

Abstract
The inflammasome is a complex of proteins that plays a critical role in mounting an inflammatory response in reply to a harmful stimulus that compromises the homeostatic state of the tissue. The NLRP3 inflammasome, which is found in a wound-like environment, is comprised of three components: the NLRP3, the adaptor protein ASC and caspase-1. Interestingly, while ASC levels do not fluctuate, caspase-1 levels are elevated in both physiological and pathological conditions. Despite the observation that merely raising caspase-1 levels is sufficient to induce inflammation, the crucial question regarding the mechanism governing its expression is unexplored. We find that in an inflammatory microenvironment, caspase-1 is regulated by NFκB. Consistent with this association, the inhibition of caspase-1 activity parallels the effects on wound-healing caused by the abrogation of NFκB activation. Surprisingly, not only does inhibition of the NFκB/caspase-1 axis disrupt the inflammatory phase of the wound-healing program, it also impairs the stimulation of cutaneous epithelial stem cells of the proliferative phase. These data provide a mechanistic basis for the complex interplay between different phases of the wound-healing response in which the downstream signaling activity of immune cells can kindle the amplification of local stem cells to advance tissue repair.
Introduction
The major function of the skin is to provide a physical barrier between the body and the external environment. Consequently, it is frequently damaged and mounts a complex wound-healing program to restore tissue homeostasis. This wound-healing response is organized into three interdependent phases: an inflammatory phase leading to the recruitment and activation of immune cells; a proliferative phase comprising the expansion of the progenitor cell pool, angiogenesis and re-epithelialization (migration of keratinocytes to close the wound); and the remodeling phase wherein alterations in tissue architecture and the extracellular matrix is carried out (Gurtner et al., 2008). Hindering the mechanistic understanding of this process is the fact that signaling by cells in one phase of the wound-healing program impact the behavior of cells associated with another phase.
In response to many different external stimuli, inflammation can be mediated by a multiprotein complex known as an inflammasome, which promotes the maturation and secretion of proinflammatory cytokines. The inflammasome can be comprised of an NLR (nucleotide-binding domain and leucine-rich repeat containing) protein, an adaptor protein ASC (apoptosis-associated speck-like protein) and pro-caspase-1. The NLR proteins are a family of intracellular sensors that can detect the presence of pathogens and also recognize danger or stress signals (Schroder and Tschopp, 2010). Whereas the levels of the adaptor protein Asc remains unchanged, we found that the stress kinase p38 MAPK is responsible for the upregulation of NLRP3 in a wound-like environment (Lee et al., 2009), but the regulation of caspase-1 expression remains poorly understood.
Caspase-1 is a cysteine protease that is synthesized as a zymogen. The assembly of NRLP3 inflammasome induces the cleavage and activation of procaspase-1 which can then process interleukin-1 (IL-1) family members such as IL-1α (Keller et al., 2008), IL-1β and IL-18 into their mature forms (Yu and Finlay, 2008). In the context of wounding (Lee et al., 2009) as well as diseases with chronic inflammation such as psoriasis (Johansen et al., 2007) heart disease (Merkle et al., 2007), gout and arthritis (Church et al., 2008), there is an upregulation of procaspase-1 expression. Despite these observations, both the mechanism and the functional relevance of increasing the expression of procaspase-1 has failed to garner much attention. A clue to the importance of the upregulation of procaspase-1 was the development of cutaneous inflammation simply by overexpressing pro-caspase-1 in epidermal keratinocytes (Yamanaka et al., 2000).
A prime candidate for the transcriptional regulator of caspase-1 is the transcription factor nuclear factor-kappa B (NFκB), a well-known mediator of the inflammatory response (Karin, 2009). This dimeric complex is sequestered in the cytosol via its association with IκB. Activation of NFκB is accomplished through the IκB kinase (IKK), which can phosphorylate IκB and lead to its dissociation from NFκB. Upon activation, NFκB dimers are translocated into the nucleus where they modulate the expression of numerous genes involved in the immune response. To investigate the potential link between NFκB and caspase-1 in restoring tissue homeostasis, we exploited the conditional deletion of epidermal caspase-8 (C8cKO) as a model of wound-healing and in which caspase-1 levels are elevated throughout the epidermis (Lee et al., 2009).
Results
The expression of caspase-1 is regulated by NFκB
To test the hypothesis that NFκB regulates procaspase-1 expression, epidermal explants from wild-type and caspase-8 conditional knockout (C8KO) mice were treated with DMSO (vehicle control) or a pharmacological inhibitor of NFκB activation. Inhibition of NFκB activation was accomplished with a chemical inhibitor of the upstream activator of NFκB (IKK inhibitor [IKK-I]). In the C8KO epidermis, the increased procaspase-1 is diminished to near wild type levels with the treatment with IKK-I at both the RNA (Figure 1A) and protein level. This effect is both dose-dependent (Supplementary Figure 1) and reversible (Figure 1A). Interestingly, IKK-I did not affect the expression of NLRP3 despite suggestions to the contrary (Bauernfeind et al., 2009) (Figure 1B). On the other hand, p38 MAPK regulates NLRP3 in the wound-like epidermis (Figure 1B), but did not affect the increase in procaspase-1 (Figure 1A). The third component of many NLRP3-containing inflammasomes is Asc, whose levels were constant in wild type and C8KO epidermis (Figure 1C). Altogether, these inhibitor studies suggest that expression of the components of the NLRP3 inflammasome is under the regulation of at least two convergent signaling pathways – p38 MAPK regulates NLRP3 expression, while NFκB controls the expression of procaspase-1.
Figure 1. NFκB regulates caspase-1 expression.
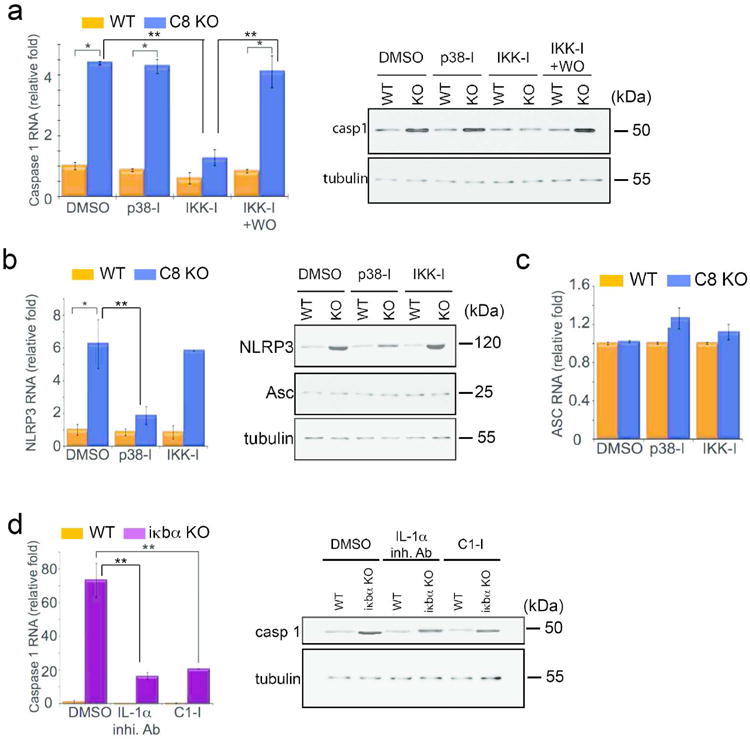
Epidermal explants from wild type (WT) and caspase-8 KO (C8 KO) were treated with DMSO, or inhibitors for p38 MAPK (p38-I) or IKK (IKK-I) and qPCR and Western blots were performed for (A) caspase-1, (B) NLRP3, and (C) Asc expression. WO = inhibitor washed-out. (D) qPCR of caspase-1 expression in epidermis of newborn wild type or iκbα knockout mice treated with DMSO, IL-lα inhibitory antibody (IL-1α inhi. Ab), or caspase-1 inhibitor (C1-I). For all experiments n=5 biological replicates and each sample was run in triplicate for qPCR; error bars are SEM, * denotes p<.001, and ** denotes p<.00001. β-tubulin level was used as a loading control for Western blots.
