

Abstract
The understanding of ionic mechanisms underlying cardiac rhythm disturbances (arrhythmias) is an issue of significance in the medical science community. Several advances in molecular, cellular and optical techniques in the past few decades have substantially increased our knowledge of ionic mechanisms that are thought to underlie the arrhythmias. The application of these techniques in the study of ion channel biophysics and regulatory properties has provided a wealth of information, with some important therapeutic implications for dealing with the disease. In this review we briefly consider the cellular and tissue manifestations of a number of cardiac rhythm disturbances, while focusing on our current understanding of the ionic current mechanisms that have been implicated in such rhythm disturbances.
1. Introduction
An abnormal rhythm of the heart (arrhythmia) is a significant cause of concern in the medical community, and in its most severe manifestation, i.e., fibrillation, the abnormality has devastating consequences. Ventricular fibrillation (VF) is the most common cause of >250,000 sudden cardiac deaths each year, and atrial fibrillation (AF) affects ∼2 million people in the United States alone, increasing their risk for stroke and heart failure [1, 2]. In the past few decades, advances in a variety of techniques, including molecular and cellular electrophysiology and optical mapping of electrical excitation have led to a substantial increase in our knowledge of ionic mechanisms underlying cardiac arrhythmias. We have come to better understand that these abnormalities in rhythm may occur as a result of inherited genetic disorders [3], acquired cardiac disease conditions such as heart failure [4], or may be due to side-effects of drugs [5]. Regardless of the underlying cause, certain unifying principles have been found to underlie the initiation and maintenance of arrhythmias, which can be broadly classified as occurring at the cell and tissue levels (Fig. 1). At the cellular level, arrhythmogenesis can occur as a result of abnormal oscillations (automaticity), and early or delayed depolarizations. i.e. EADs and DADs respectively [6]. At the tissue level, vortex shedding and reflection can play a role in the initiation of arrhythmogenesis, whereas reentry in the form of spiral waves or rotors is now thought to play an important role in sustaining arrhythmias including AF and VF [7]. This review briefly describes these two levels (cellular and tissue) by summarizing the main ionic current determinants that are thought to underlie the various arrhythmia mechanisms.
Fig. 1. Mechanisms of Arrhythmias.

The various cellular and tissue mechanisms of arrhythmias are depicted.
2. Automaticity
The primary pacemaker of the heart, the sinoatrial node (SAN), generates spontaneous action potentials (normal automaticity), and in doing so is able to suppress the latent or subsidiary pacemakers such as the AV node, as well as the Purkinje fibers [6]. The ionic mechanisms governing the SAN automaticity have been studied in substantial details, and it is thought that this oscillatory nature is governed by multiple ionic mechanisms, which are shown in Fig. 2A [8]. The diastolic depolarization is thought to result from the inward currents generated by the hyperpolarization-activated funny current, If, the Na+-Ca2+ exchanger current, INaCa, the T-type Ca2+ current, ICaT, and the concomitant decay of the outward repolarizing K+ currents (the rapid and slow delayed rectifiers IKr and IKs, as well as the transient outward current, Ito) [8]. The exact contribution of If versus the so-called Ca2+ clock in driving pacemaking activity via INaCa though remains unresolved [9]. The pacemaking activity can be enhanced due to sympathetic regulation (Fig. 2B), causing a faster diastolic depolarization, and sinus tachycardia. This occurs mainly due to adrenergic effects on multiple ion channels, which includes gating shifts in the activation of If (to more depolarized voltages), and the well-known effects of enhancing Ca2+ cycling, as well as the densities of the L-type Ca2+ current, ICaL, IKs, and the Na+-K+ pump current, INaK [8]. Similar ionic mechanisms (albeit with different precise contribution of various ion channels) contribute to the other pacemaking sites in the heart [10]. However, abnormal automaticity can also arise from normally well polarized atrial and ventricular cells, and is often seen in the setting of ischemia, infarction or stretch [11, 12]. This phenomenon was elegantly demonstrated in single isolated rabbit Purkinje cells, which were coupled to a ventricular cell “model” wherein the resting potential (Vrest), and the coupling resistance could be varied in a controlled manner [13]. At a Vrest of -50 mV, and a moderate coupling resistance of 400 MΩ, which mimicked inducing injury currents, spontaneous oscillations, or “abnormal automaticity” could be induced in normally quiescent Purkinje cells (Fig. 2C). A similar abnormal automaticity has been observed in depolarized ventricular cells when the inward rectifier K+ current, IK1, was suppressed by means of adenovirus [14]. These abnormal automaticities could underlie ventricular tachycardia seen in the setting of ischemia/infarction [15]. Further, abnormal automaticity originating in the pulmonary vein region is now thought to underlie the initiation of AF, predominantly in the paroxysmal form [16].
Fig. 2. Automaticity.
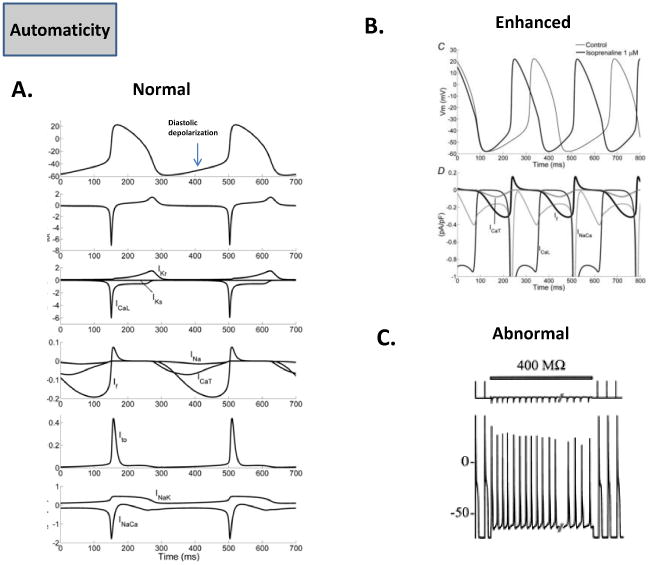
(A.) The normal SAN pacemaker activity and the main underlying ionic currents are shown [8]. (B.) Enhanced automaticity in the SAN pacemaker during adrenergic stimulation, and underlying ionic determinants are shown [8]. (C.) Shows coupling of Purkinje cell to a ventricular cell model via a resistance of 400 MΩ. The ventricular cell is clamped at Vrest of -50 mV, which induces abnormal automaticity in the Purkinje cell [13].
