

Abstract
Severe chronic active Epstein-Barr virus (CAEBV) disease is defined as a severe progressive illness lasting 6 months or longer with infiltration of tissues with EBV-positive lymphocytes, markedly elevated levels of EBV DNA in the blood, and no known immunodeficiency such as HIV. These patients usually have fever, splenomegaly, lymphadenopathy, and may have markedly elevated EBV antibody titers to viral capsid antigen. Although the cause of most cases of severe CAEBV is unknown, one well-documented case was associated with compound heterozygous mutations in PRF1 (perforin 1). Here we report a patient with prolonged severe CAEBV who underwent bone marrow transplant for his disease and subsequently was found to have compound heterozygous mutations in STXBP2 (MUNC18-2) as well as a heterozygous mutation in PRF1 (perforin 1).
Keywords: Epstein-Barr virus, chronic active Epstein-Barr virus, MUNC18-2, STXBP2, perforin 1
Case Report
A 24 year old Caucasian man presented to National Institutes of Health (NIH) with lymphadenopathy, pancytopenia, progressive encephalitis, and hemophagocytosis. The patient was previously reported in brief [1]. In 1991 he presented with fatigue, myalgias, fever, pancytopenia; he was treated with antibiotics. In 1993 he had recurrent fatigue, myalgias, fever, night sweats, pancytopenia, hepatitis, and proteinuria; a bone marrow biopsy showed lymphoid aggregates. In 1994 he had recurrent fever, pancytopenia, hepatitis, splenomegaly, encephalitis, peripheral neuropathy, and facial palsy. The cerebrospinal fluid (CSF) showed pleocytosis, and an MRI showed enhancement of frontal lobe. The EBV IgM was <10; EA IgG 1:640; EBNA IgG >1:5, and VCA IgG 1:2,560. He was treated with acyclovir and intravenous immunoglobulin, and then corticosteroids. In 1995 he again had recurrent fatigue and fevers, pancytopenia, splenomegaly, and he improved on corticosteroids. In 1996 he again had fever, fatigue, splenomegaly, progressive neurological deficits (cerebellar signs, diplopia, peripheral neuropathy); magnetic resonance imaging (MRI) showed multiple cerebellar and cerebral lesions. He was treated with corticosteroids, and interferon-β. In 1997 he had fever, fatigue, malaise, splenomegaly, hepatitis, lymphadenopathy, pancytopenia, paresthesias, and numbness. MRI showed demyelinating lesions in the thoracic spine. He developed zoster. A splenectomy showed a massively enlarged spleen with marked hemophagocytosis and scattered EBV encoded RNA (EBER) positive lymphocytes and hemophagocytosis (Fig. 1). EBV was in B cells based on combined staining for EBER and CD20 or CD3. He was treated with acyclovir, corticosteroids, cyclosporine, and splenectomy. In 1998 a bone marrow biopsy showed hemophagocytosis. MRI showed periventricular white matter changes. EBV PCR of the blood was positive and a lymph node and liver biopsy were positive for EBV RNA. He was treated with corticosteroids and azathioprine. In 1999 (eight years after his initial presentation) he had fatigue, weight loss, night sweats, fever, and anemia. A bone marrow and liver biopsy showed an atypical lymphohistiocytic T cell infiltrate that contained scattered EBER-positive lymphocytes.
Fig. 1.
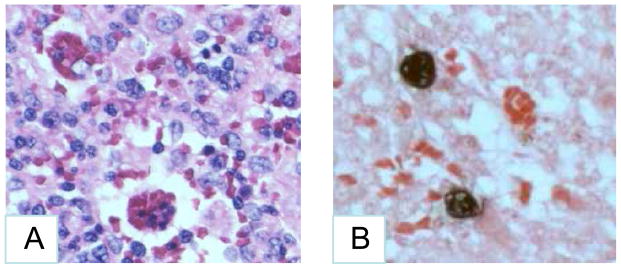
Pathology of tissue at splenectomy. a Hemophagocytosis. Original magnification is 400x. b In situ hybridization shows lymphocytes containing EBV encoded RNA (EBER). Original magnification is 1000x.
