 61. Berthoux L, Towers GJ, Gurer C, Salomoni P, Pandolfi PP, et al. (2003) As(2)O(3) enhances retroviral reverse transcription and counteracts Ref1 antiviral activity. J Virol 77: 3167–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Saenz DT, Teo W, Olsen JC, Poeschla EM (2005) Restriction of feline immunodeficiency virus by Ref1, Lv1, and primate TRIM5alpha proteins. J Virol 79: 15175–15188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kaul A, Stauffer S, Berger C, Pertel T, Schmitt J, et al. (2009) Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog 5: e1000546 10.1371/journal.ppat.1000546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ (2005) A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci USA 102: 13212–13217. 10.1073/pnas.0506306102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP (1993) Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73: 1067–1078. [DOI] [PubMed] [Google Scholar]
- 66. Berthoux L, Sebastian S, Sokolskaja E, Luban J (2005) Cyclophilin A is required for TRIM5{alpha}-mediated resistance to HIV-1 in Old World monkey cells. Proc Natl Acad Sci USA 102: 14849–14853. 10.1073/pnas.0505659102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Neagu MR, Ziegler P, Pertel T, Strambio de Castillia C, Grütter C, et al. (2009) Potent inhibition of HIV-1 by TRIM5-cyclophilin fusion proteins engineered from human components. J Clin Invest 119: 3035–3047. 10.1172/JCI39354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mitrophanous K, Yoon S, Rohll J, Patil D, Wilkes F, et al. (1999) Stable gene transfer to the nervous system using a non-primate lentiviral vector. Gene Ther 6: 1808–1818. 10.1038/sj.gt.3301023 [DOI] [PubMed] [Google Scholar]
- 69. Butler SL, Hansen MS, Bushman FD (2001) A quantitative assay for HIV DNA integration in vivo. Nat Med 7: 631–634. 10.1038/87979 [DOI] [PubMed] [Google Scholar]
- 70. De Iaco A, Luban J (2011) Inhibition of HIV-1 infection by TNPO3 depletion is determined by capsid and detectable after viral cDNA enters the nucleus. Retrovirology 8: 98 10.1186/1742-4690-8-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liu GE, Alkan C, Jiang L, Zhao S, Eichler EE (2009) Comparative analysis of Alu repeats in primate genomes. Genome Res 19: 876–885. 10.1101/gr.083972.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. De Iaco A, Santoni F, Vannier A, Guipponi M, Antonarakis S, et al. (2013) TNPO3 protects HIV-1 replication from CPSF6-mediated capsid stabilization in the host cell cytoplasm. Retrovirology 10: 20 10.1186/1742-4690-10-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Morozova KS, Piatkevich KD, Gould TJ, Zhang J, Bewersdorf J, et al. (2010) Far-red fluorescent protein excitable with red lasers for flow cytometry and superresolution STED nanoscopy. Biophys J 99: L13–L15. 10.1016/j.bpj.2010.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Manel N, Hogstad B, Wang Y, Levy DE, Unutmaz D, et al. (2010) A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature 467: 214–217. 10.1038/nature09337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Simon JH, Gaddis NC, Fouchier RA, Malim MH (1998) Evidence for a newly discovered cellular anti-HIV-1 phenotype. Nat Med 4: 1397–1400. 10.1038/3987 [DOI] [PubMed] [Google Scholar]
- 76. Varthakavi V, Smith RM, Bour SP, Strebel K, Spearman P (2003) Viral protein U counteracts a human host cell restriction that inhibits HIV-1 particle production. Proc Natl Acad Sci USA 100: 15154–15159. 10.1073/pnas.2433165100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bates P, Young JA, Varmus HE (1993) A receptor for subgroup A Rous sarcoma virus is related to the low density lipoprotein receptor. Cell 74: 1043–1051. [DOI] [PubMed] [Google Scholar]
- 78. Schmitz C, Marchant D, Neil SJD, Aubin K, Reuter S, et al. (2004) Lv2, a novel postentry restriction, is mediated by both capsid and envelope. J Virol 78: 2006–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pineda MJ, Orton BR, Overbaugh J (2007) A TRIM5alpha-independent post-entry restriction to HIV-1 infection of macaque cells that is dependent on the path of entry. Virology 363: 310–318. 10.1016/j.virol.2007.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. McKnight A, Griffiths DJ, Dittmar M, Clapham P, Thomas E (2001) Characterization of a late entry event in the replication cycle of human immunodeficiency virus type 2. J Virol 75: 6914–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Boone LR, Innes CL, Heitman CK (1990) Abrogation of Fv-1 restriction by genome-deficient virions produced by a retrovirus packaging cell line. J Virol 64: 3376–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Towers G, Collins M, Takeuchi Y (2002) Abrogation of Ref1 retrovirus restriction in human cells. J Virol 76: 2548–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kahl CA, Cannon PM, Oldenburg J, Tarantal AF, Kohn DB (2008) Tissue-specific restriction of cyclophilin A-independent HIV-1- and SIV-derived lentiviral vectors. Gene Ther 15: 1079–1089. 10.1038/gt.2008.50 [DOI] [PubMed] [Google Scholar]
- 84. Logue EC, Taylor KT, Goff PH, Landau NR (2011) The cargo-binding domain of transportin 3 is required for lentivirus nuclear import. J Virol 85: 12950–12961. 10.1128/JVI.05384-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, et al. (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472: 481–485. 10.1038/nature09907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kapahi P, Takahashi T, Natoli G, Adams SR, Chen Y, et al. (2000) Inhibition of NF-kappa B activation by arsenite through reaction with a critical cysteine in the activation loop of Ikappa B kinase. J Biol Chem 275: 36062–36066. 10.1074/jbc.M007204200 [DOI] [PubMed] [Google Scholar]