3.EAD and DAD
Initiation
Afterdepolarizations (EADs and DADs) are abnormal transmembrane electrical activities that are always associated with a preceding cardiac action potential (atrial or ventricular) [6]. A typical normal ventricular action potential is generated due to the activation/inactivation of inward or depolarizing currents such the Na+ current, INa, and ICaL, and a number of repolarizing K+ currents (Ito, IKr, IKs, IK1) [17]. Further, a number of electrogenic exchanger (INaCa) and pump (INaK) currents also contribute, and some of these are shown in Fig. 3A [17]. An EAD occurs, when the repolarization phase is prolonged either in acquired disease conditions such as heart failure [4] or inherited Long QT syndromes (LQT1, LQT2, LQT3) [3]. The prolongation of repolarization can occur as a result of a downregulation in various K+ currents, or sustained opening of inward currents such as the late Na+ current [3,4]. Other conditions that can contribute to a prolongation of the action potential and lead to EADs, include electrolyte abnormalities such as hypokalemia, acidosis, slow heart rate, catecholamines, or drugs that block K+ currents [6]. The prolonged repolarization at depolarized potentials leads to a reactivation of ICaL, which then turns on and off, leading to small membrane depolarizations, also known as EADs [18]. Fig. 3B shows simulation results from modeling of LQT2 [19]. When the ventricular cell that incorporates a reduced IKr density (LQT2) is continuously paced at 500 ms basic cycle length (BCL), the action potential is prolonged, but repolarizes normally (pre-pause). However, when an action potential is evoked after a long pause of 1500 ms, the repolarization is abnormally prolonged, and displays EADs. The underlying Ca2+ transient is seen to be normal, but the EADs are driven by repeated re-activation of ICaL [19]. EADs are thought to play an important role in the genesis of Torsade de Pointes (TdP) [6]. A DAD occurs after the action potential is fully repolarized [20], and its genesis is generally attributed to the occurrence of Ca2+ overload in a cardiac cell [21], which may occur in heart failure [4], due to digitalis toxicity [20], or even due to mutations in Ca2+ cycling proteins such as ryanodine, which lead to catecholaminergic polymorphic tachycardia (CPVT) [22]. Ca2+ overload causes calcium waves, and can generate a transient inward current (Iti), which depolarizes and causes DADs [6]. It has been proposed that Iti is generated either by the activation of an electrogenic INaCa [23], a nonspecific cationic current [23], or the calcium-activated chloride current [24]. A computer simulation of a DAD in a ventricular cell is shown in Fig. 3C; in this case Ca2+ overload due to a calsequestrin mutation is mimicked [25]. After the last paced beat, a spontaneous Ca2+ release leads to a DAD, which is also converted to a spontaneous action potential or triggered activity; this is because in addition to a larger inward INaCa, the IK1 current density is also downregulated in this simulation [25]. Thus prolonged repolarization and ICaL reactivation is key for EADs, and abnormal Ca2+ cycling, INaCa, and IK1 are seen to be important ionic determinants of a DAD.
Fig. 3. Afterdepolarizations.
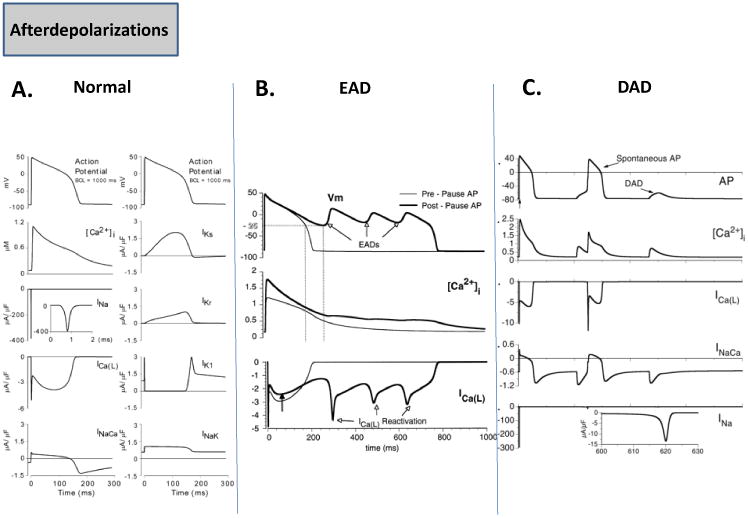
(A.) The normal ventricular action potential and underlying ionic currents are depicted [17]. (B.) Representative simulation of an action potential in LQT2 at a basic cycle length of 500 ms (pre-pause AP), and an EAD occurring after a pause of 1500 ms is shown. Also shown is the underlying Ca2+ transient as well as the repetitive reactivation of ICaL [19]. (C.) Representative simulation of a DAD and a spontaneous action potential during Ca2+ overload, and the underlying Ca2+ transient, ionic currents (ICaL, INaCa, INa), are shown [25].
Propagation into Tissue
In order to initiate arrhythmias, propagation of an EAD/DAD into tissue is necessary. An important factor that controls the spread of excitation via an EAD/DAD is intercellular coupling. A decrease in cell-cell coupling is commonly observed in diseases such as heart failure and ischemia (which gives rise to EADs and DADs), and has also been shown to be associated with slower impulse propagation [17]. Slower impulse propagation can lead to reentry (see next section). In experimental and theoretical studies of coupled cardiac cells and tissue, it was shown that strong intercellular coupling suppressed EAD/DAD formation, whereas in the presence of moderate uncoupling, these afterdepolarizations could propagate into the tissue; in the case of severe uncoupling however, the impulse propagation failed [26-28].