In order to examine whether NFκB activity is sufficient to induce procaspase-1, we utilized newborn epidermal explants lacking Iκbα, a negative regulator of NFκB. The basal level of NFκB activity in wild-type and Iκbα knockout mice are the same, but there is a prolonged activation of NFκB signaling with a pulse of a stimulus such as IL-1 or TNF in iκbα null cells (Klement et al., 1996). A ∼13-fold increase in the level of a bona fide NFκB target gene, RANTES, verified that the loss of Iκbα induces an extended activation of NFκB signaling in epidermal explants (Supplementary Figure 2). Under these conditions, procaspase-1 expression remains ∼70 fold higher in the Iκbα knockout epidermis relative to the wild type control (Figure 1D) but there is no appreciable effect on NLRP3 or Asc (data not shown). Interestingly, despite the ∼70 fold increase in caspase-1 RNA levels in the iκbα KO relative to the wild type control, the increase of caspase-8 protein level, though significant, was only ∼4 fold. Secreted IL-1α is not only the product of caspase-1 activity but is also involved in a positive feedback loop to activate NFκB in an autocrine or paracrine fashion (Vallabhapurapu and Karin, 2009). In order to determine how much of the increase in caspase-1 expression is due to this positive feedback loop we tested the effect of blocking the signaling of IL-1α with a neutralizing antibody. Neutralizing extracellular IL-1α dramatically reduces the induction of procaspase-1 expression in the iκbα null background (Figure 1D). The remaining ∼15 fold increase in caspase-1 levels in the iκbα knockout epidermis may reflect the direct intracellular signaling events linking NFκB to caspase-1. This theory was substantiated by the effect of the caspase-1 inhibitor, which would effectively short circuit the positive feedback loop resulting from the caspase-1 dependent secretion of IL-1α. The treatment of iκbα null epidermis with the caspase-1 inhibitor likewise results in a ∼15-fold difference in caspase-1 RNA levels compared to the wild type explants (Figure 1D). The decrease in caspase-1 expression by the IL-1α inhibitor antibody or the caspase-1 inhibitor in the iκbα KO skin is also reflected at the protein level (Figure 1D).
Reconstitution of caspase-1 regulatory control in vitro
In contrast to the heterogeneous population of cells that comprise the epidermis used above, we investigated whether the NFκB-caspase-1 relationship is valid within wounded epidermal keratinocytes in vitro. 16 hours after applying a scratch wound on a confluent sheet of keratinocytes, a decrease in caspase-8 protein levels was only observed in cells migrating from the wound edge (Figure 2A). This occurred in tandem with the activation of NFκB (Figure 2B and Supplementary Figure 3A). Consistent with our proposed pathway, the decrease in caspase-8 in epidermal keratinocytes leading to the activation of NFκB resulted in an increase in caspase-1 expression (Figure 2C and Supplementary Figure 3B). Interestingly, the increased in caspase-1 protein levels was observed in both the cytosol and nucleus of the migrating cells (Supplementary Figure 3C and D). This nuclear translocation of caspase-1 has previously been described upon exposure of cells to TNF(Mao et al., 1998) though the function remains unknown. We then examined whether activation of NFκB is sufficient to induce caspase-1 expression in vitro by transfecting primary keratinocytes with a constitutively active mutant of IKK2 (IKK2-CA). Twelve hours after transfection, about 10% of the cultured cells expressed the IKK2-CA construct leading to the activation of NFκB (Figure 2D and Supplementary Figure 3E) and a ∼2.5 fold increase in caspase-1 RNA (Figure 2E) and protein (Supplementary Figure 3F) expression. Moreover, activation of NFκB with IL-1α can elevate the amount of procaspase-1 protein, and this effect can be blocked by inhibiting NFκB activation with the IKK inhibitor or with glyburide, which blocks inflammasome function (Figure 2F, Supplementary Figure 3G).
Figure 2. Regulation of caspase-1 by NFκB in wounded cells.
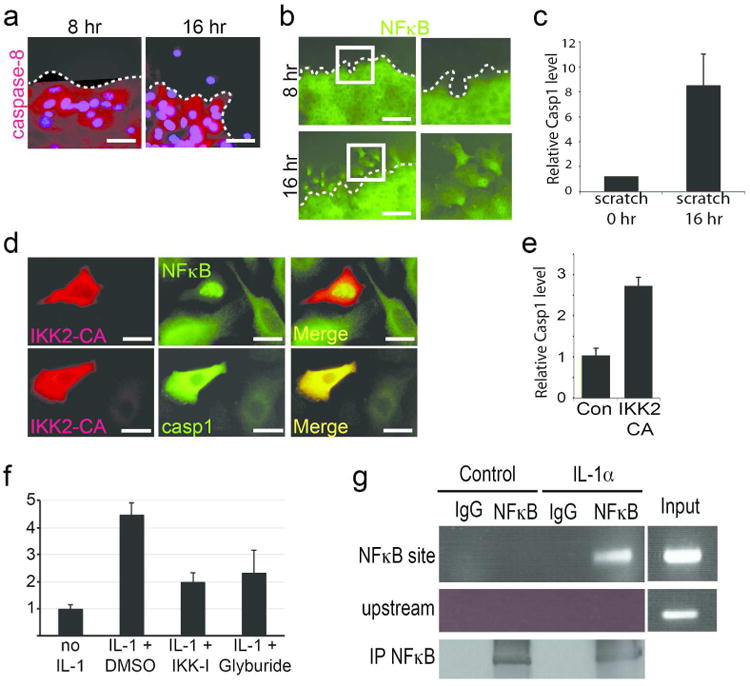
(A) Immunofluorescence of (A) caspase-8 (red), DAPI (blue) and (B) NFκB (green) in primary keratinocytes after 8 or 16 hours of applying a scratch wound (dotted line denotes wound margin). Right panels in (B) are the magnified views of the boxed areas in the left panels. (C) qPCR of caspase-1 RNA levels at 0 and 16 hours after applying scratch wound. (D) Primary keratinocytes were transfected with constitutively active HA-tagged IKK2 (IKK2-CA) and stained for active IKK2 (red) and NFκB (green) or caspase-1 (green). Cells expressing IKK2 have nuclear NFκB and higher levels of caspase-1. (E) qPCR of relative levels of caspase-1 RNA in keratinocytes transfected with empty vector (Con; normalized to 1) or IKK2-CA. (F) Relative caspase-1 protein levels in keratinocytes exposed to IL-1 and DMSO, IKK-I, or glyburide. Caspase-1 protein levels in cells without IL-1α stimulation is normalized to 1 and data are presented as fold difference. Data is the mean of 3 biological replicates and error bars are SEM. (G) ChIP of NFκB upon stimulation with IL-1α and immunoprecipitation with IgG control antibody or an antibody recognizing the p65 subunit of NFκB. * = p<.001. For qPCR experiments (C & E), results are from 3 biological replicates with each PCR reaction performed in triplicate (error bars are SEM).
