

Abstract
A novel rapid self-integrating, injectable, and bio-erodible hydrogel is developed for tissue complex regeneration. The figure shows the self-integration of the hydrogel pieces to form various structures. In the figure, some hydrogel disks were dyed pink with rodamine and the others were left with the original light yellow color to visualize the interfaces (scale bar=5mm). This hydrogel is demonstrated to engineer cartilage-bone complex.
Keywords: Supramolecular, Hydrogel, Tissue engineering, Tissue complex, Self-healing
Hydrogels are increasingly used as tissue-engineering scaffolds and drug carriers.[1, 2] Compared to porous scaffolding materials, hydrogels are appealing in filling irregular tissue defects due to their pre-gelling fluidity, which circumvents the pre-shaping and invasive surgery. Traditional hydrogels are usually made by using reactive chemical reagents or applying ultra-violet (UV) light to form permanent covalent crosslinks between hydrophilic polymers in an aqueous solution. The process and chemicals might harm cells and delicate biological substances [3], and the resulting stable hydrogels lose the fluidity and cannot be reshaped.
In contrast to the formation of covalent crosslinks, supramolecular hydrogels self-assemble via cooperative and highly specific physical interactions that transiently crosslink the hydrophilic polymer chains.[4, 5–7, 8] The transient supramolecular crosslinks can be temporally interrupted by an external stimulus (such as a mechanical force) and can rebuild after the removal of the stimulus. This unique characteristic brings about the shear-thinning property (viscosity decreases with an increasing rate of shear stress) and thus allows the formed hydrogel to be injectable.[5, 9] Therefore, shear-thinning hydrogels offer benefits over other hydrogels in that the already formed gel can be injected and can recover the gel state in a different shape (a defect or mold) after injection, which prevents the cells from sedimentation as in liquid gel precursors or/and loss of the biomolecules through leakage.[8–10]
Despite the potential advantages of mild processing conditions and shear-thinning property, supramolecular hydrogels have not been widely used in tissue engineering.[5, 6, 8] Scaffolding materials for tissue engineering have to be biocompatible and are desired to degrade/erode in a proper time period, which are often not easy to achieve in many supramolecular hydrogels.[5, 7]
The regeneration of tissue complexes, where the intimate integration between different adjacent tissue types plays a key role in their function, has been a long-standing challenge in the field of tissue engineering. For example, well-integrated bone-cartilage complex is essential for the function of joints. Such structural complexity posts a significant technical barrier to tissue engineers.[11, 12] A scaffolding material designed to regenerate one specific tissue type is unlikely optimal for regenerating another tissue type, and therefore often fails to regenerate a functional multi-tissue complex.[12, 13]
The transient nature of certain supramolecular interactions allows a hydrogel to self-heal or self-integrate, i.e., separate hydrogel pieces can be joined to form an integrated construct.[6, 7, 14] Previous reports on self-healing materials focused on the role of self-healing property in extending the service life of structural materials, which are neither biocompatible nor biodegradable/erodible.[15] We hypothesize that a supramolecular hydrogel with a rapid self-integrating property may provide a novel solution to the regeneration of tissue complexes if it is biocompatible and biodegradable or erodible. In such an approach, separate pieces of a hydrogel, each carrying the respective cell types and signaling biomolecules, can be integrated into a two- or multiple-tissue construct, where the component tissues are regenerated in spatially defined regions and are seamlessly integrated at their interfaces.
To achieve the above-mentioned goal, the ideal hydrogel should be able to rapidly self-integrate in its fully gelled form without any external intervention. We therefore developed a novel rapid self-integrating and shear-thinning hydrogel by grafting a significant number of multiple-hydrogen-bond units on a biocompatible hydrophilic polymer (Scheme 1a), and investigated the regeneration of a cartilage-bone tissue complex in vivo using the novel hydrogel and a subcutaneous implantation model in nude mice (Scheme 1b).
Scheme 1.