4. Spiral Waves: Ionic Bases of Initiation and Maintenance
Initiation
The concept of reentry has been around for almost a century, and the cardiac muscle was shown to sustain circulating excitatory wavefronts by early pioneering investigators, such as Mayer, Mines and Lewis; this topic is briefly reviewed in a recent article [7]. Beginning in the 1970s, the concept of leading circle driving reentry was put forward [29], whereas others, using both theoretical concepts, and experiments using voltage sensitive dyes in cardiac tissue postulated that spiral waves were driving functional reentry [30,31]. However, the precise mechanisms of fibrillation, i.e. small number of rotors, or multiple random wavelets, remain unresolved [32]. Despite this, the concept of spirals as underlying drivers of fibrillation has found support in both experiments and simulations [7], and recently in the clinic [33]. A spiral wave is a form of functional reentry in which the curved wavefront and wavetail meet each other, at a singularity point (white asterisk, Fig. 4A) [34], and the tissue at the center is not refractory, unlike the leading circle theory [7]. The wavefront represents an area of depolarized cells as the cardiac impulse travels, and the wavetail is made up of cells that are returning to rest (repolarization). A stationary spiral spins around a circular trajectory forming the “core” (Fig. 4A); however, the core can take various complex shapes, depending upon the tissue excitability. The concept of wavelength (λ) is defined as the distance between the wavefront and wavetail, and is measured by the product of refractory period (RP) and the conduction velocity (CV), i.e. λ = RP × CV, and is applicable for a planar wavefront of cardiac impulse propagation [17]. In contrast, there is no “fixed” wavelength for spiral waves, but the distance between the wavefront and the wavetail varies, increasing as a function of the distance from the singularity point (Fig. 4A) [7]. This is because electrotonic gradients established between cells at the core and cells in the near vicinity shorten significantly the action potentials of cells near the core. Fig. 4B shows a snapshot of a simulated rotor generated using a computer ionic model of a 2D sheet of atrial cells in persistent AF [35]. The top panel shows membrane voltage distribution, and the bottom panel shows a plot of the product of the variables “h.j”, which represent the fast (h) and slow inactivation variables (j) of the Na+ current, INa. These variables represent the availability of INa, and when “h.j” is 0.0, no INa is available, and the tissue in unexcitable (the white area in the bottom panel of Fig. 4B), whereas a value of 1.0 means that the tissue is fully available for excitation. All other values between 1.0 and 0.0 represent the “excitable tissue or gap”, where INa is available [7]. A spiral wave is initiated when cardiac impulse propagation is blocked, in part due to tissue heterogeneities in excitability, repolarization or conduction velocity, or even dynamical properties such as action potential alternans, and is analogous to the formation of eddies, when a flow of water waves is broken when it interacts with an obstacle, and rooted in the concept of critical curvature [7]. The concept is explained in the schema shown in Fig. 4C. An artificial linear obstacle is etched into the ventricular muscle sheet (red line), and the curvature of the wavefront about to proceed is shown (Fig. 4C). The white curve bounds the area adjacent to the obstacle, which needs to propagate laterally in order to circumnavigate the obstacle. The radius of this area R is comparable to the width of the wavefront, which determines whether that wavefront (i.e., the source) will be able to excite the tissue ahead of it (i.e., the sink). The ability of the source (or propagating wavefront) to provide enough current in order to elicit a response in the area ahead (the sink) is what allows the impulse to propagate safely (“safety factor”). On the other hand, if the source is unable to provide the necessary current, the safety factor is compromised, and the resulting “source-sink mismatch” can lead to conduction blocks [17]. As shown in the top panel of Fig. 4C (condition I), when the excitability of the tissue is normal and a wave is initiated by a point stimulus near the lower right margin of the obstacle (asterix), the wavefront proceeds to circumnavigate the obstacle without detaching and activates the entire sheet [7]. Here, R is larger than the minimum radius of excitation RCr necessary to sustain normal propagation. In the lower panel of Fig. 4C, when the excitability of the tissue is diminished (condition II), R < RCr, and the wavefront now detaches from the obstacle, curls and begins to rotate around its broken tip, generating a spiral [7]. These concepts were illustrated in the combined experimental-theoretical studies of Cabo et al., where excitability was manipulated by blocking INa [36]. This scenario can exist in pathophysiological conditions such as ischemia or an infarct in the ventricles, or atrial remodeling due to persistent AF, where both INa density and excitability are reduced and numerous obstacles in the form of patchy fibrotic tissue exist, thereby reducing the safety factor for impulse propagation (resulting in slow conduction), and set the stage for initiation of the spirals that maintain tachycardia or fibrillation [7]. The ionic mechanisms underlying the safety factor have been investigated in a series of elegant studies [17,37]. The results showed that when excitability was reduced by gradual reduction in the availability of INa, the safety factor was also reduced monotonically, and at a certain level, when the safety factor was <1, the impulse propagation ceased. In contrast, as the intercellular coupling due to gap junctions was reduced, although the CV was also reduced, the safety factor was in fact increased, and very slow propagation could be sustained [17,37]. This also has implications for maintenance of spiral waves (see next section). In this regard, an interesting and important development in recent years has been the realization that proteins/ion channels thought to act independently such as the Na+ channel, are in fact interacting with other proteins including gap junctions, in a macromolecular complex [38]. For example, it was shown that loss of Cx43 expression also results in a reduction in the density of INa [39]. Similarly, an overexpression of SCN5A that underlies INa in turn has been shown to influence the density of the inward rectifier, IK1, and vice versa [40]. Thus, our concepts regarding a safety factor for propagation will have to be revisited to take into account these newly described ion-channel interactions.
Fig. 4. Spiral Waves, and their Initiation.
