

To the editor
Basal cell carcinomas (BCCs) are locally invasive epithelial tumors that are caused by activating mutations in the Hedgehog (HH) pathway, typically through the loss of the receptor Patched1 or by activating the G-protein coupled receptor Smoothened (SMO). Genomic analysis by our group and others have revealed that BCCs are typically diploid and carry a high frequency of non-silent single nucleotide variants (SNVs) compared to other cutaneous and non-cutaneous tumors (Alexandrov et al. 2013; Atwood et al. 2014; Atwood et al. 2015; Jayaraman et al. 2014). Given their high mutational load, how these variants confer selective tumor growth without deleterious effects remains poorly understood. We previously identified and functionally validated nine SMO mutations that drive the majority of drug resistance in BCC through two distinct mechanisms that maintain HH signaling in the presence of drug: induction of constitutive activity or disruption of ligand binding (Atwood et al. 2015). However, SMO mutations with unclear function are frequently found across many HH and non-HH dependent cancers with drug-resistant BCCs bearing the highest rate of recurrent mutations at 66% (Figure 1a).
Figure 1. Mutational profile of SMO in advanced basal cell carcinoma.
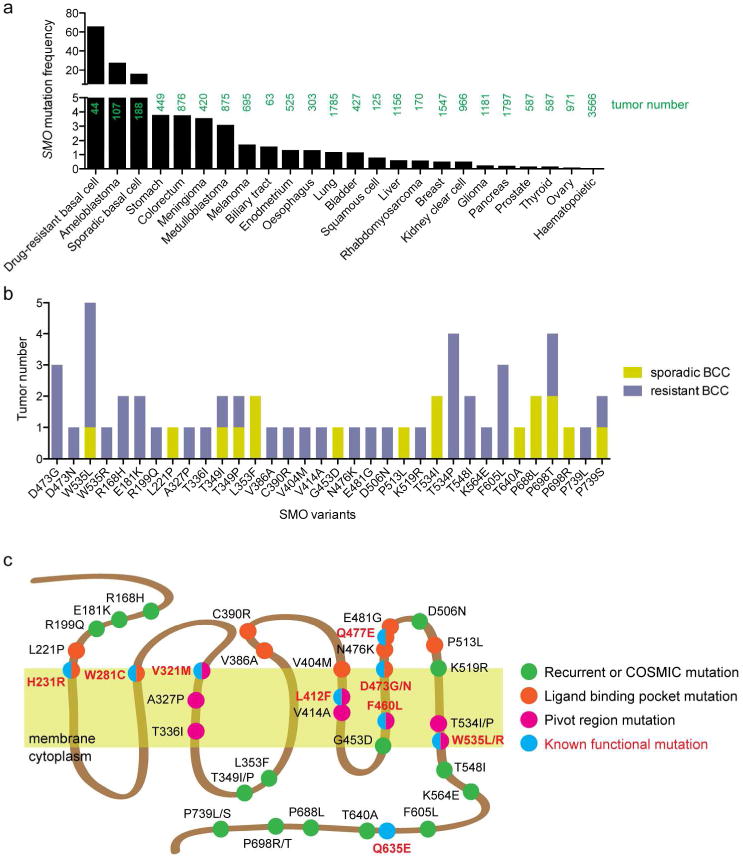
(a) Frequency of SMO mutations in various cancers from the COSMIC database and current literature (Atwood et al. 2015; Kool et al. 2014; Lee et al. 2014; Sweeney et al. 2014). (b) Number of tumors bearing recurrent, COSMIC database, or regional-specific (ligand binding pocket or pivot region) mutations. (c) Schematic of SMO showing location of mutations.
