

Abstract
The present study explored whether the intercalated cell Cl−/HCO3− exchanger pendrin modulates epithelial Na+ channel (ENaC) function by changing channel open probability and/or channel density. To do so, we measured ENaC subunit subcellular distribution by immunohistochemistry, single channel recordings in split open cortical collecting ducts (CCDs), as well as transepithelial voltage and Na+ absorption in CCDs from aldosterone-treated wild-type and pendrin-null mice. Because pendrin gene ablation reduced 70-kDa more than 85-kDa γ-ENaC band density, we asked if pendrin gene ablation interferes with ENaC cleavage. We observed that ENaC-cleaving protease application (trypsin) increased the lumen-negative transepithelial voltage in pendrin-null mice but not in wild-type mice, which raised the possibility that pendrin gene ablation blunts ENaC cleavage, thereby reducing open probability. In mice harboring wild-type ENaC, pendrin gene ablation reduced ENaC-mediated Na+ absorption by reducing channel open probability as well as by reducing channel density through changes in subunit total protein abundance and subcellular distribution. Further experiments used mice with blunted ENaC endocytosis and degradation (Liddle's syndrome) to explore the significance of pendrin-dependent changes in ENaC open probability. In mouse models of Liddle's syndrome, pendrin gene ablation did not change ENaC subunit total protein abundance, subcellular distribution, or channel density, but markedly reduced channel open probability. We conclude that in mice harboring wild-type ENaC, pendrin modulates ENaC function through changes in subunit abundance, subcellular distribution, and channel open probability. In a mouse model of Liddle's syndrome, however, pendrin gene ablation reduces channel activity mainly through changes in open probability.
Keywords: epithelial sodium channel, pendrin
pendrin, encoded by Slc26a4, is an aldosterone-sensitive, electroneutral, Na+-independent Cl−/HCO3− exchanger that is expressed in the apical regions of renal intercalated cells (1, 14, 28, 30, 31, 36, 39, 40). With increased circulating aldosterone, increased pendrin-mediated Cl− absorption and HCO3− secretion are observed, which secondarily stimulates Na+ absorption, thereby contributing to the pressor response to this steroid hormone (36). Conversely, pendrin gene ablation (Slc26a−/−, Pds−/−) reduces renal Na+ and Cl− absorption, resulting in natriuresis and chloriuresis and a fall in blood pressure (15, 37).
In the absence of pendrin-mediated Cl− absorption (pendrin-null mice), natriuresis is observed after dietary NaCl restriction (15). However, since pendrin does not transport Na+, our laboratory explored why renal Na+ absorption is impaired in pendrin-null mice. Among the major mechanisms of renal Na+ absorption, we observed that pendrin-null mice have a selective reduction in the abundance and function of the epithelial Na+ channel (ENaC) (15). To examine the effect of pendrin gene ablation on ENaC function, transepithelial voltage (VT) was measured in cortical collecting ducts (CCDs) perfused in vitro before and after the application of an ENaC inhibitor (benzamil) to the luminal fluid (15). CCDs from aldosterone-treated or furosemide-treated wild-type mice (WT) had a substantial lumen-negative VT, which was greatly reduced with benzamil application to the luminal fluid (15, 25). However, in CCDs from pendrin-null mice, VT was low and changed much less with benzamil (15, 25). These observations are consistent with robust ENaC-mediated Na+ absorption in WT mice and with low ENaC-mediated transport in CCDs from pendrin-null mice. However, the effect of pendrin gene ablation on ENaC-mediated Na+ absorption has not been tested directly.
To explore how pendrin gene ablation reduces ENaC function, we examined ENaC subunit abundance in kidneys from WT and pendrin-null mice. While ENaC abundance is similar in kidneys from WT and pendrin-null mice after a NaCl-replete diet (15), α-, β-, and γ-ENaC subunit protein abundance are reduced in kidneys from pendrin-null (Slc26a4−/−, Pds−/−) mice after aldosterone administration or a NaCl-restricted diet (15, 25). The reduced ENaC abundance and function observed in pendrin-null mice appears limited to the kidney and is not explained by altered endocrine function (15).