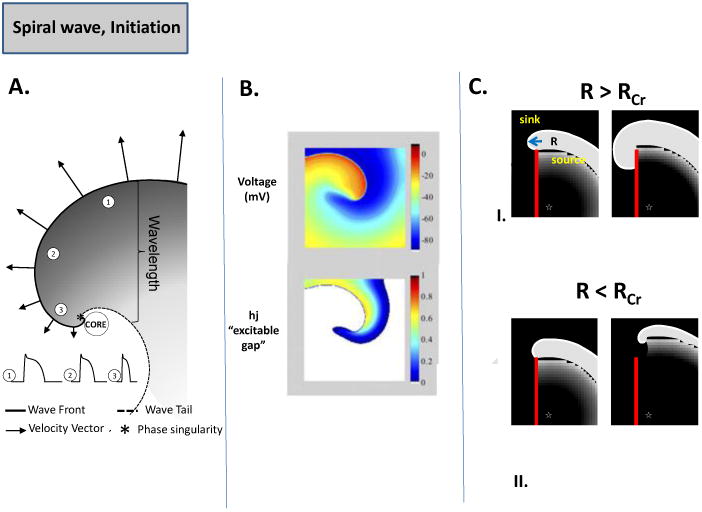
(A.) Schematic of the spiral wave: Electrotonic effects of the core decrease conduction velocity (arrows), action potential duration (representative examples shown from positions 1, 2 and 3) and wavelength (the distance from the wave front (black line) to the wave tail (dashed line) [7]. As CV decreases and wavefront curvature becomes more pronounced, a point singularity occurs as the wave front and tail meet “*” [34]. (B.) Computer simulation of spiral wave. Top panel: snapshot of the transmembrane voltage distribution during simulated reentry in chronic AF conditions in a 2D sheet incorporating human atrial ionic math models [35]. Bottom panel: snapshot of inactivation variables of sodium current, “h.j” during reentry. (C.) Schematic of spiral wave initiation. As a wave progresses along an obstacle (red line) the curvature of the wave at the edge of the obstacle will determine if the wave detaches. If the curvature of the wavefront (R) at the edge of the obstacle is greater than the critical curvature for detachment (RCr) the wave remains attached; if R is less than RCr, when excitability is reduced (due to reduced INa, increased IK1, or enhance fibrosis) the wave will detach from the obstacle and initiate reentry [7].
Maintenance
Earlier studies in guinea pig hearts suggested an important role of IK1 in VF, which was terminated by BaCl2 at low concentrations [41]. The chamber-specific, left-to-right atrial spatiotemporal gradient of AF frequencies in the presence of acetylcholine was shown to be attributable to different densities of another inward rectifier K+ channel, the acetylcholine-activated K+ current, IKACh [42]. The density of IK1 was also found to be upregulated in atrial myocytes isolated from chronic AF patients [2]. When simulations were conducted to examine the consequences of IK1 increase, the spirals were found to be faster, and the core area was smaller, thus stabilizing fibrillation [35]. The simulations suggested, that in addition to causing a shorter action potential duration (APD), IK1 accelerated spirals by increasing the availability of INa; this was possible due to the hyperpolarization of the resting membrane potential (Vrest) by IK1. In transgenic mice where IK1 was overexpressed, it was possible to induce sustained spirals (> 1 hour) in isolated hearts, which were extremely fast (∼45-60 Hz), compared to wild-type hearts, where spirals were transient, and slower (∼20-25 Hz) [43]. In Fig. 5A, the activation map of a spiral observed experimentally in a TG mouse with IK1 overexpression (right) versus a WT heart (left) is shown [43]. It can be seen that the spiral completes one rotation much earlier in the TG mouse [43]. To study the role of the two main repolarizing K+ currents in the human ventricle, i.e. the fast and the slow delayed rectifier K+ currents, viz. IKr and IKs [44], we utilized monolayers of confluent, electrically coupled neonatal rat ventricular myocytes (NRVMs) [45,46]. The density of either IKs or IKr was increased by adenoviral overexpression of either KvLQT1-minK, or hERG, respectively in NRVM monolayers. We found that IKs overexpression did not increase spiral frequency (∼5-13 Hz in both cases), but over time, caused an increasing number of wavebreaks to occur, as shown in Fig 5B by the representative phase maps in a control monolayer, and a monolayer in which IKs was increased [45]. Experiments in isolated HEK cells and suggested that the increased wavebreaks was because of the phenomenon of post-repolarization refractoriness, occurring due to residual outward IKs current, following the action potential [45]. In contrast to IKs, a larger density of IKr caused the spiral to accelerate significantly compared to control (∼21 Hz in the former, versus ∼9 Hz in the latter case) [46]. Both experiments and simulations showed that the faster spiral was due to APD shortening, as well as transient Vrest hyperpolarization, which again would indirectly affect spiral frequency by modifying INa availability [46]. Ionic currents affecting intracellular Ca2+ cycling ([Ca2+]i), such as L-type Ca2+ current, ICaL, also influence the APD, and are thus likely to influence the spiral wave dynamics. In a previous study, verapamil (a partial blocker of ICaL), reduced the excitation frequency of VF, the core meander was increased, and VF was converted to ventricular tachycardia (VT) [47]. However, these results must be interpreted with caution, since at the concentrations of verapamil used in this study, verapamil blocks both ICaL, as well as IKr [48]. The role of [Ca2+]i in sustaining VF/spirals remains controversial. One report suggested that [Ca2+]i -action potential dissociation during VF was a consequence, not a cause of wavebreaks in VF, and that no spontaneous voltage-independent [Ca2+]i waves could be seen [49]; however, this finding remains controversial [50]. The Na+ current, INa is a key determinant of excitability and the upstroke of the cardiac action potentials [17]. In presence of TTX, which blocks INa, the excitation frequency of the spiral wave was reduced, and the core meander was increased [51]. The role of other ionic currents such as Ito, the atrial-specific delayed rectifier IKur and ICaT in maintaining spirals is less clear. Recent experiments have also shed light on the important role of intercellular coupling in the maintenance of spiral waves. In the presence of Carbenoxolone, a relatively selective inhibitor of intercellular coupling, spiral wave reentry was stabilized, and the incidence of pacing-induced sustained ventricular tachycardia (>30 seconds) was substantially increased in rabbit hearts (85+4%), compared to controls (38+4%) [52].
Fig. 5. Maintenance of Spiral Waves.