He was treated with azathioprine, corticosteroids, intravenous immunoglobulin, and then was referred to the Baylor College of Medicine and Houston Methodist Hospital for transplant. He received etoposide, but still had residual disease, and then received conditioning with cytosine arabinoside, cyclophosphamide, anti-thymocyte globulin, and total body irradiation followed by a T cell depleted bone marrow from a 5/6 matched unrelated donor. He received additional prophylaxis for graft-versus host disease (GVHD) with cyclosporine. His post-transplant course was complicated by stage 1 skin GVHD, and subsequently he had zoster which resolved with acyclovir. He had a mild CSF pleocytosis that resolved without therapy. He is currently 15 years post-transplant and working full time. He is EBV seronegative, his blood EBV PCR is negative, and he has complete engraftment.
First-generation (Sanger) and next-generation (Agilent HaloPlex Target Enrichment and Life Technologies Ion Proton) genomic DNA sequencing from pre-transplant peripheral blood mononuclear cells detected two heterozygous mutations in STXBP2 (which encodes syntaxin binding protein or MUNC18-2), one at nucleotide 1247 G>C at a splice acceptor site of exon 15, and a second at nucleotide 1621 G>A resulting in a glycine to serine change at amino acid 541 in exon 18 (nucleotide numbering based on NCBI reference sequence NM_006949.2). The patient also had a heterozygous mutation in PRF1 (perforin 1) at nucleotide 272 C>T, resulting in an alanine to valine change at amino acid 91 (nucleotide numbering based on NCBI reference sequence NM_001083116.1). All coding exons and adjacent splicing sites within these genes were PCR-amplified and verified by Sanger sequencing. Sanger sequencing of genomic DNA from the patient’s mother showed only the glycine to serine mutation at amino acid 541 in exon 18 of STXBP2. Research studies were approved by the Institutional Review Board at the National Institute of Allergy and Infectious Diseases, NIH and the patient provided written informed consent. An immunoblot showed no detectable MUNC18-2 in the patient’s stimulated PBMCs prior to transplant, but normal levels of the protein after transplant and in a healthy blood bank donor (Fig. 2A). Stimulation of PBMCs with phorbol 12-myristate 13-acetate (PMA) and staining for cell surface CD107a, a marker of degranulation, showed reduced CD107a in the patient before transplant compared to healthy donors, and restoration of the level of CD107a to the normal level after transplant (Fig. 2B). Thus, the patient’s cells were impaired for cytotoxic granule trafficking and/or degranulation before transplant.
Fig. 2.

Immunoblot for MUNC18-2 and cell surface levels of CD107a to measure degranulation. a Immunoblot shows lack of MUNC18-2 in the patient prior to transplant, and normal levels of the protein after transplant. Cryopreserved peripheral blood mononuclear cells were stimulated with 1 ug/mL CD3 + CD28 for 48 hours in RPMI complete medium. Cells were then expanded in RPMI complete media with 100 U/mL IL-2 for 7 days. The expanded cells were lysed in RIPA buffer containing protease inhibitor, and after gel electrophoresis and transfer to a nylon membrane, the membrane was blotted with anti-MUNC18-2 antibody (LifeSpan Biosciences, Inc.). b Peripheral blood mononuclear cells were cultured in RPMI complete medium with 10 U/mL IL-2 and anti-CD107a-AF647 (BioLegend) in the presence or absence of PMA and ionomycin for one hour. Monensin (2 uM) was added for an additional 4 hours, after which the cells were washed and stained with CD8-FITC, and CD8 and CD107a was measured by flow cytometry. The experiment was performed twice with patient cells and at least three healthy controls each time; the results shown are pooled from both experiments and the means and standard error are indicated.