Full ENaC activity, however, depends not only on subunit abundance but also on subunit subcellular distribution and channel open probability, the latter being dependent on subunit processing. The α-and γ-subunits are fully active following two cleavage events, which reduce the apparent mobility of the α-subunit from 85 to 65 kDa (5) and the γ-subunit from 93 to 70 kDa (5). These α- and γ-ENaC cleavage events stimulate ENaC channel activity through increased channel open probability (5, 6, 23). Since pendrin gene ablation in aldosterone-treated mice reduces the abundance of the 70-kDa more than the 85-kDa γ-ENaC fragment (15), aldosterone-induced γ-ENaC cleavage may be blunted in the mutant mice, which should reduce ENaC channel open probability. Because the α-ENaC antibody employed in that study (15) recognizes an α-ENaC epitope (22) that is removed with these cleavage events (6), whether pendrin gene ablation alters α-ENaC cleavage events is unknown.
ENaC activity (NPo) is the product of channel open probability (Po) and membrane channel density [number of channels (N)]. Channel density depends not only on subunit total protein abundance but also on subunit subcellular distribution (34). As such, pendrin gene ablation may not reduce ENaC function exclusively through the changes in subunit total protein abundance we previously reported (15). Whether pendrin gene ablation changes ENaC subcellular distribution or intrinsic channel properties remains to be determined. Thus, the purpose of the present study was to determine 1) if pendrin gene ablation modulates Na+ absorption in the CCD; 2) if the fall in ENaC function observed with pendrin gene ablation is rescued with application of an ENaC channel-cleaving protease, such as trypsin; and 3) if pendrin gene ablation modulates ENaC channel activity through changes in intrinsic channel properties (open probability) or changes in channel subunit subcellular distribution.
METHODS
Animals.
We studied pendrin-null (Slc26a4−/− or Pds−/−) mice developed by Everett et al. (9) as well as mouse models of Liddle's mice (LL mice) previously reported. We generated mice that were both homozygous pendrin-null (Pds−/−) and homozygous for Liddle's mutation (ENaCLL mice) (26). To do so, mice homozygous for Liddle's mutation (on a C57Bl6/J background), as reported previously (26), were bred with mice on a 129S6SvEvTac background over 10 generations. The resultant mice, homozygous for the Liddle's mutation on a 129S6SvEv Tac background (9), were bred to pendrin-null mice on the same background to produce littermates that were 1) homozogous pendrin null and homozygous for Liddle's mutation (ENaCLL, Pds−/−; LL/KO mice), 2) homozygous for Liddle's mutation but expressing WT pendrin (ENaCLL, Pds+/+; LL/WT mice), 3) homozygous pendrin null with WT ENaC [ENaCWT, Pds−/−; knockout (KO) mice], and 4) WT mice (ENaCWT, Pds+/+; WT mice). Mouse genotype was determined from tail biopsies using real-time PCR with specific probes designed for each gene (Transnetyx, Cordova, TN). Age- and sex-matched male and female mice from each group were studied.
Animal conditioning.
Unless otherwise stated, mice ate a balanced diet (no. 53881300, Zeigler Brothers) prepared as a gel (0.6% agar, 74.6% water, and 24.8% mouse chow) supplemented with NaCl (∼ 0.8 meq NaCl/day) (15) and received aldosterone by minipump (250 μg·kg body wt−1·day−1) for 5–7 days before study.
The Institutional Animal Care and Use Commmittee of Emory University approved all treatment protocols.
Immunoblot analysis.
Semiquantitative immunoblots of kidney lysates from aldosterone-treated wild-type, pendrin null, Liddle’s, and Liddle’s/pendrin null mice were performed as reported previously (15, 42). Lysates were separated by SDS-PAGE on 10% gels and then electroblotted onto polyvinylidene difluoride membranes (Immobilon, Millipore, Bedford, MA). Blots were blocked with 5% nonfat dry milk on PBS-T and then incubated with primary antibodies overnight at 4°C. Rabbit, anti-rat α-, β-, and γ-ENaC antibodies used for immunoblots and immunohistochemistry, described previously (22), were purchased from StressMarq (catalog nos. SPC-403D, SPC-404D, and SPC-405D). Attached primary antibodies were identified by the use of Alexa Fluor 680-linked anti-rabbit IgG (Molecular Probes, Eugene, OR) and visualized by infrared detection with the LICOR Odyssey protein analysis system (Lincoln, NE). Equal protein loading was confirmed in gels run in parallel stained with Coomassie blue dye.