Design of a DEX-UPy hydrogel for multi-tissue complex regeneration. (a) Schematic illustration of DEX-UPy hydrogel formation and the mechanisms of the shear-thinning and self-recovery properties. Top: multiple hydrogen bonds of UPy and their dynamic interactions. Bottom: hydrogel network formation through UPy hydrogen bonds, the shear-thinning under shear stress and self-recovering of the hydrogel. (b) Schematic illustration of self-integration and the application in cartilage-bone tissue complex regeneration. Hydrogels encapsulating chondrocytes (blue) and BMSCs/BMP-2 (red) were integrated into a construct and was then implanted subcutaneously in a nude mouse to form the cartilage-bone tissue complex.
The chosen multiple-hydrogen-bond unit is a quadruple hydrogen-bond array named UPy (Ureido-pyrimidinone) because it has a dramatically higher intermolecular bonding strength than a single hydrogen bond.[16] The chosen polymer is dextran (DEX) because it is an FDA approved polysaccharide and has numerous functional groups to allow the intended covalent grafting of many UPy units along the DEX backbone. Utilizing the hydroxyl groups on DEX, we grafted many UPy units onto DEX to form the UPy-bearing polymers (Supporting Information Scheme S1). The UPy graft density could be controlled simply by changing the feeding ratio of UPy to dextran. 1H NMR analysis was carried out to confirm the structure of the DEX-UPy and the UPy-dimmer formation (Supporting Information Figure S1). The density of substitution (DS, the number of UPy units per 100 glucose units) was estimated by calculating the ratio of the areas under the characteristic peaks.
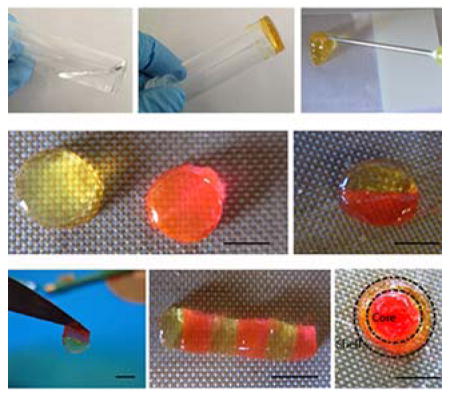
The multi-functionalized DEX-UPy polymer could form a robust hydrogel when the UPy density was sufficiently high. For example, DEX-UPy2 (DS 5.5, 10% w/w) could be dissolved in water at elevated temperature (around 70°C) and form a stable hydrogel after being cooled down to room temperature (Figure 1a, panel 3), while pure DEX and DEX-UPy with a very low DS (DEX-UPy1, DS 2.8, 10% w/w) formed clear solutions under the same conditions (Figure 1a, panels 1&2). Polymers with too high a density of substitution (DS ≥ 8.1) had poor water solubility and thus could not be used for hydrogel preparation. With an appropriate UPy content, a DEX-UPy polymer could form a hydrogel, be loaded into a syringe and be subsequently injected through a needle (Figure 1a, panel 4). The hydrogel behaved like a liquid under the shear stress during injection and quickly solidified after the injection, which is a shear-thinning behavior. Different shapes of the hydrogel could be fabricated by injecting it into differently shaped molds. To illustrate the self-integration capacity, the hydrogel disks were cut into different parts and were subsequently brought together. One hydrogel was dyed with rodamine (pink) and the other was left with the original color (light yellow) to examine the integration between the hydrogel pieces. The hydrogel was found to integrate within minutes after they were brought into contact (Figure 1b). Different patterns could be achieved, such as a rod of joined disks and an integrated disk consisting of an inner core and an outer ring. Previously, a telechelic PEG-based co-polymer was functionalized with UPy at their two ends, and such modified PEG was shown to form a hydrogel and used for protein delivery.[17] However, hydrogel formation of this polymer required hydrophobic segments and urea groups, which help the nanofiber formation to stabilize the gel. While the PEG-UPy hydrogels were able to self-heal macroscopically, the process took days because of the slow kinetics of the nanofiber reformation at the interface (see Basting et al, on page 7 of SI),[18] and therefore not able to quickly integrate into a stable 3D structure for our aimed tissue repair/regeneration applications.
Figure 1.