These results provide evidence that NFκB is involved in the regulation of caspase-1 expression, but it is unclear whether NFκB is the direct upstream transcriptional activator. To address this question, we cloned a 1kb fragment of the proximal promoter of caspase-1, which contains a conserved NFκB binding motif (Supplementary Figure 4A), and found that mutating this consensus sequence significantly reduced the ability of active NFκB to induce caspase-1 transcription (Supplementary Figure 4B). Moreover, we tested whether this likewise occurs on the conserved motif on the endogenous caspase-1 promoter in primary keratinocytes via chromatin immunoprecipitation. After a 4-hour stimulation with IL-1α (Supplementary Figure 4C), we were able to co-precipitate the NFκB binding motif of the caspase-1 promoter with the p65 subunit of the NFκB complex (Figure 2G), suggesting that this transcription factor is a direct activator of caspase-1 expression.
Caspase-1 amplifies NFκB signaling in the epidermis
Upon establishing the NFκB/caspase-1 regulatory relationship in vitro, we then focused our attention on its regulation and function within a tissue. We incubated wild type or C8KO epidermal explants in media with vehicle control (DMSO) or IKK inhibitor and assayed for the amount of secreted IL-1, which is a read-out for activated caspase-1. We previously found that the levels of IL-`α in the C8KO skin is higher than wild type, but its secretion was still dependent on caspase-1 activity (Lee et al., 2009). Reducing caspase-1 expression in the C8KO epidermis through the inhibition of NFκB also reduces the secretion of IL-1α to wild type levels (Figure 3A). This occurs concomitant with a blockage of proteolytic cleavage of caspase-1 into its active p20 fragment (Supplementary Figure 5A). Furthermore, we observed that the prolonged activity of NFκB in the IκBα deficient epidermis results in the higher expression of procaspase-1 (Figure 1D), prompting us to examine whether this likewise translates into an increase in IL-1α secretion. The prolonged NFκB signaling in the IκBα KO resulted in a substantial increase activated caspase-1 (Supplementary Figure 5B) and in IL-1α secretion (Figure 3B) compared to the wild type epidermis. Reducing the activity of caspase-1 in the IκBα knockout epidermis (Supplementary Figure 5B) dramatically reduced the amount of secreted IL-1α (Figure 3B). This effect of activated NFκB on raising IL-1α secretion can be reconstituted in vitro by transfecting keratinocytes with the IKK2-CA construct (Figure 3C). Moreover, pharmacological inhibition of caspase-1 activity can also abrogate the stimulation of IL-1α secretion. The secretion of IL-1α has been shown to be dependent upon caspase-1 activity (Keller et al., 2008), and the NFkB-caspase-1 signaling pathway also yields similar results when tested on the intracellular levels of IL-1α (Supplementary Figure 5C-F) as well as the secretion of IL-1β, a classical readout of caspase-1 function (Supplementary Figure 5G-I).
Figure 3. Caspase-1 propagates NFκB signaling in epidermal keratinocytes.
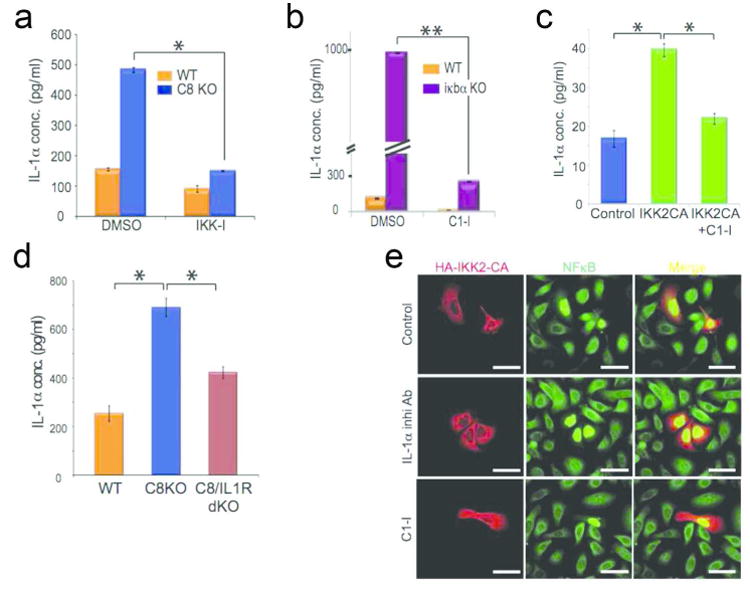
Secretion of IL-1α was measured by ELISA from different samples: (A) Wild type [orange bars] or caspase-8 null (C8KO) [blue bars] epidermis treated with DMSO or IKK-I; (B) wild type or iκbα null [purple bars] epidermis treated with DMSO or C1-I; (C) primary keratinocytes transfected with empty vector (control), IKK2-CA, or IKK2 CA and treated with C1-I; (D) epidermis from wild type, caspase-8 null, and caspase-8/IL-1R double knockout mice. ELISA results are the mean of 3 biological replicates and error bars are SEM. * denotes p<.001, and ** denotes p<.00001. (E) Primary keratinocytes were transfected with constitutively active IKK2 (HA-IKK2-CA) and treated with DMSO (control), IL-1α inhibitory antibody, or caspase-1 inhibitor (C1-I) and stained for IKK2-CA (red) and NFκB (green). Data is representative of 3 independent experiments.