Discussion
MUNC18-2 is one of several proteins important for docking of vesicles containing cytotoxic granules to the plasma membrane of cytotoxic T lymphocytes and NK cells. Docking of vesicles allows their fusion with the plasma membrane and release of cytotoxic granules, including perforin 1 and granzymes, with destruction of virus-infected cells [2]. MUNC18-2, encoded by STXBP2, binds to syntaxin 11 and to the SNARE complex to regulate SNARE-mediated fusion of membrane vesicles with the plasma membrane [3]. Mutations in MUNC18-2 impair secretion of cytotoxic granules and result in familial hemophagocytic lymphohistocytosis (FHL) type 5 [4, 5]. In three reviews of FHL5, patients presented at a median of age of 3–15 months, with a range of 3 days to 17 years of age [6, 7, 8]. Our patient presented at age 17, much older than most of the other patients. Most patients present with liver disease and about 50% have neurologic disease, often with lesions on MRI (similar to our patient). Similar lesions on MRI have also been reported in patients with severe CAEBV [9]. About one-third of patients also have an inflammatory bowel-like disease, bleeding disorders, or hypogammaglobulinemia (our patient had the latter).
Our patient had three heterozygous mutations that likely contributed to his disease. Both the mutation at the splice acceptor site of exon 15 and the missense mutation in exon 18 of STXBP2 have been reported previously in patients with FHL [8. 10]. Like other patients with mutations in the splice acceptor site of exon 15 [5,8], our patient presented at an older age and had a more prolonged course with recurrent reactivation of disease that transiently responded to corticosteroids compared with patients who have other mutations. Previously four patients with CAEBV were reported with mutations in STXBP2; one had the G541S mutation observed in our patient as well as another missense mutation; the other three patients were homozygous for the splice acceptor site of exon 15 mutation [10,11]. Two of these patients presented with hemophagocytic lymphohistiocytosis (HLH) after primary EBV infection, one presented with HLH that only met full criteria for HLH shortly after primary EBV infection, and one had been infected with EBV in the past before presenting with HLH. Tissue evidence of EBV was reported in only one of the four cases, with EBV present in the lung. Our patient also had an A91V perforin 1 mutation, which although present in up to 5% of Caucasians (Exome Aggregation Constorium population frequency), impairs the function of the protein [12, 13]. Since the mother had only the STXBP2 G541S mutation, and DNA was not available from the unaffected father (deceased), the STXBP2 splice mutation and/or the PRF1 mutation were carried by the father or were de novo mutations. When combined with mutations in other proteins associated with HLH, including MUNC18-2, the A91V perforin 1 mutation predisposes to HLH [14, 15]. Thus, we postulate that the perforin mutation likely exacerbated the effect of the compound heterozygous STXBP2 mutation reported here. We previously reported one patient with severe CAEBV and compound heterozygous mutations in PRF1 [16]. Both the latter patient and our patient had EBV predominantly in B cells in the tissues and both were Caucasian. In contrast, most patients in the literature with severe CAEBV are Asian and have EBV in T or NK cells. Thus, the finding of B cell CAEBV in the two patients with mutations in PRF1 and STXBP2 may be more related to their ethnicity than their mutations.
Our finding of mutations in SXTBP2 and PRF1 [16] in patients with severe CAEBV suggests that mutations in other proteins that contribute to HLH may also be found in patients with severe CAEBV.
Acknowledgments
This work was supported by the intramural research program of the National Institute of Allergy and Infectious Diseases.
Contributor Information
Jeffrey I. Cohen, Laboratory of Infectious Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland
Julie E. Niemela, Department of Laboratory Medicine, Clinical Center, National Institutes of Health, Bethesda, Maryland
Jennifer L. Stoddard, Department of Laboratory Medicine, Clinical Center, National Institutes of Health, Bethesda, Maryland
Stefania Pittaluga, Laboratory of Pathology, National Cancer Institute, National Institutes of Health, Bethesda, Maryland.
Helen Heslop, Center for Cell and Gene Therapy, Baylor College of Medicine, The Methodist Hospital and Texas Children’s Hospital, Houston, Texas.
Elaine S. Jaffe, Laboratory of Pathology, National Cancer Institute, National Institutes of Health, Bethesda, Maryland
Kennichi Dowdell, Laboratory of Infectious Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland.