Formation of DEX-UPy hydrogel, its injectability and self-integrating property. (a) Gel forming property of dextran (DEX) polymers with different UPy substitution densities (DS). DEX with no modification or with low UPy DS (DS 2.8, 10% w/w) was in a solution state, while a DEX-UPy polymer with an appropriate UPy DS (DS 5.5, 10% w/w) readily formed a hydrogel at room temperature and could be injectable through a 26G needle. (b) Self-integration of the hydrogel pieces to form various structures. Some hydrogel disks were dyed pink with rodamine and the others were left with the original light yellow color to visualize the interfaces. Scale bar=5mm.
In order to quantify the mechanical properties associated with shear-thinning and the subsequent recovery, we measured the rheological properties of the hydrogels. A hydrogel made of a 10% (w/w) DEX-UPy (DS 5.5) polymer solution had a storage modulus of 170 Pa, whereas the hydrogel made of the same polymer with a concentration of 12.5% (w/w) had a storage modulus of 700 Pa (Figure 2a), showing the non-linear relationship between the polymer concentration and the mechanical properties of such a polymer. To mimic the change from the statically stored gel to the mechanical injection of the gel from a syringe, a shear force was applied to the hydrogel with a 10% polymer concentration. At a critical shear stress level the hydrogel yielded and lost its mechanical integrity, where the storage modulus (G′) fell below the loss modulus (G″) (Figure 2b). This phenomenon demonstrates the shear-thinning property. Furthermore, cyclic high and low stresses were applied to the hydrogel. At a low stress level, the gel remained stable and the G′ was higher than G″. At a high stress level the gel changed into a liquid-like state with the G′ lower than G″. Upon removal of the high stress, the material recovered its gel property within seconds (Figure 2c). The hydrogel could be shear-thinned and recovered for many cycles without significant loss of the mechanical properties. The thermal stability of the hydrogel was examined by measuring the rheological properties of the hydrogel against the temperature (Figure 2d). The hydrogel was softened while being heated at a rate of 2°C/min from room temperature, but it maintained the gel state even at 70°C, which was the temperature when the polymer was initially dissolved in water.
Figure 2.

The rheological properties of DEX-UPy hydrogels. (a) The frequency spectra of the dynamic moduli of DEX-UPy (DS 5.5) hydrogels with concentrations of 12.5% and 10% w/w, where a constant stress 0.1 Pa was applied. (b) Dynamic modulus of 10% w/w hydrogel under increasing stress. Note the hydrogel yielded at a critical stress, corresponding to the state of being injected through a needle. (c) The dynamic modulus of hydrogel (DS 5.5, 10% w/w) under cyclic high (100 Pa) and low (0.1 Pa) stresses. Under the low stress DEX-UPy behaved like a gel while it turned into a sol state under the high stress. Gel state was instantly recovered after the removal of the high stress. (d) Change of the dynamic modulus of the hydrogel (DS 5.5, 10% w/w) with temperature. The modulus decreased upon heating, however remained in the gel state even at 70°C.
When a hydrogel is used as a scaffold for tissue engineering, it serves as a temporary template and therefore is desired to degrade or erode in vivo. DEX is known not to be degradable by human enzymes. Therefore, covalently crosslinked DEX hydrogels are not degradable in the human body unless degradable bonds are incorporated into the crosslinkers.[19] The DEX-UPy hydrogel disintegrated over time mainly through physical erosion, during which the hydrophilic polymer molecules disassociated from one another and diffused into the aqueous environment. The hydrogel with a concentration of 10% and a UPy content of 5.5% lost its ~20% mass during the first day and additional ~35% over a month, while the hydrogel with a concentration of 12.5% and the same UPy content eroded slower and lost ~45% mass in the entire time period (Supporting Information Figure S2). The erosion profile of this hydrogel can be adjusted to be suitable for engineering different tissue types, where a temporary template is typically needed for weeks to months.