The ability of IL-1α ligand to activate NFκB suggests that the release of this cytokine can propagate the NFκB-caspase-1-IL-1α module throughout the epidermis via a paracrine effect. We found that removal of the IL-1 receptor in the wound model diminished the level of secreted IL-1α compared to the C8cKO epidermis (Figure 3D). We further tested this paracrine model by transfecting primary keratinocytes with the constitutively active IKK mutant construct (IKK2-CA). As we have shown, keratinocytes expressing IKK2-CA displayed nuclear NFκB twelve hours after transfection (Figure 2D). However, when we allowed the cells to incubate longer (∼24 hours), cells adjacent to the transfected cells were induced to activate NFκB (Figure 3E). Use of the IL-1α neutralizing antibody or the caspase-1 inhibitor restricted NFκB activation solely to cells transfected with IKK2-CA.
To test whether this is also valid in wound-like conditions in vivo, we subcutaneously injected vehicle control (DMSO), or IKK inhibitor daily in the back skin of newborn wild type or C8cKO mice for four days. Inhibition of NFκB activation, which blocks IL-1α release (Figure 3A), suppresses the expression of active NFκB throughout the epidermis, though the dermal activation of NFκB is largely unaffected (Figure 4A). This block in epidermal activation of NFκB was also present when excisional wounds were treated with IKK inhibitor (Supplementary Figure 6). Moreover, genetic removal of caspase-1 expression from the C8cKO epidermis (Figure 4B-C) likewise reduces the expression of active NFκB throughout the epidermis.
Figure 4. Caspase-1 expands NFκB activity throughout the epidermis.
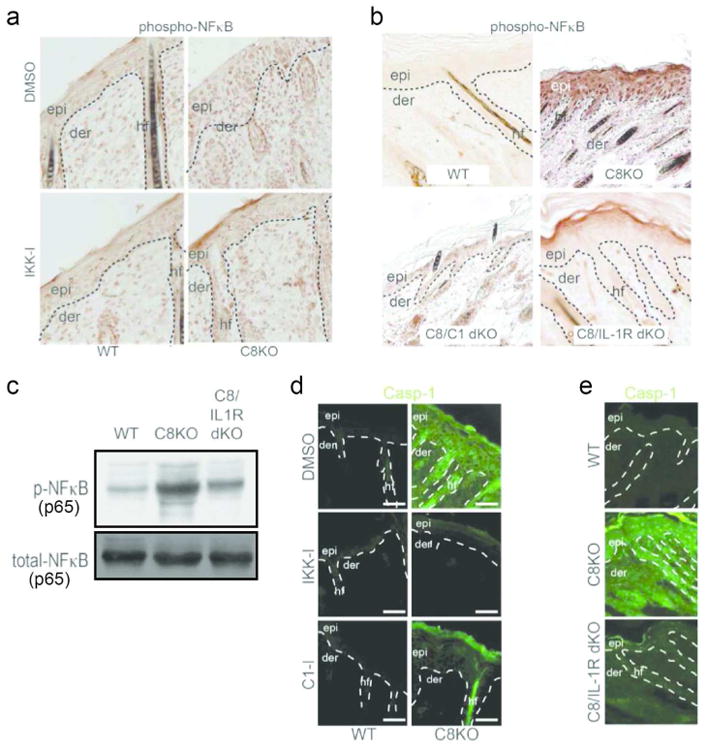
Immunohistochemistry of phosphorylated NFκB in (A) wild type and caspase-8 knockout skin injected with DMSO or IKK-I, and (B) in skin lacking both caspase-8/caspase-1 (C8/C1 dKO) or skin that are null for caspase-8 and IL-1 receptor (C8/IL-1R dKO). (C) Level of phosphorylated NFκB, with total NFκB as loading control. (D) Caspase-1 expression in wild type or C8KO epidermis treated with vehicle control or chemical inhibitors. (E) Caspase-1 expression in wild type, caspase-8 null and caspase-8/IL-1R double knockout skin samples.
Since the inhibition of NFκB activity and caspase-1 expression both prevented the spreading of active NFκB in the C8cKO epidermis, we predicted that the expression of caspase-1 would likewise be adversely affected. Caspase-1 is expressed at a low basal level in wild type epidermis, is significantly upregulated in the C8cKO tissue, and this induction is lost in the presence of IKK-I (Figure 4D). Interestingly, application of a pharmacological inhibitor of caspase-1 activity (C1-I) results in a dramatic reduction in the expression of caspase-1 throughout the tissue and the remaining protein is restricted to the upper layers of the epidermis (Figure 4D). Moreover, in the absence of IL-1 signaling, caspase-1 expression is not highly induced in the C8cKO epidermis (Figure 4E). Therefore, abrogation of NFκB activation inhibits the upregulation of caspase-1 expression and inhibition of the latter's proteolytic activity can effectively block the spreading of NFκB and caspase-1 expression from the surface of the epidermis to the lower layers of this tissue, which occurs through IL-1 paracrine signaling.
Inhibition of NFκB and caspase-1 delays wound closure rates
We then focused our attention on the functional contribution of these proteins to the wound healing response. Excisional wounds were made on the back of wild type mice treated with DMSO or IKK-I, caspase-1 knockout mice and IL-1R KO mice (Figure 5). Interestingly, inhibition of NFκB activation or caspase-1 expression significantly delayed the closure rate of an excisional wound compared to control samples (Figure 5A). This delay in wound closure by 2 or more days parallels the effects seen when other important factors for wound healing are deleted (Devalaraja et al., 2000; Echtermeyer et al., 2001; Jameson et al., 2002). We verified that both the inhibition of NFκB activation and caspase-1 expression indeed impacted inflammation as they were able to decrease the recruitment of immune cells to the skin. For instance, the number of macrophages and T-cells recruited to the skin in the C8cKO (Figure 5 B and C) or to wounded skin (Supplementary Figure 7 A and B) is severely reduced when NFκB activity or caspase-1 expression is suppressed. The failure to recruit immune cells to the skin under these conditions is also reflected in the diminished expression of inflammatory genes (Figure 5D). Together these data highlight the critical role for the NFκB/caspase-1/IL-1 axis in the establishment of the inflammatory phase of the cutaneous wound healing response.
Figure 5. Effect of NFκB and caspase-1 on wound closure.