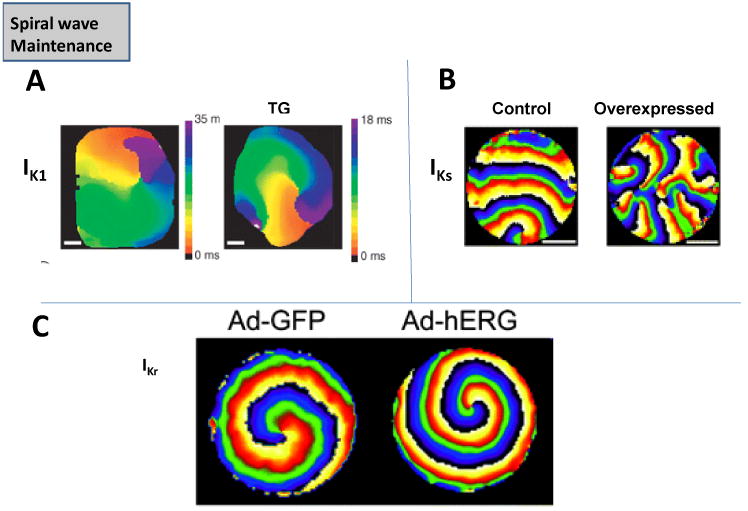
(A.) Spirals in wild-type (WT) mouse hearts and transgenic (TG) mice where IK1 was overexpressed [43]. (B.) Spirals two-dimensional layers of rat neonatal ventricular myocytes in control, and where IKs was overexpressed via adenovirus [45]. (C.) Spirals in two-dimensional layers of rat neonatal ventricular myocytes in control, and where IKr was overexpressed via adenovirus [46].
Termination
Our previous experimental and theoretical studies have explored the mechanism(s) underlying the termination of sustained spiral waves [7], either by simulating drug block of K+ channels [35], or by a reduction in excitability by pathophysiological mechanisms such as hyperkalemia [53].
The results suggested that spiral waves can terminate either by collision with each other, or at the tissue boundary, whereby they are extinguished [35, 53]. A reduction in excitability [53] or an increased plateau phase of the action potential [35] resulted in an increase in the spiral tip or core meander, which in turn increased the possibility of the spiral waves to be pushed toward the boundaries and to self-terminate. Similar mechanisms have been proposed to explain the success of (i) defibrillation in terminating VF, where it has been suggested that shock results in the unpinning or destabilization of spiral waves [54], and (ii) termination of sustained tachycardia in the presence of rotigaptide, which enhances intercellular coupling [52].
5. Reflection
One of the earliest studies on reflection was conducted by Antzelevitch and colleagues, and is shown in Fig 6A [55]. Canine false tendons were placed in a three chamber solution, where the middle chamber was perfused with sucrose of high K+, to stop conduction. Transmembrane potential recordings were made in the proximal (P) and distal (D) chambers, and the sucrose gap was shunted by means of an extracellular resistance. At the right values of this resistance, the stimulus in the proximal segment elicited an action potential in this segment, and with an appropriate delay in the distal segment as well. Interestingly, once the proximal chamber action potential had recovered, the electrotonic currents flowing from the distal to the proximal direction could invoke an action potential in the P region as well (reflection); see Fig 6A [55]. More recently, a similar phenomenon was found in 2D tissue of NRVM monolayers, where proximal and distal rectangular patches were connected via a narrow isthmus (top left panel, Fig. 6B) [56]. When the proximal region was stimulated, and the propagation was plotted as a function of time across a narrow line through the isthmus and patches (bottom left panel, Fig. 6B), the impulse first propagated from the proximal side to the distal side, and then was reflected into the proximal side again. The time dependent action potentials can be seen in the right panel (Fig. 6B). In this monolayer, INa was overexpressed via adenovirus, which led to prolonged APD/EAD on the distal side; the isthmus provided a delay for the impulse propagation due to the source-sink mismatch, which allowed the proximal side to recover, and upon receiving electrotonic current from the isthmus, fire a reflected action potential [56]. Thus, reflection is a complex mechanism driven by delays and electrotonic current induced via source-sink mismatch, and is suggested to underlie either ventricular extrasystole or bigeminy [6].
Fig. 6. Reflection.
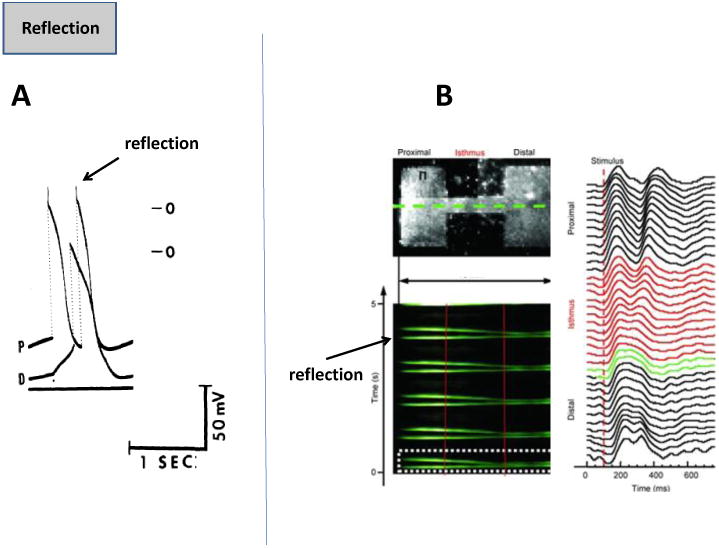
(A.) Shows transmembrane potentials in proximal (P) and distal (D) canine false tendons, with reflection giving rise to a second action potential in the P region [55]. (B.) The top left panel shows two rectangular patches of NRVM connected via a narrow isthmus. The bottom left panel shows time-space plot (Y axis, time, X axis-space) of impulse propagation (green) along the dotted line shown in the above panel. The right panel shows time plot of action potentials along the green line [56].
6. Other Mechanisms
There are other arrhythmias, which indeed can be classified as a subset of the mechanisms described above. One such phenomenon is phase 3 EADs [57], or phase-2 reentry [58]. In the case of the former, the EAD is thought to occur due to an abbreviated APD but a normal Ca2+ transient, and an inward current principally mediated via INaCa [57]. It has been proposed that phase-2 reentry is important in mediating J waves [6] and Brugada syndromes [58], and is caused via an Ito-mediated shortening of the action potential, which causes a re-excitaion of the epicardium, and subsequently leads to reentry [58]. Other mechanisms that predispose to arrhythmias include alternans (voltage or calcium), which is again due to insufficient recovery of either inward currents ICaL or INa, or abnormal Ca2+ cycling mechanisms [59]. It is thought that alternans are precipitated at either high frequencies of pacing, or pathophysiological conditions such as ischemia, and give rise to reentrant tachycardia/VF [60]. Other forms of reentry have also been proposed, such as Figure of 8 reentry [61], or anisotropic reentry [62]. Anisotropic cellular coupling and microfibrosis has been proposed to underlie the latter [62], although further studies are necessary to examine their precise ionic determinants.