Quantitative analysis of immunohistochemistry.
Kidneys were fixed in situ with 2% paraformaldehyde-lysine-periodate and embedded in paraffin or polyester wax [polyethylene glycol 400 distearate (Polysciences, Warrington, PA) and 10% 1-hexadecanol] as described previously (15, 36). β- and γ-ENaC immunoreactivity was detected using immunoperoxidase procedures. For qualitative observations, 2-µm-thick paraffin sections were exposed to 3% H2O2 in methanol for 30 min to block endogenous peroxidase activity, followed by protein blocking using 1% BSA, 0.2% gelatin, 0.05% saponin solution. The primary antibody was diluted in PBS. Sections were rinsed with PBS supplemented with 0.1% BSA, 0.05% saponin, 0.2% gelatin, exposed to horseradish peroxide-conjugated goat anti-rabbit secondary antibody (1:200, Dako), then washed and reacted with diaminobenzidine. Sections were washed with distilled water and counterstained with hematoxylin. For quantitative immunohistochemistry, 3-µm-thick polyester wax sections mounted on triple chrome-alum gelatin-coated slides were used. Antigen retrieval was accomplished by immersing slides in Trilogy (Cell Marque, Rocklin, CA) and heating to 88–96°C for 1 h. Endogenous peroxidase activity was blocked by incubating the sections in 3% H2O2 in distilled water for 45 min. The sections were then washed, blocked for 15 min with Serum-Free Protein Block (Dako Cytomation), and then incubated at 4°C overnight with primary antibody diluted in Dako antibody diluent. The sections were washed in PBS and incubated for 30 min with polymer-linked, peroxidase-conjugated goat anti-rabbit IgG (Vector ImmPRESS, Vector Laboratories, Burlingame, CA), again washed with PBS, then exposed to diaminobenzidine (DAB substrate kit, Vector) for 5 min. The sections were washed in distilled water, dehydrated with xylene, mounted, and observed by light microscopy.
The subcellular distribution of β- and γ-ENaC was quantified as previously described in bright-field light micrographs (13). High-resolution digital micrographs were taken of defined tubular segments using a Nikon E600 microscope and a DXM1200F digital camera (36-megapixel images, ×40 objective) and ACT-1 software (Nikon) or a Leica DM2000 microscope and a Leica DFC425 digital camera (14.4-megapixel images, ×63 objective) and Leica DFC Twain software and the LAS application suite (Leica Microsystems, Buffalo Grove, IL). Pixel intensity was quantified across a line drawn from the tubule lumen through the center of individual cells without a visible nucleus and adjacent to the nucleus in cells with a visible nucleus using National Institutes of Health ImageJ (version 1.34s) software. The apical and basolateral edges were determined as the point at which the intensity diverged from baseline. Initial experiments used the Nikon microscope. Background pixel intensity was calculated as the mean pixel intensity outside the cell and subtracted from pixel intensity at each point. In later experiments, a Leica microscope was used and background pixel intensity was calculated as the mean pixel intensity in the basal 30% of the cell, where ENaC is not expressed, and subtracted from the pixel intensity at each point. Total cellular expression was determined by integrating net pixel intensity across the entire cell. Cell height was determined as the distance in pixels between the apical and basolateral edges of the cell. Immunoreactivity expressed at the apical 20% of the cell was determined by integrating pixel intensity at this region. The individual performing the microscopy and quantifying the results was blinded as to the treatment group of each animal. Each value reported reflects the mean of measurements made in at least 10 tubules that were selected at random. Data from all cells in the CCD were averaged for each animal and used in the statistical analysis.
In vitro perfusion of isolated CCDs.
CCDs were dissected from medullary rays and perfused at flow rates of 2–3 nl/min in the presence of symmetric HCO3−-buffered physiological solution containing (in mM) 125 NaCl, 24 NaHCO3, 2.5 K2HPO4, 2 CaCl2, 2.2 MgSO4, and 5.5 glucose. Tubules were equilibrated at 37°C for 30 min before the collections began. A stock solution of benzamil hydrochloride (3 × 10−3 M) was prepared in water and applied to the perfusate solution in a 1:1,000 dilution. Sample collections and VT measurements were begun 5–10 min after the application of benzamil. All chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Measurement of net transepithelial Na+ flux.