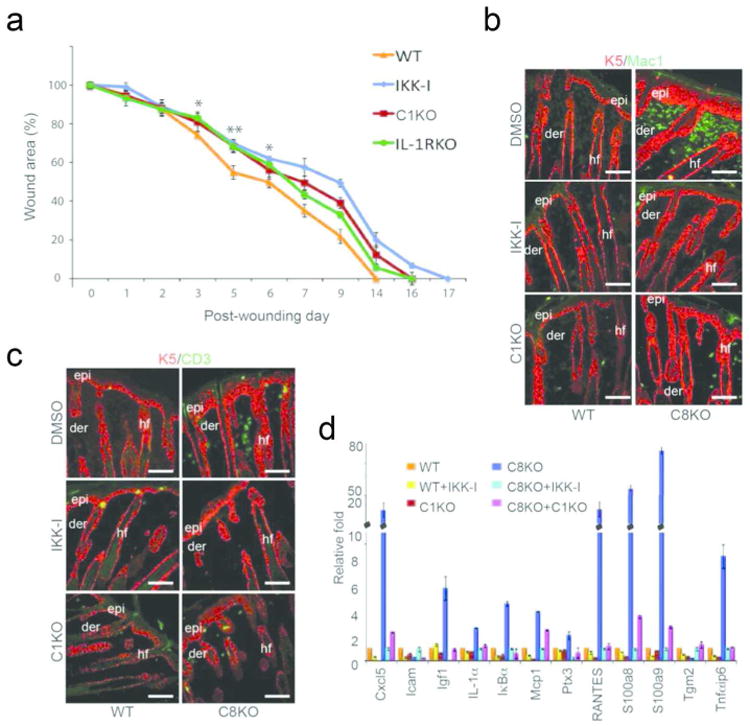
(A) Rates of wound closure were measured in wild type mice treated daily with DMSO or IKK-I, or in mutant mice lacking either caspase-1 (C1KO) or IL-1 receptor (IL-1RKO). Data points are averages of 6 mice with two wounds/mouse (* denotes p<.001, and ** denotes p<.00001 when comparing DMSO vs. IKK-I kinetics in wild type mice). (B) Recruitment of macrophages (green) in the wild type and caspase-8 null skin treated with DMSO (control) or IKK-I or in skin lacking caspase-1 and -8. (C) Recruitment of T-cells (green) monitored using the pan T cell marker CD3. The epidermis (epi) and hair follicles (hf) in B & C are noted in red with an antibody detecting keratin-5. Scale bar: 50 μm; der = dermis (D) Inflammatory gene expression in the wounded/wound-like skin. RNA was extracted from the wounded skin of wild type mice (treated with DMSO [orange bars] or IKK-I [yellow bars]) or caspase-1 KO mice (C1KO; red bars) 3 days post excisional wounding. RNA was also extracted from the skin of P5 caspase-8 knockout (C8KO) mice (treated with DMSO [blue] or IKK-I [light blue]) or the caspase-8/caspase-1 (C1KO) double knockout mouse [magenta]. Results are from 3 biological replicates for each mouse genotype/treatment and each sample was repeated in triplicate. Error bars are SEM.
NFκB/Caspase-1 signaling affects proliferation during the wound healing response
Closer examination of the effects of the NFκB and caspase-1 inhibition on the rate of wound closure reveals an affect during the proliferative phase of the wound response, which commences around day three post-wounding. This effect may be attributable to the role that NFκB and caspase-1 play in the secretion of IL-1. We previously demonstrated the IL-1 mediated epithelial-mesenchymal signaling loop that stimulates keratinocytes proliferation in a wound is recapitulated in the C8cKO model (Lee et al., 2009). One outcome of this pro-proliferation signaling network is the thickening of the epidermis (epidermal hyperplasia).
Consistent with this model, inhibition of NFκB activation or loss of caspase-1 expression in the C8cKO skin significantly reduced epidermal hyperplasia (Figure 6A and Supplementary Figure 8A). This phenomenon was also seen in response to excisional wounds (Supplementary Figure 8B). The thickening of the epidermis in the C8cKO mouse is fueled by an increase in the number of proliferating progenitor cells found in the basal layer of the epidermis and the hair follicle (Figure 6B). Both are reduced in the presence of the inhibitor of NFκB activation or loss of caspase-1 expression (Figure 6B-D). However, these effects are not applicable to all the phenomena associated with the proliferative phase of the wound healing response. For instance, new blood vessel formation is a feature of this phase of the wound-healing program, but the injection of IKK inhibitor or the loss of caspase-1 expression do not qualitatively decrease angiogenesis in the skin (Figure 6E). Altogether, these observations link a functional inflammatory phase of the wound healing response with the optimal stimulation of cell proliferation that occurs during a wound healing response.
Figure 6. Hyperproliferation of the epidermis in caspase-8 KO and wounded skin.
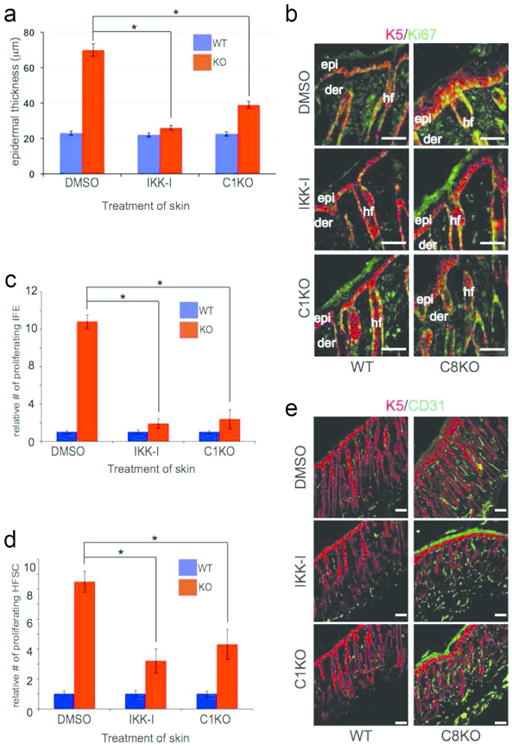
(A) Measurement of epidermal thickness of wild type (WT) or caspase-8 knockout (KO) skin treated with DMSO, or IKK-I, or skin also lacking caspase-1. (B) Proliferating cells were marked by Ki67 (green) and the epidermis and hair follicle are marked by keratin 5 (red) in mice treated as in (A). Scale bar: 50 μm. Quantification of proliferating (C) interfollicular epidermal (IFE) stem cells and (D) hair follicle stem cell (HFSC) in skin sections from (B). Results are the average of 6 skins (5 sections/skin) for each treatment and error bars are SEM, (* denote p<.001). (E) Angiogenesis monitored in wild type or knockout mice treated with DMSO, or IKK-I, or skin also lacking caspase-1 via staining with CD31 (green).
Discussion
A major obstacle to understanding wound healing is the untangling of the extensive interdependence of the three classical phases of this process. Using the ablation of epidermal caspase-8 as a model of wound healing, we have uncovered a molecular mechanism synchronizing these complex reactions. Upon wounding, caspase-8, which normally resides in the granular layer near the surface of the epidermis is downregulated and initiates a biochemical program to promote tissue repair (Lee et al., 2009). One effect of this downregulation is the increase in IL-1α RNA expression as well as the release of the preexisting reservoir of this cytokine from epidermal keratinocytes (Lee et al., 2009). IL-1α has a plethora of effects ranging from the promotion of an inflammatory microenvironment to promoting a growth arrest and survival model in suprabasal keratinocytes to the stimulation of stem cell proliferation. Given the profound impact of IL-1α signaling in the framework of wound healing, the mechanism governing its release is an important issue to resolve. We found that at least two different signaling pathways impact the components of the NLRP3 inflammasome – p38 MAPK regulates NLRP3 expression (Lee et al., 2009) and NFκB influences caspase-1 expression levels (Supplementary Figure 9). Together these pathways lead to IL-1α secretion that can operate in an autocrine fashion to potentiate the NFκB signaling cascade or function in a paracrine manner to propagate NFκB signaling to different layers of keratinocytes in the epidermis. This exemplifies the central and complex role that IL-1α and NFκB play in regulating epithelial homeostasis (Swamy et al., 2010).