Drugs or proteins could be encapsulated in the hydrogel and released over time in durations from days to months (Supporting Information Figure S3), which is dependent on the size and characteristics of the drug or protein. Doxycycline was used as a model drug of small molecules, and it was nearly completely released in vitro during the first week. Since most growth factors for tissue morphogenesis are protein-based, we also encapsulated a model protein, bovine serum albumin (BSA), in the hydrogels and measured the release kinetics. BSA could be released for more than a month and there was no significant burst release (only about 10 percent of BSA was released during the first day). A sustained release was achieved for BSA during the entire experimental duration of 4 weeks. Such sustained release profile for proteins/growth factors is highly desired for tissue engineering applications.[2, 20]
The DEX-UPy hydrogel remained a viscous liquid for minutes after being cooled down to room temperature, thus it was possible to mix and encapsulate cells in the hydrogel. Different types of cells, including chondrocytes and bone marrow stem cells (BMSCs) were individually encapsulated and cultured in the hydrogel. Under confocal microscope, uniform cell distribution in each hydrogel was observed (Supporting Information Figure S4). The live-dead assay confirmed that both chondrocytes and BMSCs maintained a high viability in the hydrogel as examined after in culture for two weeks (Supporting information Figure S4), indicating that the DEX-UPy hydrogel is highly compatible with mammalian cells.
Due to the injectability and self-integrating property of DEX-UPy hydrogel, we were able to fabricate a self-integrated scaffold to engineer bone-cartilage tissue complex. Chondrocytes for cartilage formation and BMSCs plus bone morphogenetic protein 2 (BMP-2) for bone regeneration were encapsulated separately into two vials of the hydrogel (Figure S5). Each cell-encapsulated gel was subsequently injected into one side of a circular disk-shaped PDMS (polydimethylsiloxane) mold separated by a Teflon film. Without external intervention, intimate integration of the two components was achieved immediately after the baffle film was removed. The self-integrated hydrogel construct was subcutaneously implanted in a nude mouse to evaluate the potential of osteochondral tissue complex regeneration. Three groups of cell-gel constructs were implanted, including chondrocytes alone, BMSCs/BMP-2, and self-integrated constructs with the two types of cells encapsulated on two sides of the gel disk. The tissue formation was examined histologically after 8 weeks of implantation. Positive staining of mineralized tissue (Alizarin red, for bone) and sulfated glycosaminoglycan (Alcian blue, for cartilage) in the histological sections validated the formation of cartilage and bone within the single cell-type groups respectively (Figure 3a, 3b). The results demonstrate the capability of the new hydrogel in supporting the growth of both bone and cartilage tissues. The self-integrated osteochondral implants were stained using both Alcian blue and Alizarin red (Figure 3c). Both bone and cartilage tissues were identified within their spatially defined regions. While the chondrocytes side (left) was stained positive for cartilage only, the BMSCs/BMP-2 side (right) was stained positive for bone only. The regenerated cartilage and bone tissues were intimately integrated as shown in the magnified image (Figure 3d). We quantified the volumes of cartilage and bone tissues using 10 sections from 4 different samples (Figure 3e). Cartilage occupied a larger volume (~70%) than bone (~30%). Several studies have shown that co-culture of BMSCs with chondrocytes, either in a mixture or in close contact, would induce the differentiation of BMSCs to chondrocytes.[21] We suspected that some BMSCs at the interface region was induced into chondrocytes, therefore resulting in cartilage formation across the original boundary, leading to more cartilage than bone formation and the seamless integration between these two types of tissues. If it is desired to generate equal volumes of bone and cartilage, a smaller initial volume of chondrocyte-containing hydrogel could be used to counter balance the additional chondrocytes induced from BMSCs at the interface.
Figure 3.

Subcutaneous implantation of the cell-gel constructs. (a) A section of BMSCs/BMP-2 only group, stained with Alizarin red (positive staining represents mineralized bone tissue); (b) A section of chondrocytes only group, stained with Alcian blue (positive staining represents cartilage tissue); (c) A section of the self-integrated group, stained with both Alizarin red and Alcian blue; (d) A magnified image of the interface region of image c. (e) Quantification of bone and cartilage volumes using an image analysis software (Adobe Photoshop). There was significantly more cartilage (~70%) than bone (~30%) in volume after subcutaneous implantation for 8 weeks. Scale bar=5mm in (a), (b) and (c); scale bar=100um in (d).