Na+ concentration in collected perfusate samples was measured based on previously reported methods (41) that used a continuous-flow fluorimeter and the Na+-sensitive dye sodium green (Molecular Probes). The flowing reagent stream contained 64 μM sodium green tetramethylammonium salt (S 6900, Molecular Probes) and 20 mM MOPS titrated to pH 7.4 with 2-amino-2-(hydroxymethyl)-1,3 propanediol (Tris-HCl, Sigma). The sample was pipetted through a mineral oil seal into the center of the flowing reagent stream. The sample was carried in the flowing stream through a 10-cm length of Silastic laboratory tubing (inner diameter: 0.3 mm, no. 508-001, Dow Corning) at a flow rate of 3.25 μl/min to reach the cuvette 2.5 min after injection. Light emitted from a 75-W xenon short arc lamp (75 XE, Ushio, Tokyo, Japan) passed through three heat filters (infrared filters, KG1, Omega) through a band-pass filter (490/20 nm) and then through an optical fiber to illuminate the cuvette containing the sample in the flowing stream. Light emitted from the cuvette passed through a band-pass filter (535/40 nm) and was detected by a photomultiplier tube.
Transepithelial Na+ flux (JNa) was calculated according to the following equation:
where Co and CL are perfusate and collected fluid Na+ concentrations, respectively, Q is flow rate (in nl/min), and L is tubule length. Net fluid transport was taken to be zero since net fluid flux has not been observed in CCDs when perfused in vitro in the presence of symmetric solutions and in the absence of vasopressin (16, 17). JNa was expressed as picomoles per millimeter per minute.
VT was measured in the perfusion pipette connected to a high-impedance electrometer through an agar bridge saturated with 0.16 M NaCl and a calomel cell as previously described (38). The reference was an agar bridge from the bath to a calomel cell.
Isolated, split-open collecting duct preparation.
Mice were euthanized by overdose of pentobarbital followed by cervical dislocation, and the kidneys were immediately removed. CCDs were dissected as described above and put in ice-cold physiological saline solution containing (in mM) 140 NaCl, 5 KCl, 1 CaCl2, and 10 HEPES titrated to pH 7.4 with NaOH, as previously described (3). Isolated collecting ducts were allowed to settle onto 5 × 5-mm coverglasses coated with poly-l-lysine. Coverglasses containing tubules were placed in a perfusion chamber mounted on an inverted Nikon Eclipse TE2000 microscope and superfused with room temperature HEPES-buffered (pH 7.4) saline solution. To gain access to the apical membrane of principal cells, tubules were split open with a sharpened micropipette controlled with a micromanipulator. Isolated, split-open tubules were used for patch-clamp analysis within 2 h after isolation.
Patch-clamp electrophysiology.
A microelectrode was filled with physiological buffer solution in which lithium was substituted for sodium, which contained (in mM) 140 LiCl, 5 KCl, 1 CaCl2, and 10 HEPES titrated to pH 7.4 with NaOH, as previously described (3). Gap-free single channel current data from gigaohm seals (recording pipette resistance 7–8 MΩ) were acquired (and subsequently analyzed) with an Axopatch 1D (Axon Instruments) patch-clamp amplifier interfaced via a Digidata 1420 (Axon Instruments) to a PC running the pCLAMP 10.3 suite of software (Axon Instruments). Currents were low-pass filtered at 100 Hz with an eight-pole Bessel filter (Frequency Devices). Unitary current was determined from all-point amplitude histograms fitted with single- or multi-Gaussian curves using the standard 50% threshold criterion to differentiate between events. Channel NPo can be calculated from the single channel record without any assumptions about the total number of channels in a patch or open probability of a single channel using the following relationship:
where T is the total recording time and i is the number of channels open. Open probability was calculated by dividing NPo by the number of active channels within a patch as defined by all-point amplitude histograms.
Statistics.
All data are presented as means ± SE; n used in the statistical analysis represents data from separate mice. To test for statistical significance between two groups, a paired or an unpaired Student's t-test was used. When results from more than two groups were compared, ANOVA was used followed by Tukey's protected t-test. The criterion for statistical significance was P < 0.05.