To determine how these additional SMO mutations promote tumor growth, we identified 28 mutations through our genomic analysis of 44 drug-resistant and 36 sporadic BCC that were either recurrent, found to overlap with the COSMIC database, or were regional-specific (ligand binding pocket or pivot region) and interrogated their ability to promote HH signaling (Figure 1b, c). We expressed wildtype human SMO (SMO-WT) or SMO mutants in Smo-null mouse embryonic fibroblasts (MEFs) to assess the ability of these variants to activate the HH pathway with or without ligand. SMO-W535L is a known constitutively active mutant that was present in many of our tumor samples and significantly increased basal HH activity in the absence of HH ligand as determined by mRNA levels of the HH target gene Gli1 (Figure 2a). No other SMO variant induced constitutive activity, including SMO-WT and the known ligand binding pocket mutant SMO-D473G (Yauch et al. 2009), suggesting these variants could not confer tumor growth by themselves. This was surprising as several of the residues (A327P, T336I, V414A, and T534I) lie in the pivot regions of transmembrane helices 3, 5, and 7 that control SMO activation (Figure 1c) and correspond to residues 320-340, 410-415, and 530-540 from the SMO crystal structure (Atwood et al. 2015; Wang et al. 2013). Addition of HH ligand revealed a range of responses from the SMO variants to activate the pathway. No SMO mutation conferred a statistically significant increase in SMO activity with the majority of variants acting as passenger mutations (Figure 2b). However, 13 variants disrupted SMO activity by 50% or more with 7 of the variants effectively abolishing activity. How the tumor could withstand the loss of SMO activity remains unclear, although only one functional copy of SMO is necessary to transduce HH signal.
Figure 2. Variation in the response of SMO mutations to Hedgehog ligand.
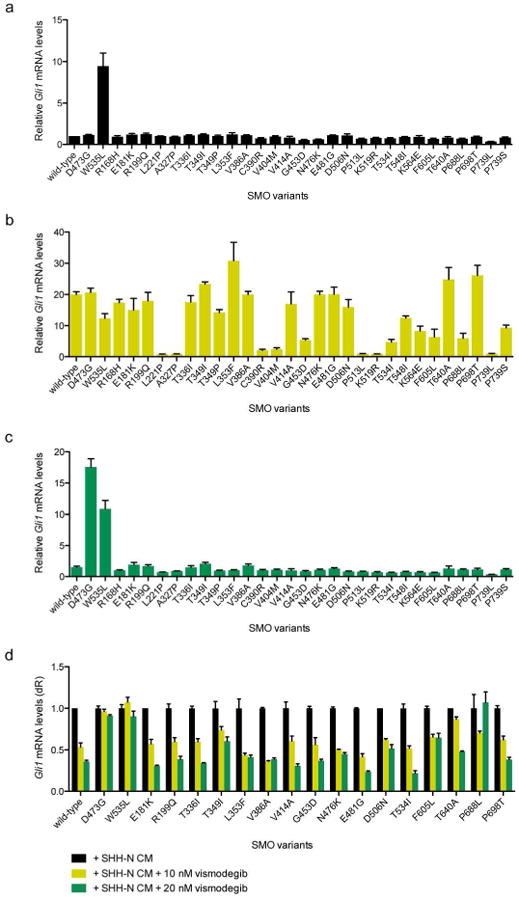
(a) SMO variants expressed in Smo-null MEFs under serum-starvation conditions demonstrating baseline HH pathway activity. Note that previously characterized SMO mutations conferring vismodegib resistance (Atwood et al. 2015) were excluded from this analysis. (b) SMO variant expressing cells treated with SHH-N CM reveal a range of SMO activity to promote HH target gene induction. (c) SMO variant expressing cells treated with SHH-N CM and 100 nM vismodegib or (d) with or without 10 or 20 nM vismodegib show no apparent resistance to drug.
To assess the ability of the SMO variants to confer drug resistance to vismodegib, the current FDA-approved SMO antagonist, we added both HH ligand and 100 nM vismodegib to the Smo-null MEFs and assayed for HH activation. SMO-D473G and SMO-W535L both resisted SMO inhibition and displayed robust activation as expected (Figure 2c). However, no other SMO variant conferred vismodegib resistance, suggesting these mutations could not confer drug resistance and that the resistance mechanism in these tumors would be independent of SMO. This is also surprising as seven of these variants (L221P, V386A, C390R, V404M, N476K, E481G, and P513L) lie in the ligand binding pocket of SMO where vismodegib sits to repress protein function (Figure 1c). Because the concentration of vismodegib in our screening assay was roughly 12-fold above the IC50 and data from our previous studies demonstrated that even small changes in IC50 appeared to provide a growth advantage (Atwood et al. 2015), we assessed vismodegib sensitivity of the SMO mutants at low drug concentrations near the IC50 of 10 nM and 20 nM. Using this more sensitive assay, SMO-D473G and SMO W535L maintained Gli1 mRNA levels as expected, however the other SMO mutants displayed a vismodegib response similar to SMO-WT (Figure 2d).