Recently, however, work from David Wallach's group has raised doubts about the role of IL-1α in mediating inflammation in the epidermal caspase-8 conditional knockout mouse (Kovalenko et al., 2009). This is based on their finding that genetic ablation of both IL-1α and IL-1β did not abrogate the cutaneous inflammation stimulated by the loss of epidermal caspase-8. However, as we observed, removal of one signaling pathway such as IL-1 can delay the kinetics of wound closure but not completely abrogate it. The ≥ 2 day delay in wound healing by blocking components of the NFκB/caspase-1/IL-1 axis is consistent with the delay in wound-healing found when other important components are deleted such as γδ T-cells (Jameson et al., 2002), syndecan-4 (Echtermeyer et al., 2001), and CXCR2 (Devalaraja et al., 2000). We also observed that the C8cKO/IL1 receptor double knockout transiently dampens the phenotype of the C8cKO mouse, which is likely due to the fact that other components in the inflammatory phase can compensate for the loss of one constituent. Further support for IL-1 signaling in a wound-like environment is provided by the finding of increased nuclear NFkB when the IL-1 receptor antagonist is missing (Ishida et al., 2006). Thus we propose that the large reservoir of IL-1α released from damaged keratinocytes, is amongst the earliest responders to epidermal trauma to stimulate the wound healing response. In particular, IL-1α can induce the inflammatory phase and thereby promote the proliferative phase and this role is assumed by infiltrating immune cells, which secretes the more potent IL-1β.
An important question that arises from our model is the link between the downregulation of caspase-8 and the activation of NFκB prior to the positive feedback loop mediated by IL-1α secretion. Of particular relevance are reports that deletion of RIP3 rescues the caspase-8 knockout phenotype, including cutaneous inflammation (Gunther et al., 2011; Kaiser et al., 2011; Welz et al., 2011). Interestingly, more recent studies have demonstrated that RIP3 can also activate NLRP3 and caspase-1 (Kang et al., 2013; Vince et al., 2012). Thus, the loss of caspase-8 may liberate RIP3 to interact with different partners to stimulate inflammasome activation. Further delineating the details of inflammasome activation will contribute important new insights on how to combat inflammatory diseases when these signaling pathways are perturbed.
Materials and Methods
Reagents
The list of antibodies, inhibitors and primers and their usage are detailed in the Supplementary information section.
Mice and cell/tissue culture
Generation of C8KO (Lee et al., 2009) and iκbα KO (Klement et al., 1996) mice have previously been reported. Wild-type primary mouse keratinocytes are obtained from the epidermis of newborn pups and cultured in low-calcium mouse keratinocyte media as described previously (Lee et al., 2009). Epidermal explants were taken from P0-P3 skin by chemically dissociating it from the dermis with dispase for 1 hour at 37°C. All animal work was carried out in accordance with the policies and approved procedures of the Institutional Animal Care and Use Committee of UC San Diego.
Tissue preparation
Epidermal explants were incubated with p38-I and IKK-I overnight and collected in Trizol (Invitrogen) for RNA extraction or in sample buffer for protein extraction. The wash-out of IKK-I treated epidermis was done by washing with PBS once and transferred to fresh mouse keratinocyte media and incubated for another 6 hours. Glyburide was incubated with the epidermis for 1 hour, and transferred to fresh media with glyburide for another hour. IL-1α (2 ng/ul) was added to WT and iκbα KO epidermis for 2 hours, and replaced with fresh media and incubated for another 6 hours prior to sample collection.
Wounding and injection of mice
DMSO, IKK-I, and C1-I were injected to the back skin of WT and caspase 8 KO daily for 4 days starting at P0 prior to the onset of the phenotype, and back skin were harvested at the 5th day for RNA extraction and OCT embedding. In vivo wounding was performed on the back skin of adult WT mice with 5mm-punch biopsy, and DMSO and inhibitors were applied to the wound site daily for 3 days. Wounded skin was collected for RNA extraction and OCT embedding. Data presented are averages of 2 wound per mouse (n=6 mice) +/- SEM for each treatment. For in vitro scratch wounds, primary mouse keratinocytes were cultured on coverslips to confluency. The scratch wound was performed on the cell monolayer with a p200 pipet tip. Cells were incubated at 37°C for up to 16 hours and then fixed for immunofluorescence or harvested for RNA extraction.
Supplementary Material
Supplementary Figure 1. The effect of IKK inhibitor (IKK-I) on caspase-1 expression is dose dependent. (B&C) Epidermal explants from Figure 1 were subjected to RNA extraction for quantitative RT-PCR to examine the expression of NLRP3 (B) and ASC (C). n=5 biological replicates performed in triplicate. * denotes p<.001, and error bars are SEM.
Supplementary Figure 2. Prolonged activation of NFkB signaling in the iκbα null epidermis. Measurement of a bona fide NFκB target, RANTES, expression by qPCR in wild type and iκbα knockout epidermis. * denotes p <.001. The data are from three independent experiments performed in triplicate. Error bars are SEM.
Supplementary Figure 3. Activation of NFkB signaling and caspase-1 expression in in vitro scratch wounds. (A) Relative qPCR measurement of RANTES (a bona fide target of NFkB) expression following a scratch wound of cultured primary keratinocytes. RANTES levels at 0 hours post wounding was normalized to 1. * denotes p <.001. Results are from 3 independent experiments performed in triplicate, and error bars are SEM. (B) Caspase-1 full-length protein expression levels in cells 0 hours and 16 hours post scratch wound. Tubulin is the loading control. (C) Immunofluorescence of caspase-1 (green) levels and localization after in vitro scratch wounding. (D) Cell fractionation. Cellular cytoplasm (cyto) was separated from the nucleus (nuc) and probed by Western blot for lamin B (marker of the nucleus), tubulin (marker of the cytoplasm), and caspase-1 (casp-1) in unwounded (UW) cells or following a scratch wound (W). (E) The expression of RANTES was investigated in IKK2-CA transfected primary keratinocytes by PCR. RT=reverse transcriptase. (F) Caspase 1 levels in cells transfected with control vector (con) or with the constitutively active IKK2 (IKK2 CA). Tubulin is the loading control. (G) Levels of caspase-1 full length protein in the absence or presence of IL-1 (treated with DMSO buffer control or chemical inhibitors). This is a representative western blot that comprises one of the biological replicates that comprise Figure 2F.
Supplementary Figure 4. NFκB regulates the promoter activity of caspase-1. (A) The conserved NFκB binding motif searched by sequence analysis on Genomatix website. (B) WT and mutant mouse caspase-1 (WT mC1 and mut-mC1) luciferase reporter constructs were co-transfected with IKK-CA respectively into primary keratinocytes to examine the promoter activity. (C) Primary keratinocytes were treated with IL-1α for 6 hours. Samples of RNA were extracted every hour and processed for caspase-1 expression by qPCR. Results are from 3 independent experiments performed in triplicate, and error bars are SEM.