References
- 1.Cohen JI, Jaffe ES, Dale JK, Pittaluga S, Heslop HE, Rooney CM, et al. Characterization and treatment of chronic active Epstein-Barr virus disease: a 28-year experience in the United States. Blood. 2011;117:5835–49. doi: 10.1182/blood-2010-11-316745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Filipovich AH. The expanding spectrum of hemophagocytic lymphohistiocytosis. Curr Opin Allergy Clin Immunol. 2011;11:512–6. doi: 10.1097/ACI.0b013e32834c22f5. [DOI] [PubMed] [Google Scholar]
- 3.Hackmann Y, Graham SC, Ehl S, Höning S, Lehmberg K, Aricò M, et al. Syntaxin binding mechanism and disease-causing mutations in Munc18-2. Proc Natl Acad Sci U S A. 2013;110:E4482–91. doi: 10.1073/pnas.1313474110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.zur Stadt U, Rohr J, Seifert W, Koch F, Grieve S, Pagel J, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet. 2009;85:482–92. doi: 10.1016/j.ajhg.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Côte M, Ménager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. 2009;119:3765–73. doi: 10.1172/JCI40732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meeths M, Entesarian M, Al-Herz W, Chiang SC, Wood SM, Al-Ateeqi W, et al. Spectrum of clinical presentations in familial hemophagocytic lymphohistiocytosis type 5 patients with mutations in STXBP2. Blood. 2010;116:2635–43. doi: 10.1182/blood-2010-05-282541. [DOI] [PubMed] [Google Scholar]
- 7.Cetica V, Santoro A, Gilmour KC, Sieni E, Beutel K, Pende D, et al. STXBP2 mutations in children with familial haemophagocytic lymphohistiocytosis type 5. J Med Genet. 2010;47:595–600. doi: 10.1136/jmg.2009.075341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pagel J, Beutel K, Lehmberg K, Koch F, Maul-Pavicic A, Rohlfs AK, et al. Distinct mutations in STXBP2 are associated with variable clinical presentations in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL5) Blood. 2012;119:6016–24. doi: 10.1182/blood-2011-12-398958. [DOI] [PubMed] [Google Scholar]
- 9.Ohga S, Sanefuji M, Ishimura M, Nomura A, Torisu H, Kira R, et al. Epstein-Barr virus load in cerebrospinal fluid of patients with chronic active Epstein-Barr virus infection. Pediatr Infect Dis J. 2008;27:1027–30. doi: 10.1097/INF.0b013e318178d21e. [DOI] [PubMed] [Google Scholar]
- 10.Rohr J, Beutel K, Maul-Pavicic A, Vraetz T, Thiel J, Warnatz K, et al. Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica. 2010;95:2080–7. doi: 10.3324/haematol.2010.029389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beutel K, Gross-Wieltsch U, Wiesel T, Stadt UZ, Janka G, Wagner HJ. Infection of T lymphocytes in Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in children of non-Asian origin. Pediatr Blood Cancer. 2009;53:184–90. doi: 10.1002/pbc.22037. [DOI] [PubMed] [Google Scholar]
- 12.Trambas C, Gallo F, Pende D, Marcenaro S, Moretta L, De Fusco C, et al. A single amino acid change, A91V, leads to conformational changes that can impair processing to the active form of perforin. Blood. 2005;106:932–7. doi: 10.1182/blood-2004-09-3713. [DOI] [PubMed] [Google Scholar]
- 13.Voskoboinik I, Sutton VR, Ciccone A, House CM, Chia J, Darcy PK, et al. Perforin activity and immune homeostasis: the common A91V polymorphism in perforin results in both presynaptic and postsynaptic defects in function. Blood. 2007;110:1184–90. doi: 10.1182/blood-2007-02-072850. [DOI] [PubMed] [Google Scholar]
- 14.Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood. 2011;118:5794–8. doi: 10.1182/blood-2011-07-370148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang K, Chandrakasan S, Chapman H, Valencia CA, Husami A, Kissell D, et al. Synergistic defects of different molecules in the cytotoxic pathway lead to clinical familial hemophagocytic lymphohistiocytosis. Blood. 2014;124:1331–4. doi: 10.1182/blood-2014-05-573105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katano H, Ali MA, Patera AC, Catalfamo M, Jaffe ES, Kimura H, et al. Chronic active Epstein-Barr virus infection associated with mutations in perforin that impair its maturation. Blood. 2004;103:1244–52. doi: 10.1182/blood-2003-06-2171. [DOI] [PubMed] [Google Scholar]