RESULTS
Pendrin gene ablation reduces the benzamil-sensitive component of Na+ absorption.
To explore the effect of pendrin gene ablation on ENaC-mediated transport, we measured Na+ concentration in collected fluid samples using continuous flow fluorimetry and the Na+-sensitive dye sodium green (41). Using this method, 2.4 nmol NaCl samples gave a coefficient of variation of 1% and a resolution of 2.3 mM, which compares with a resolution of 1 mM, as previously reported (41). This assay equally detects Na+ and Ca2+ but only weakly detects H+, NH4+, and K+ and is insensitive to pH over a range of 6.8–7.6 (Table 1). Because the Na+ absorption observed in this study was so much greater than the expected rate of Ca2+ absorption (4), Ca2+ flux should contribute a very small component of the signal detected. Moreover, because Na+ flux is net absorptive, whereas Ca2+ flux is net secretory (4), detection of Ca2+ by the Na+ assay would slightly underestimate true net Na+ absorption.
Table 1.
Sodiometer responses to various chloride salts
| Compound | Peak Height, cm | n | Na+ Concentration at Peak Height, mM |
|---|---|---|---|
| 10 mM NaCl | 0.7 | 2 | 10 |
| 10 mM CaCl2 | 0.74 ± 0.05 | 3 | 10.5 ± 0.8 |
| 10 mM NH4Cl | 0.08 ± 0.03 | 3 | 1.2 ± 0.5 |
| 10 mM HCl | 0.08 ± 0.03 | 3 | 1.2 ± 0.5 |
| 10 mM KCl | 0.28 ± .04 | 3 | 4.0 ± 0.6 |
Values are means ± SE.
Since pendrin gene ablation reduces benzamil-sensitive VT, in CCDs from aldosterone-treated mice (15, 25), we explored the effect of pendrin gene ablation on ENaC-mediated Na+ transport. To do so, Na+ absorption was measured directly in CCDs from aldosterone-treated WT and pendrin-null mice before and after application of benzamil (3 μM) to the perfusate. As shown in Fig. 1, A and B, benzamil reduced Na+ absorption by ∼90% in CCDs from both WT and pendrin-null mice. However, pendrin gene ablation reduced both total and benzamil-sensitive Na+ absorption by ∼50%, which reflects a fall in ENaC-mediated Na+ absorption (Fig. 1, C and D). We conclude that pendrin gene ablation reduces ENaC-mediated Na+ absorption.
Fig. 1.

Effect of pendrin gene ablation on total and benzamil-sensitive Na+ absorption [Na+ flux (JNa)]. JNa was measured in cortical collecting ducts (CCDs) taken from aldosterone-treated wild-type (WT) and pendrin-null mice. As shown, benzamil reduced JNa by ∼90% in CCDs from both groups (A and B). However, both total JNa (C) and the benzamil-sensitive component of JNa (D) were lower in CCDs from pendrin-null relative to WT mice. *P < 0.05.
Application of an ENaC-cleaving protease stimulates ENaC activity in pendrin-null mice.
While pendrin gene ablation reduces the abundance of all three ENaC subunits, abundance of the 70-kDa fragment of γ-ENaC falls most markedly (15), which raises the possibility that pendrin gene ablation blunts aldosterone-induced γ-ENaC cleavage. Since trypsin application increases γ-ENaC cleavage both in native CCDs perfused in vitro and in heterologous expression systems (5, 23), we tested the hypothesis that trypsin will increase ENaC activity in CCDs from aldosterone-treated pendrin-null mice by cleaving ENaC subunits not fully processed. We further hypothesized that trypsin will not change ENaC activity in CCDs from aldosterone-treated WT mice because ENaCs in these mice are fully cleaved.
Since lumen-negative VT observed in the CCD occurs primarily through ENaC-mediated Na+ absorption, the lumen-negative VT should rise with increased ENaC-mediated Na+ absorption. Therefore, we examined the effect of trypsin on VT in CCDs from aldosterone-treated WT and pendrin-null mice. As shown in Fig. 2, in CCDs from WT mice, the lumen-negative VT was unchanged with trypsin application, as expected with fully processed ENaC subunits. However, in CCDs from aldosterone-treated pendrin-null mice, we observed a roughly 3-mV increase in lumen-negative VT with trypsin application. One interpretation of these data is that pendrin gene ablation blunts aldosterone-induced ENaC cleavage, which is partially rescued with the application of a channel-cleaving protease.