Supplementary Figure 5. Activation of caspase-1 by NFκB. Epidermal explants from (A) Wild type (WT) or caspase-8 conditional knockout (C8cKO) were treated with DMSO vehicle control or an inhibitor of NFκB activation (IKK-I). Lysates were generated from the explants and probe via Western blot for full length pro-caspase1 (full length casp1) or the activated proteolytic p20 fragment of caspase1 (cleaved casp1). (B) Wild type or iκBα knockout (iκBα KO) were treated with DMSO or caspase-1 inhibitor (C1-I). Lysates were generated and probed as in (A). Tubulin was used as a loading control. Intracellular levels of IL-1α was measured by ELISA from different samples: (C) Wild type [orange bars] or caspase-8 null (C8KO) [blue bars] epidermis treated with DMSO or IKK-I; (D) wild type or iκbα null [purple bars] epidermis treated with DMSO or C1-I; (E) primary keratinocytes transfected with empty vector (control), IKK2-CA, or IKK2 CA and treated with C1-I; (F) epidermis from wild type, caspase-8 null, and caspase-8/IL-1R double knockout mice. Secretion of IL-1β was measured by ELISA from different samples: (G) Wild type (orange) or caspase-8 null (C8KO, blue) epidermis treated with DMSO or IKK-I; (H) wild type or iκbα null (purple) epidermis treated with DMSO or C1-I; (I) Epidermis from wild type, caspase-8 null, and caspase-8/IL-1R double knockout mice. All ELISA results are the mean of 3 biological replicates and error bars are SEM. * denotes p<.001, and ** denotes p<.00001.
Supplementary Figure 6. Phosphorylation of NFκB is induced in wounded skin. Immunohistochemistry of phosphorylated NFκB in wounded wild type skin treated with DMSO, IKK inhibitor, and caspase-1 inhibitor. Proximal area: <1mm from the wound. Distal area: ∼5mm from the wound.
Supplementary Figure 7. Recruitment of immune cells to wounded skin. Recruitment of (A) T-cells (green) monitored using the pan T cell marker CD3 and (B) macrophages (green) were examined in wounded skin treated with DMSO, IKK inhibitor, or caspase-1 inhibitor. The epidermis (epi) and hair follicle (hf) are labeled with keratin-5 (red) and arrowheads denote wound site. Der = dermis. Proximal wound area: <1mm from the wound. Distal wound area: ∼5mm from the wound.
Supplementary Figure 8. Proliferation and inflammation is regulated by NFκB. (A) Epidermal thickness was measured in wild type and caspase-8 knockout skins, injected with DMSO, IKK inhibitor, or caspase-1 inhibitor. (B) Epidermal thickness in excisional wounds treated with DMSO (control), IKK-I or C1-I. Values are relative to the thickness of the epidermis of unwounded skin normalized to 1. For each set of skins, values are the mean of 3 different wounds and error bars are SEM.
Supplementary Figure 9. Model of caspase-8 dependent NFκB-caspase-1 pathway mediating IL-1α secretion. Our data suggests a model in which wounding in the skin results in the downregulation of caspase-8 in granular layer of epidermis, and in turn activates NFκB to initiate the transcription of procaspase-1. Increased procaspase-1 assembles with ASC and NLRP3 to form NLRP3 inflammasomes and facilitates the maturation of caspase-1. Active caspase-1 releases the reservoir of IL-1α and then IL-1α serves as a signal amplifier to activate NFκB-caspase-1 signaling pathway spinous layer.
Acknowledgments
We thank members of the Jamora lab for insightful discussions and suggestions. P.L. is supported by a predoctoral fellowship from the NIH (5F31AR056593). This work was supported by a Hellman Faculty Fellowship (C.J.) and grants from the NIH/NIAMS (Grant 5R01AR053185-03) and the American Cancer Society (Grant 115457-RSG-08-164-01-DDC) to CJ.
Footnotes
Conflict of Interest: The authors state no conflict of interest
References
- Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–91. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church LD, Cook GP, McDermott MF. Primer: inflammasomes and interleukin 1beta in inflammatory disorders. Nat Clin Pract Rheumatol. 2008;4:34–42. doi: 10.1038/ncprheum0681. [DOI] [PubMed] [Google Scholar]
- Devalaraja RM, Nanney LB, Du J, et al. Delayed wound healing in CXCR2 knockout mice. J Invest Dermatol. 2000;115:234–44. doi: 10.1046/j.1523-1747.2000.00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echtermeyer F, Streit M, Wilcox-Adelman S, et al. Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J Clin Invest. 2001;107:R9–R14. doi: 10.1172/JCI10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther C, Martini E, Wittkopf N, et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–9. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature. 2008;453:314–21. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- Ishida Y, Kondo T, Kimura A, et al. Absence of IL-1 receptor antagonist impaired wound healing along with aberrant NF-kappaB activation and a reciprocal suppression of TGF-beta signal pathway. J Immunol. 2006;176:5598–606. doi: 10.4049/jimmunol.176.9.5598. [DOI] [PubMed] [Google Scholar]
- Jameson J, Ugarte K, Chen N, et al. A role for skin gammadelta T cells in wound repair. Science. 2002;296:747–9. doi: 10.1126/science.1069639. [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Long AB, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–72. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TB, Yang SH, Toth B, et al. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013;38:27–40. doi: 10.1016/j.immuni.2012.09.015. [DOI] [PubMed] [Google Scholar]
- Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1:a000141. doi: 10.1101/cshperspect.a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller M, Ruegg A, Werner S, et al. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–31. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- Klement JF, Rice NR, Car BD, et al. IkappaBalpha deficiency results in a sustained NF-kappaB response and severe widespread dermatitis in mice. Mol Cell Biol. 1996;16:2341–9. doi: 10.1128/mcb.16.5.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko A, Kim JC, Kang TB, et al. Caspase-8 deficiency in epidermal keratinocytes triggers an inflammatory skin disease. J Exp Med. 2009;206:2161–77. doi: 10.1084/jem.20090616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Lee DJ, Chan C, et al. Dynamic expression of epidermal caspase 8 simulates a wound healing response. Nature. 2009;458:519–23. doi: 10.1038/nature07687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao PL, Jiang Y, Wee BY, et al. Activation of caspase-1 in the nucleus requires nuclear translocation of pro-caspase-1 mediated by its prodomain. J Biol Chem. 1998;273:23621–4. doi: 10.1074/jbc.273.37.23621. [DOI] [PubMed] [Google Scholar]
- Merkle S, Frantz S, Schon MP, et al. A role for caspase-1 in heart failure. Circ Res. 2007;100:645–53. doi: 10.1161/01.RES.0000260203.55077.61. [DOI] [PubMed] [Google Scholar]
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Swamy M, Jamora C, Havran W, et al. Epithelial decision makers: in search of the ‘epimmunome’. Nat Immunol. 2010;11:656–65. doi: 10.1038/ni.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- Vince JE, Wong WW, Gentle I, et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36:215–27. doi: 10.1016/j.immuni.2012.01.012. [DOI] [PubMed] [Google Scholar]
- Welz PS, Wullaert A, Vlantis K, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:330–4. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Tanaka M, Tsutsui H, et al. Skin-specific caspase-1-transgenic mice show cutaneous apoptosis and pre-endotoxin shock condition with a high serum level of IL-18. J Immunol. 2000;165:997–1003. doi: 10.4049/jimmunol.165.2.997. [DOI] [PubMed] [Google Scholar]
- Yu HB, Finlay BB. The caspase-1 inflammasome: a pilot of innate immune responses. Cell Host Microbe. 2008;4:198–208. doi: 10.1016/j.chom.2008.08.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. The effect of IKK inhibitor (IKK-I) on caspase-1 expression is dose dependent. (B&C) Epidermal explants from Figure 1 were subjected to RNA extraction for quantitative RT-PCR to examine the expression of NLRP3 (B) and ASC (C). n=5 biological replicates performed in triplicate. * denotes p<.001, and error bars are SEM.