In summary, we have successfully developed a novel injectable (shear-thinning) and self-integrating hydrogel that is biocompatible, bio-erodible and capable of releasing biomolecules sustainably. The cartilage-bone tissue complex was successfully regenerated in a subcutaneous implantation model using the self-integrating hydrogel with the selected cells and biomolecules. We have proved the concept of engineering a multi-tissue complex using the novel supramolecular hydrogel with shear-thinning and self-integrating properties. The hydrogel may also be utilized to regenerate other tissue complexes by combining different types of cells and biomolecules.
Materials and Methods
Detailed materials and methods can be found in the supporting information.
Supplementary Material
Acknowledgments
The authors are grateful to Danielle Zurcher and Prof. Anne McNeil (Department of Chemistry) for their assistance in rheological measurements. The authors gratefully acknowledge the financial support from the NIH (NIDCR DE022327, DE015384, and HL114038), DOD (W81XWH-12-2-0008), and NSF (DMR-1206575). S.H. acknowledges the scholarship from China Scholarship Council.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Sen Hou, Center for Biomedical Engineering and Regenerative Medicine, Frontier Institute of Science and Technology, Xi’an Jiaotong University, Xi’an 710054, China. Department of Biologic and Materials Sciences, University of Michigan, Ann Arbor, MI, 48109, USA.
Dr. Xuefei Wang, Department of Biologic and Materials Sciences, University of Michigan, Ann Arbor, MI, 48109, USA
Sean Park, Department of Biologic and Materials Sciences, University of Michigan, Ann Arbor, MI, 48109, USA.
Dr. Xiaobing Jin, Department of Biologic and Materials Sciences, University of Michigan, Ann Arbor, MI, 48109, USA
Prof. Peter X. Ma, Email: mapx@umich.edu, Center for Biomedical Engineering and Regenerative Medicine, Frontier Institute of Science and Technology, Xi’an Jiaotong University, Xi’an 710054, China. Department of Biomedical Engineering, Macromolecular Science and Engineering Center, Department of Materials Science and Engineering, University of Michigan, Ann Arbor, MI 48109, USA
References
- 1.Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Advanced Materials. 2006;18:1345. [Google Scholar]
- 2.Lutolf MP, Hubbell JA. Nature Biotechnology. 2005;23:47. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 3.Hennink WE, van Nostrum CF. Advanced Drug Delivery Reviews. 2002;54:13. doi: 10.1016/s0169-409x(01)00240-x. [DOI] [PubMed] [Google Scholar]
- 4.a) Wei Z, Yang JH, Zhou J, Xu F, Zrinyi M, Dussault PH, Osada Y, Chen YM. Chemical Society Reviews. 2014;43:8114. doi: 10.1039/c4cs00219a. [DOI] [PubMed] [Google Scholar]; b) Boekhoven J, Stupp SI. Advanced Materials. 2014;26:1642. doi: 10.1002/adma.201304606. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hunt JN, Feldman KE, Lynd NA, Deek J, Campos LM, Spruell JM, Hernandez BM, Kramer EJ, Hawker CJ. Advanced Materials. 2011;23:2327. doi: 10.1002/adma.201004230. [DOI] [PubMed] [Google Scholar]; d) Sun TL, Kurokawa T, Kuroda S, Ihsan AB, Akasaki T, Sato K, Haque MA, Nakajima T, Gong JP. Nat Mater. 2013;12:932. doi: 10.1038/nmat3713. [DOI] [PubMed] [Google Scholar]
- 5.Appel EA, del Barrio J, Loh XJ, Scherman OA. Chemical Society Reviews. 2012;41:6195. doi: 10.1039/c2cs35264h. [DOI] [PubMed] [Google Scholar]
- 6.Park KM, Yang JA, Jung H, Yeom J, Park JS, Park KH, Hoffman AS, Hahn SK, Kim K. ACS Nano. 2012;6:2960. doi: 10.1021/nn204123p. [DOI] [PubMed] [Google Scholar]