Acknowledgments
Funding for this work was provided by NHLBI Grants 1R56HL124319-01, American Heart Association G-I-A 14GRNT19710006 (JMA) and HL118304, HL122352 (SVP). Also, we would like to thank the investigators at the Center for Arrhythmia Research, University of Michigan, Ann Arbor, for their useful discussions.
Footnotes
Conflicts of Interest: JM Anumonwo: None, SV Pandit: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005 doi: 10.1172/JCI26381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heijman J, Voigt N, Nattel S, Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res. 2014 Apr 25;114(9):1483–99. doi: 10.1161/CIRCRESAHA.114.302226. [DOI] [PubMed] [Google Scholar]
- 3.Cerrone M, Cummings S, Alansari T, Priori SG. A clinical approach to inherited arrhythmias. Circ Cardiovasc Genet. 2012 Oct 1;5(5):581–90. doi: 10.1161/CIRCGENETICS.110.959429. Review. [DOI] [PubMed] [Google Scholar]
- 4.Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res. 2004 Oct 15;95(8):754–63. doi: 10.1161/01.RES.0000145047.14691.db. Review. [DOI] [PubMed] [Google Scholar]
- 5.Behr ER, Roden D. Drug-induced arrhythmia: pharmacogenomic prescribing? EurHeart J. 2013 Jan;34(2):89–95. doi: 10.1093/eurheartj/ehs351. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antzelevitch C, Yan GX. In: Ionic and Cellular Basis for Arrhythmogenesis From Contemporary Cardiology: Management of Cardiac Arrhythmias. Yan GX, Kowey PR, editors. Humana Press; 2011. [Google Scholar]
- 7.Pandit SV, Jalife J. Rotors and the dynamics of cardiac fibrillation. Circ Res. 2013 Mar 1;112(5):849–62. doi: 10.1161/CIRCRESAHA.111.300158. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Severi S, Fantini M, Charawi LA, DiFrancesco D. An updated computational model of rabbit sinoatrial action potential to investigate the mechanisms of heart rate modulation. J Physiol. 2012 Sep 15;590(Pt 18):4483–99. doi: 10.1113/jphysiol.2012.229435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lakatta EG, DiFrancesco D. What keeps us ticking: a funny current, a calcium clock, or both? J Mol Cell Cardiol. 2009 Aug;47(2):157–70. doi: 10.1016/j.yjmcc.2009.03.022. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schram G, Pourrier M, Melnyk P, Nattel S. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res. 2002 May 17;90(9):939–50. doi: 10.1161/01.res.0000018627.89528.6f. Review. [DOI] [PubMed] [Google Scholar]
- 11.Katzung BG, Hondeghem LM, Grant AO. Letter: Cardiac ventricular automaticity induced by current of injury. Pflugers Arch. 1975 Oct 28;360(2):193–7. doi: 10.1007/BF00580542. [DOI] [PubMed] [Google Scholar]
- 12.Janse MJ, van Capelle FJ. Electrotonic interactions across an inexcitable region as a cause of ectopic activity in acute regional myocardial ischemia. A study in intact porcine and canine hearts and computer models. Circ Res. 1982 Apr;50(4):527–37. doi: 10.1161/01.res.50.4.527. [DOI] [PubMed] [Google Scholar]
- 13.Huelsing DJ, Spitzer KW, Pollard AE. Spontaneous activity induced in rabbit Purkinje myocytes during coupling to a depolarized model cell. Cardiovasc Res. 2003 Sep 1;59(3):620–7. doi: 10.1016/s0008-6363(03)00507-8. [DOI] [PubMed] [Google Scholar]
- 14.Miake J, Marbán E, Nuss HB. Biological pacemaker created by gene transfer. Nature. 2002 Sep 12;419(6903):132–3. doi: 10.1038/419132b. [DOI] [PubMed] [Google Scholar]
- 15.Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev. 1989 Oct;69(4):1049–169. doi: 10.1152/physrev.1989.69.4.1049. Review. [DOI] [PubMed] [Google Scholar]
- 16.Haïssaguerre M, Jaïs P, Shah DC, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Métayer P, Clémenty J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998 Sep 3;339(10):659–66. doi: 10.1056/NEJM199809033391003. [DOI] [PubMed] [Google Scholar]
- 17.Kléber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. 2004 Apr;84(2):431–88. doi: 10.1152/physrev.00025.2003. Review. [DOI] [PubMed] [Google Scholar]
- 18.January CT, Riddle JM. Early afterdepolarizations: mechanism of induction and block. A role for L-type Ca2+ current. Circ Res. 1989 May;64(5):977–90. doi: 10.1161/01.res.64.5.977. [DOI] [PubMed] [Google Scholar]
- 19.Viswanathan PC, Rudy Y. Pause induced early afterdepolarizations in the long QT syndrome: a simulation study. Cardiovasc Res. 1999 May;42(2):530–42. doi: 10.1016/s0008-6363(99)00035-8. [DOI] [PubMed] [Google Scholar]
- 20.Ferrier GR, Saunders JH, Mendez C. A cellular mechanism for the generation of ventricular arrhythmias by acetylstrophanthidin. Circ Res. 1973 May;32(5):600–9. doi: 10.1161/01.res.32.5.600. [DOI] [PubMed] [Google Scholar]
- 21.Bers DM. Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol. 2014;76:107–27. doi: 10.1146/annurev-physiol-020911-153308. Review. [DOI] [PubMed] [Google Scholar]