Supplementary Figure 2. Prolonged activation of NFkB signaling in the iκbα null epidermis. Measurement of a bona fide NFκB target, RANTES, expression by qPCR in wild type and iκbα knockout epidermis. * denotes p <.001. The data are from three independent experiments performed in triplicate. Error bars are SEM.
Supplementary Figure 3. Activation of NFkB signaling and caspase-1 expression in in vitro scratch wounds. (A) Relative qPCR measurement of RANTES (a bona fide target of NFkB) expression following a scratch wound of cultured primary keratinocytes. RANTES levels at 0 hours post wounding was normalized to 1. * denotes p <.001. Results are from 3 independent experiments performed in triplicate, and error bars are SEM. (B) Caspase-1 full-length protein expression levels in cells 0 hours and 16 hours post scratch wound. Tubulin is the loading control. (C) Immunofluorescence of caspase-1 (green) levels and localization after in vitro scratch wounding. (D) Cell fractionation. Cellular cytoplasm (cyto) was separated from the nucleus (nuc) and probed by Western blot for lamin B (marker of the nucleus), tubulin (marker of the cytoplasm), and caspase-1 (casp-1) in unwounded (UW) cells or following a scratch wound (W). (E) The expression of RANTES was investigated in IKK2-CA transfected primary keratinocytes by PCR. RT=reverse transcriptase. (F) Caspase 1 levels in cells transfected with control vector (con) or with the constitutively active IKK2 (IKK2 CA). Tubulin is the loading control. (G) Levels of caspase-1 full length protein in the absence or presence of IL-1 (treated with DMSO buffer control or chemical inhibitors). This is a representative western blot that comprises one of the biological replicates that comprise Figure 2F.
Supplementary Figure 4. NFκB regulates the promoter activity of caspase-1. (A) The conserved NFκB binding motif searched by sequence analysis on Genomatix website. (B) WT and mutant mouse caspase-1 (WT mC1 and mut-mC1) luciferase reporter constructs were co-transfected with IKK-CA respectively into primary keratinocytes to examine the promoter activity. (C) Primary keratinocytes were treated with IL-1α for 6 hours. Samples of RNA were extracted every hour and processed for caspase-1 expression by qPCR. Results are from 3 independent experiments performed in triplicate, and error bars are SEM.
Supplementary Figure 5. Activation of caspase-1 by NFκB. Epidermal explants from (A) Wild type (WT) or caspase-8 conditional knockout (C8cKO) were treated with DMSO vehicle control or an inhibitor of NFκB activation (IKK-I). Lysates were generated from the explants and probe via Western blot for full length pro-caspase1 (full length casp1) or the activated proteolytic p20 fragment of caspase1 (cleaved casp1). (B) Wild type or iκBα knockout (iκBα KO) were treated with DMSO or caspase-1 inhibitor (C1-I). Lysates were generated and probed as in (A). Tubulin was used as a loading control. Intracellular levels of IL-1α was measured by ELISA from different samples: (C) Wild type [orange bars] or caspase-8 null (C8KO) [blue bars] epidermis treated with DMSO or IKK-I; (D) wild type or iκbα null [purple bars] epidermis treated with DMSO or C1-I; (E) primary keratinocytes transfected with empty vector (control), IKK2-CA, or IKK2 CA and treated with C1-I; (F) epidermis from wild type, caspase-8 null, and caspase-8/IL-1R double knockout mice. Secretion of IL-1β was measured by ELISA from different samples: (G) Wild type (orange) or caspase-8 null (C8KO, blue) epidermis treated with DMSO or IKK-I; (H) wild type or iκbα null (purple) epidermis treated with DMSO or C1-I; (I) Epidermis from wild type, caspase-8 null, and caspase-8/IL-1R double knockout mice. All ELISA results are the mean of 3 biological replicates and error bars are SEM. * denotes p<.001, and ** denotes p<.00001.
Supplementary Figure 6. Phosphorylation of NFκB is induced in wounded skin. Immunohistochemistry of phosphorylated NFκB in wounded wild type skin treated with DMSO, IKK inhibitor, and caspase-1 inhibitor. Proximal area: <1mm from the wound. Distal area: ∼5mm from the wound.
Supplementary Figure 7. Recruitment of immune cells to wounded skin. Recruitment of (A) T-cells (green) monitored using the pan T cell marker CD3 and (B) macrophages (green) were examined in wounded skin treated with DMSO, IKK inhibitor, or caspase-1 inhibitor. The epidermis (epi) and hair follicle (hf) are labeled with keratin-5 (red) and arrowheads denote wound site. Der = dermis. Proximal wound area: <1mm from the wound. Distal wound area: ∼5mm from the wound.
Supplementary Figure 8. Proliferation and inflammation is regulated by NFκB. (A) Epidermal thickness was measured in wild type and caspase-8 knockout skins, injected with DMSO, IKK inhibitor, or caspase-1 inhibitor. (B) Epidermal thickness in excisional wounds treated with DMSO (control), IKK-I or C1-I. Values are relative to the thickness of the epidermis of unwounded skin normalized to 1. For each set of skins, values are the mean of 3 different wounds and error bars are SEM.
Supplementary Figure 9. Model of caspase-8 dependent NFκB-caspase-1 pathway mediating IL-1α secretion. Our data suggests a model in which wounding in the skin results in the downregulation of caspase-8 in granular layer of epidermis, and in turn activates NFκB to initiate the transcription of procaspase-1. Increased procaspase-1 assembles with ASC and NLRP3 to form NLRP3 inflammasomes and facilitates the maturation of caspase-1. Active caspase-1 releases the reservoir of IL-1α and then IL-1α serves as a signal amplifier to activate NFκB-caspase-1 signaling pathway spinous layer.