Altogether, our results reveal a surprising frequency of neutral and inactivating SMO variants in our drug-resistant BCC tumor population that provides a broader view to our recently described set of variants that confer drug resistance (Atwood et al. 2015). Our data supports a model where tumors are permissive to genetic mutations, generating many genetically diverse clones that compete as a way to grow. This ability to “roll the genetic dice” allows many mutations in key genes like SMO that would have activating, neutral, or negative effects on the cell. However, a small percentage of clones fortunate enough to contain activating mutations would continue to divide and contribute to a larger fraction of the tumor mass. Interestingly, SMO loss-of-function mutations would have no adverse effect on tumor growth as only one normal SMO gene is necessary to confer HH pathway activation, essentially making loss-of-function alleles similar to neutral mutations. Our functional studies included many variants that are recurrent in other genomic databases and argue against recurrent alleles necessarily imparting functional relevance. Rather, asymmetric distribution of variations could reflect bias in genome-wide chromatin accessibility or DNA repair mechanisms. On a cellular level, this suggests that individual tumor cells can be genetically distinct from each other and harbor many mutations, even in drivers like SMO, and have no negative effects on the growth of the tumor. Although our results focus on the SMO locus, similar strategies may be operative at other genetic loci and tumors with high SNV frequencies may generate drug-resistance at a higher rate. Moreover, as we expand our use of high-throughput sequencing of tumors for personalized medicine, our results present a cautionary tale to functionally validate any mutation before concluding their ability to exert oncogenic effects.
Acknowledgments
The work was funded by the V Foundation Translational Award and NIAMS 5ARO54780, 2AR046786 (AEO), NIH Pathway to Independence Award 1K99CA176847 (SXA), the Damon Runyon Clinical Investigator Award (JT), the American Skin Association Clinical Scholar Award (AC), the Stanford Cancer Institute Grant and the Dermatology Foundation Career Development Award (KS).
Abbreviations
- BCC
basal cell carcinoma
- HH
Hedgehog
- SMO
Smoothened
- SNV
single nucleotide variant
Footnotes
Conflict of Interest: AO and AC are investigators in Genentech, Novartis, and Eli Lilly clinical trials. JT is a consultant to Genentech.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342–53. doi: 10.1016/j.ccell.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood SX, Whitson RJ, Oro AE. Advanced treatment for basal cell carcinomas. Cold Spring Harb Perspect Med. 2014;4:a013581. doi: 10.1101/cshperspect.a013581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman SS, Rayhan DJ, Hazany S, et al. Mutational landscape of Basal cell carcinomas by whole-exome sequencing. Journal of Investigative Dermatology. 2014;134:213–20. doi: 10.1038/jid.2013.276. [DOI] [PubMed] [Google Scholar]
- Kool M, Jones DTW, Jäger N, et al. Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer Cell. 2014;25:393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Bhaduri A, Mah A, et al. Recurrent point mutations in the kinetochore gene KNSTRN in cutaneous squamous cell carcinoma. Nat Genet. 2014;46:1060–2. doi: 10.1038/ng.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney RT, McClary AC, Myers BR, et al. Identification of recurrent SMO and BRAF mutations in ameloblastomas. Nat Genet. 2014;46:722–5. doi: 10.1038/ng.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wu H, Katritch V, et al. Structure of the human smoothened receptor bound to an antitumour agent. Nature. 2013;497:338–43. doi: 10.1038/nature12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauch RL, Dijkgraaf GJP, Alicke B, et al. Smoothened mutation confers resistance to aHedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572–4. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]