- 22.Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O'Connell R, Berenfeld O, Anumonwo J, Pandit SV, Vikstrom K, Napolitano C, Priori SG, Jalife J. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007 Nov 9;101(10):1039–48. doi: 10.1161/CIRCRESAHA.107.148064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kass RS, Tsien RW, Weingart R. Ionic basis of transient inward current induced by strophanthidin in cardiac Purkinje fibres. J Physiol. 1978 Aug;281:209–26. doi: 10.1113/jphysiol.1978.sp012417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laflamme MA, Becker PL. Ca2+-induced current oscillations in rabbit ventricular myocytes. Circ Res. 1996 Apr;78(4):707–16. doi: 10.1161/01.res.78.4.707. [DOI] [PubMed] [Google Scholar]
- 25.Faber GM, Rudy Y. Calsequestrin mutation and catecholaminergic polymorphic ventricular tachycardia: a simulation study of cellular mechanism. Cardiovasc Res. 2007 Jul 1;75(1):79–88. doi: 10.1016/j.cardiores.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huelsing DJ, Spitzer KW, Pollard AE. Electrotonic suppression of early afterdepolarizations in isolated rabbit Purkinje myocytes. Am J Physiol Heart Circ Physiol. 2000 Jul;279(1):H250–9. doi: 10.1152/ajpheart.2000.279.1.H250. [DOI] [PubMed] [Google Scholar]
- 27.Pollard AE, Cascio WE, Fast VG, Knisley SB. Modulation of triggered activity by uncoupling in the ischemic border. A model study with phase 1b-like conditions. Cardiovasc Res. 2002 Dec;56(3):381–92. doi: 10.1016/s0008-6363(02)00598-9. [DOI] [PubMed] [Google Scholar]
- 28.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010 Sep 8;99(5):1408–15. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allessie MA, Bonke FI, Schopman FJ. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. III. The “leading circle” concept: a new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ Res. 1977 Jul;41(1):9–18. doi: 10.1161/01.res.41.1.9. [DOI] [PubMed] [Google Scholar]
- 30.Winfree AT. When Time Breaks Down. Princeton: Princeton University Press; 1987. [Google Scholar]
- 31.Davidenko JM, Kent PF, Chialvo DR, Michaels DC, Jalife J. Sustained vortex-like waves in normal isolated ventricular muscle. Proc Natl Acad Sci USA. 1990;87:8785–8789. doi: 10.1073/pnas.87.22.8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jalife J, Berenfeld O, Skanes A, Mandapati R. Mechanisms of atrial fibrillation: mother rotors or multiple daughter wavelets, or both? J Cardiovasc Electrophysiol. 1998 Aug;9(8 Suppl):S2–12. Review. [PubMed] [Google Scholar]
- 33.Narayan SM, Krummen DE, Rappel WJ. Clinical mapping approach to diagnose electrical rotors and focal impulse sources for human atrial fibrillation. J Cardiovasc Electrophysiol. 2012 May;23(5):447–54. doi: 10.1111/j.1540-8167.2012.02332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gray RA, Jalife J, Panfilov AV, Baxter WT, Cabo C, Davidenko JM, Pertsov AM. Mechanisms of cardiac fibrillation. Science. 1995 Nov 17;270(5239):1222–3. [PubMed] [Google Scholar]
- 35.Pandit SV*, Berenfeld O*, Anumonwo JMB, Zaritski RM, Kneller J, Nattel S, Jalife J. Ionic Determinants of Functional Reentry in a 2-D Model of Human Atrial Cells During Simulated Chronic Atrial Fibrillation. Biophys J. 2005(88):3806–3821. doi: 10.1529/biophysj.105.060459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cabo C, Pertsov AM, Davidenko JM, Baxter WT, Gray RA, Jalife J. Vortex shedding as a precursor of turbulent electrical activity in cardiac muscle. Biophys J. 1996 Mar;70(3):1105–11. doi: 10.1016/S0006-3495(96)79691-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997 Nov;81(5):727–41. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- 38.Agullo-Pascual E, Cerrone M, Delmar M. Arrhythmogenic cardiomyopathy and Brugada syndrome: diseases of the connexome. FEBS Lett. 2014 Apr 17;588(8):1322–30. doi: 10.1016/j.febslet.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jansen JA, Noorman M, Musa H, Stein M, de Jong S, van der Nagel R, Hund TJ, Mohler PJ, Vos MA, van Veen TA, de Bakker JM, Delmar M, van Rijen HV. Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm. 2012 Apr;9(4):600–7. doi: 10.1016/j.hrthm.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Milstein ML, Musa H, Balbuena DP, Anumonwo JM, Auerbach DS, Furspan PB, Hou L, Hu B, Schumacher SM, Vaidyanathan R, Martens JR, Jalife J. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A. 2012 Jul 31;109(31):E2134–43. doi: 10.1073/pnas.1109370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Warren M, Guha PK, Berenfeld O, Zaitsev A, Anumonwo JM, Dhamoon AS, Bagwe S, Taffet SM, Jalife J. Blockade of the inward rectifying potassium current terminates ventricular fibrillation in the guinea pig heart. J Cardiovasc Electrophysiol. 2003 Jun;14(6):621–31. doi: 10.1046/j.1540-8167.2003.03006.x. [DOI] [PubMed] [Google Scholar]
- 42.Sarmast F, Kolli A, Zaitsev A, Parisian K, Dhamoon AS, Guha PK, Warren M, Anumonwo JM, Taffet SM, Berenfeld O, Jalife J. Cholinergic atrial fibrillation: I(K,ACh) gradients determine unequal left/right atrial frequencies and rotor dynamics. Cardiovasc Res. 2003 Oct 1;59(4):863–73. doi: 10.1016/s0008-6363(03)00540-6. [DOI] [PubMed] [Google Scholar]
- 43.Noujaim SF, Pandit SV, Berenfeld O, Vikstrom K, Cerrone M, Mironov S, Zugermayr M, Lopatin AN, Jalife J. Up-regulation of the inward rectifier K+ current (IK1) in the mouse heart accelerates and stabilizes rotors. J Physiol. 2007 Jan 1;578(Pt 1):315–26. doi: 10.1113/jphysiol.2006.121475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005 Oct;85(4):1205–53. doi: 10.1152/physrev.00002.2005. Review. [DOI] [PubMed] [Google Scholar]