- 87. Uchil PD, Quinlan BD, Chan W-T, Luna JM, Mothes W (2008) TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog 4: e16 10.1371/journal.ppat.0040016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Perez-Caballero D, Hatziioannou T, Zhang F, Cowan S, Bieniasz PD (2005) Restriction of human immunodeficiency virus type 1 by TRIM-CypA occurs with rapid kinetics and independently of cytoplasmic bodies, ubiquitin, and proteasome activity. J Virol 79: 15567–15572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, et al. (2010) Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7: 221–233. 10.1016/j.chom.2010.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Pryciak PM, Varmus HE (1992) Fv-1 restriction and its effects on murine leukemia virus integration in vivo and in vitro. J Virol 66: 5959–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Burdick RC, Hu W-S, Pathak VK (2013) Nuclear import of APOBEC3F-labeled HIV-1 preintegration complexes. Proceedings of the National Academy of Sciences 110: E4780–E4789. 10.1073/pnas.1315996110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yamashita M, Perez O, Hope TJ, Emerman M (2007) Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS Pathog 3: 1502–1510. 10.1371/journal.ppat.0030156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. De Iaco A, Luban J (2014) Cyclophilin A promotes HIV-1 reverse transcription but its effect on transduction correlates best with its effect on nuclear entry of viral cDNA. Retrovirology 11: 11 10.1186/1742-4690-11-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. de Silva TI, Cotten M, Rowland-Jones SL (2008) HIV-2: the forgotten AIDS virus. Trends Microbiol 16: 588–595. 10.1016/j.tim.2008.09.003 [DOI] [PubMed] [Google Scholar]
- 95. He J, Chen Y, Farzan M, Choe H, Ohagen A, et al. (1997) CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature 385: 645–649. 10.1038/385645a0 [DOI] [PubMed] [Google Scholar]
- 96. Guyader M, Emerman M, Sonigo P, Clavel F, Montagnier L, et al. (1987) Genome organization and transactivation of the human immunodeficiency virus type 2. Nature 326: 662–669. 10.1038/326662a0 [DOI] [PubMed] [Google Scholar]
- 97. Miyamoto T, Nakayama EE, Yokoyama M, Ibe S, Takehara S, et al. (2012) The Carboxyl-Terminus of Human Immunodeficiency Virus Type 2 Circulating Recombinant form 01_AB Capsid Protein Affects Sensitivity to Human TRIM5α. PLoS ONE 7: e47757 10.1371/journal.pone.0047757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D (1997) Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 15: 871–875. 10.1038/nbt0997-871 [DOI] [PubMed] [Google Scholar]
- 99. Bock M, Bishop KN, Towers G, Stoye JP (2000) Use of a transient assay for studying the genetic determinants of Fv1 restriction. J Virol 74: 7422–7430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Harrison IP, McKnight A (2011) Cellular entry via an actin and clathrin-dependent route is required for Lv2 restriction of HIV-2. Virology 415: 47–55. 10.1016/j.virol.2011.04.001 [DOI] [PubMed] [Google Scholar]
- 101. Cosset FL, Morling FJ, Takeuchi Y, Weiss RA, Collins MK, et al. (1995) Retroviral retargeting by envelopes expressing an N-terminal binding domain. J Virol 69: 6314–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zingler K, Bélanger CA, Peters R, Agard E, Young JA (1995) Identification and characterization of the viral interaction determinant of the subgroup A avian leukosis virus receptor. J Virol 69: 4261–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Boerger AL, Snitkovsky S, Young JA (1999) Retroviral vectors preloaded with a viral receptor-ligand bridge protein are targeted to specific cell types. Proc Natl Acad Sci USA 96: 9867–9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Berthoux L, Sebastian S, Sayah DM, Luban J (2005) Disruption of human TRIM5alpha antiviral activity by nonhuman primate orthologues. J Virol 79: 7883–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sokolskaja E, Sayah DM, Luban J (2004) Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J Virol 78: 12800–12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Braaten D, Franke EK, Luban J (1996) Cyclophilin A is required for the replication of group M human immunodeficiency virus type 1 (HIV-1) and simian immunodeficiency virus SIV(CPZ)GAB but not group O HIV-1 or other primate immunodeficiency viruses. J Virol 70: 4220–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Sebastian S, Grütter C, Strambio de Castillia C, Pertel T, Olivari S, et al. (2009) An invariant surface patch on the TRIM5alpha PRYSPRY domain is required for retroviral restriction but dispensable for capsid binding. J Virol 83: 3365–3373. 10.1128/JVI.00432-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Pizzato M, Erlwein O, Bonsall D, Kaye S, Muir D, et al. (2009) A one-step SYBR Green I-based product-enhanced reverse transcriptase assay for the quantitation of retroviruses in cell culture supernatants. J Virol Methods 156: 1–7. 10.1016/j.jviromet.2008.10.012 [DOI] [PubMed] [Google Scholar]
- 109. Vermeire J, Naessens E, Vanderstraeten H, Landi A, Iannucci V, et al. (2012) Quantification of reverse transcriptase activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and retroviral vectors. PLoS ONE 7: e50859 10.1371/journal.pone.0050859 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.