- 7.a) Holten-Andersen N, Harrington MJ, Birkedal H, Lee BP, Messersmith PB, Lee KYC, Waite JH. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:2651. doi: 10.1073/pnas.1015862108. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang Q, Mynar JL, Yoshida M, Lee E, Lee M, Okuro K, Kinbara K, Aida T. Nature. 2010;463:339. doi: 10.1038/nature08693. [DOI] [PubMed] [Google Scholar]
- 8.Foo C, Lee JS, Mulyasasmita W, Parisi-Amon A, Heilshorn SC. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:22067. doi: 10.1073/pnas.0904851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan C, Altunbas A, Yucel T, Nagarkar RP, Schneider JP, Pochan DJ. Soft Matter. 2010;6:5143. doi: 10.1039/C0SM00642D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haines-Butterick L, Rajagopal K, Branco M, Salick D, Rughani R, Pilarz M, Lamm MS, Pochan DJ, Schneider JP. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7791. doi: 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Bullough P, Jagannath A. Journal of Bone & Joint Surgery, British Volume. 1983;65-B:72. doi: 10.1302/0301-620X.65B1.6337169. [DOI] [PubMed] [Google Scholar]; b) Lu HH, Subramony SD, Boushell MK, Zhang XZ. Annals of Biomedical Engineering. 2010;38:2142. doi: 10.1007/s10439-010-0046-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mikos AG, Herring SW, Ochareon P, Elisseeff J, Lu HH, Kandel R, Schoen FJ, Toner M, Mooney D, Atala A, Van Dyke ME, Kaplan D, Vunjak-Novakovic G. Tissue Engineering. 2006;12:3307. doi: 10.1089/ten.2006.12.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atala A, Kasper FK, Mikos AG. Science Translational Medicine. 2012;4 doi: 10.1126/scitranslmed.3004890. [DOI] [PubMed] [Google Scholar]
- 14.Phadke A, Zhang C, Arman B, Hsu CC, Mashelkar RA, Lele AK, Tauber MJ, Arya G, Varghese S. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:4383. doi: 10.1073/pnas.1201122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Wu DY, Meure S, Solomon D. Progress in Polymer Science. 2008;33:479. [Google Scholar]; b) Cordier P, Tournilhac F, Soulie-Ziakovic C, Leibler L. Nature. 2008;451:977. doi: 10.1038/nature06669. [DOI] [PubMed] [Google Scholar]
- 16.Sijbesma RP, Beijer FH, Brunsveld L, Folmer BJB, Hirschberg J, Lange RFM, Lowe JKL, Meijer EW. Science. 1997;278:1601. doi: 10.1126/science.278.5343.1601. [DOI] [PubMed] [Google Scholar]
- 17.Dankers PYW, Hermans TM, Baughman TW, Kamikawa Y, Kieltyka RE, Bastings MMC, Janssen HM, Sommerdijk NAJM, Larsen A, van Luyn MJA, Bosman AW, Popa ER, Fytas G, Meijer EW. Advanced Materials. 2012;24:2703. doi: 10.1002/adma.201104072. [DOI] [PubMed] [Google Scholar]
- 18.Bastings MMC, Koudstaal S, Kieltyka RE, Nakano Y, Pape ACH, Feyen DAM, van Slochteren FJ, Doevendans PA, Sluijter JPG, Meijer EW, Chamuleau SAJ, Dankers PYW. Advanced Healthcare Materials. 2014;3:70. doi: 10.1002/adhm.201300076. [DOI] [PubMed] [Google Scholar]
- 19.van Dijk-Wolthuis WNE, Hoogeboom JAM, van Steenbergen MJ, Tsang SKY, Hennink WE. Macromolecules. 1997;30:4639. [Google Scholar]
- 20.Ma PX. Advanced Drug Delivery Reviews. 2008;60:184. doi: 10.1016/j.addr.2007.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a) Wang DA, Varghese S, Sharma B, Strehin I, Fermanian S, Gorham J, Fairbrother DH, Cascio B, Elisseeff JH. Nature Materials. 2007;6:385. doi: 10.1038/nmat1890. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen WH, Lai MT, Wu ATH, Wu CC, Gelovani JG, Lin CT, Hung SC, Chiu WT, Deng WP. Arthritis and Rheumatism. 2009;60:450. doi: 10.1002/art.24265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.