- 45.Munoz V, Gzerda KR, Pandit SV, Mironov S, Taffet SM, Rohr S, Kleber AG, Jalife J. Adenoviral Expression of IKs contributes to wavebreak and fibrillatory conduction in neonatal rat ventricular cardiomyocyte monolayers. Circ Res. 2007 Aug;101(5):475–83. doi: 10.1161/CIRCRESAHA.107.149617. [DOI] [PubMed] [Google Scholar]
- 46.Hou L, Deo M, Furspan P, Pandit SV, Auerbach D, Gong Q, Zhou Z, Berenfeld O, Jalife JA. Major Role for hERG in Determining Frequency of Reentry. Circ Res. 2010 Dec 10;107(12):1503–11. doi: 10.1161/CIRCRESAHA.110.232470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Samie FH, Mandapati R, Gray RA, Watanabe Y, Zuur C, Beaumont J, Jalife J. A mechanism of transition from ventricular fibrillation to tachycardia : effect of calcium channel blockade on the dynamics of rotating waves. Circ Res. 2000 Mar 31;86(6):684–91. doi: 10.1161/01.res.86.6.684. [DOI] [PubMed] [Google Scholar]
- 48.Zhang S, Zhou Z, Gong Q, Makielski JC, January CT. Mechanism of block and identification of the verapamil binding domain to HERG potassium channels. Circ Res. 1999 May 14;84(9):989–98. doi: 10.1161/01.res.84.9.989. [DOI] [PubMed] [Google Scholar]
- 49.Warren M, Huizar JF, Shvedko AG, Zaitsev AV. Spatiotemporal relationship between intracellular Ca2+ dynamics and wave fragmentation during ventricular fibrillation in isolated blood-perfused pig hearts. Circ Res. 2007 Oct 26;101(9):e90–101. doi: 10.1161/CIRCRESAHA.107.162735. [DOI] [PubMed] [Google Scholar]
- 50.Ogawa M, Lin SF, Weiss JN, Chen PS. Calcium dynamics and ventricular fibrillation. Circ Res. 2008 Mar 14;102(5):e52. doi: 10.1161/CIRCRESAHA.108.171538. [DOI] [PubMed] [Google Scholar]
- 51.Kneller J, Kalifa J, Zou R, Zaitsev AV, Warren M, Berenfeld O, Vigmond EJ, Leon LJ, Nattel S, Jalife J. Mechanisms of atrial fibrillation termination by pure sodium channel blockade in an ionically-realistic mathematical model. Circ Res. 2005 Mar 18;96(5):e35–47. doi: 10.1161/01.RES.0000160709.49633.2b. [DOI] [PubMed] [Google Scholar]
- 52.Takemoto Y, Takanari H, Honjo H, Ueda N, Harada M, Kato S, Yamazaki M, Sakuma I, Opthof T, Kodama I, Kamiya K. Inhibition of intercellular coupling stabilizes spiral-wave reentry, whereas enhancement of the coupling destabilizes the reentry in favor of early termination. Am J Physiol Heart Circ Physiol. 2012 Sep 1;303(5):H578–86. doi: 10.1152/ajpheart.00355.2012. [DOI] [PubMed] [Google Scholar]
- 53.Pandit SV, Warren M, Mironov S, Tolkacheva EG, Kalifa J, Berenfeld O, Jalife J. Mechanisms underlying the antifibrillatory action of hyperkalemia in Guinea pig hearts. Biophys J. 2010 May 19;98(10):2091–101. doi: 10.1016/j.bpj.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ripplinger CM, Krinsky VI, Nikolski VP, Efimov IR. Mechanisms of unpinning and termination of ventricular tachycardia. Am J Physiol Heart Circ Physiol. 2006 Jul;291(1):H184–92. doi: 10.1152/ajpheart.01300.2005. [DOI] [PubMed] [Google Scholar]
- 55.Antzelevitch C, Jalife J, Moe GK. Characteristics of reflection as a mechanism of reentrant arrhythmias and its relationship to parasystole. Circulation. 1980 Jan;61(1):182–91. doi: 10.1161/01.cir.61.1.182. [DOI] [PubMed] [Google Scholar]
- 56.Auerbach DS, Grzda KR, Furspan PB, Sato PY, Mironov S, Jalife J. Structural heterogeneity promotes triggered activity, reflection and arrhythmogenesis in cardiomyocyte monolayers. J Physiol. 2011 May 1;589(Pt 9):2363–81. doi: 10.1113/jphysiol.2010.200576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Burashnikov A, Antzelevitch C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization-induced triggered activity. Circulation. 2003 May 13;107(18):2355–60. doi: 10.1161/01.CIR.0000065578.00869.7C. [DOI] [PubMed] [Google Scholar]
- 58.Di Diego JM, Cordeiro JM, Goodrow RJ, Fish JM, Zygmunt AC, Pérez GJ, Scornik FS, Antzelevitch C. Ionic and cellular basis for the predominance of the Brugada syndrome phenotype in males. Circulation. 2002 Oct 8;106(15):2004–11. doi: 10.1161/01.cir.0000032002.22105.7a. [DOI] [PubMed] [Google Scholar]
- 59.Laurita KR, Rosenbaum DS. Cellular mechanisms of arrhythmogenic cardiac alternans. Prog Biophys Mol Biol. 2008 Jun-Jul;97(2-3):332–47. doi: 10.1016/j.pbiomolbio.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clusin WT. Mechanisms of calcium transient and action potential alternans in cardiac cells and tissues. Am J Physiol Heart Circ Physiol. 2008 Jan;294(1):H1–H10. doi: 10.1152/ajpheart.00802.2007. Epub 2007 Oct 19. Review. [DOI] [PubMed] [Google Scholar]
- 61.Lazzara R, Scherlag BJ. Generation of arrhythmias in myocardial ischemia and infarction. Am J Cardiol. 1988 Jan 15;61(2):20A–26A. doi: 10.1016/0002-9149(88)90737-0. Review. [DOI] [PubMed] [Google Scholar]
- 62.Spach MS, Heidlage JF, Barr RC, Dolber PC. Cell size and communication: role in structural and electrical development and remodeling of the heart. Heart Rhythm. 2004 Oct;1(4):500–15. doi: 10.1016/j.hrthm.2004.06.010. [DOI] [PubMed] [Google Scholar]