Fig. 2.
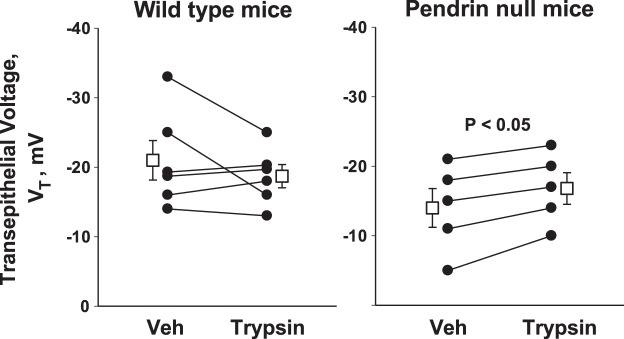
Effect of trypsin on transepithelial voltage (VT) in CCDs from aldosterone-treated pendrin-null and WT mice. Pendrin-null (n = 5) and WT (n = 6) mice consumed the gelled diet described in methods and received 200 μg·kg body wt−1·day−1 aldosterone by minipump for 4–7 days before study. VT was measured in CCDs from mice in both groups before and then 15 min after the application of 15 μg/ml trypsin (no. 1426, Sigma) to the perfusate. In CCDs from WT mice, perfusate flow rates were 2.89 ± 0.21 and 2.88 ± 0.37 nl·mm−1·min−1 before and after trypsin application, respectively, whereas in pendrin-null mice, flow rates were 2.72 ± 0.33 and 2.62 ± 0.41 nl·mm−1·min−1 before and after trypsin. Veh, vehicle.
Pendrin gene ablation reduces both ENaC channel density and ENaC open probability.
The trypsin-induced increase in ENaC activity observed in aldosterone-treated pendrin-null mice raised the possibility that pendrin gene ablation changes channel open probability (32). In further experiments, we asked if pendrin modulates ENaC activity through changes in open probability and/or channel density. To answer this question, we performed single channel recordings of principal cell apical membrane in CCDs from aldosterone-treated pendrin-null and WT mice (Fig. 3). As shown in Fig. 3B, pendrin gene ablation reduced ENaC activity in principal cells. Figure 3, C and D, shows that pendrin gene ablation reduced ENaC activity by reducing both apical plasma membrane ENaC channel density and open probability. We conclude that the fall in ENaC channel activity observed with pendrin gene ablation (Fig. 3B) occurs through changes in channel density and open probability.
Fig. 3.

Pendrin gene ablation changes intrinsic epithelial Na+ channel (ENaC) properties and ENaC channel density. A: typical single channel recordings of principal cell apical membrane taken from CCDs of aldosterone-treated pendrin-null [knockout (KO)] and WT mice. The patch on the tubule from the WT mouse has two observable current levels implying at least two channels in the patch, whereas the pendrin-null patch appears to have only one level and therefore only one channel. B: ENaC activity (NPo) in CCD principal cells from aldosterone-treated WT and pendrin-null mice. C and D: pendrin gene ablation reduced NPo by reducing both the number of channels (N; C) as well as open probability (Po; D). Numbers within the bars for NPo are the number of individual patches from 24 tubules from 8 WT mice and 27 tubules from 9 pendrin-null mice. *P < 0.05.
Pendrin gene ablation changes ENaC subcellular distribution.
We asked if the fall in apical membrane ENaC channel density observed in pendrin-null mice occurs from the fall in subunit abundance, as previously reported (15, 25), and/or through changes in ENaC subcellular distribution. To answer this question, β- and γ-ENaC immunolabeling was compared in CCDs from aldosterone-treated pendrin-null and WT mice. As shown in Figs. 4 and 5 and Table 2, both β- and γ-ENaC immunolabeling appeared weaker and more diffuse in CCDs from pendrin-null relative to WT mice. Using quantitative immunohistochemistry, we compared ENaC subunit total protein abundance per principal cell in WT and pendrin-null mice. As shown in Table 2, pendrin gene ablation reduced total β- and γ-ENaC immunolabel per cell, consistent with our previous observation that ENaC subunit abundance is lower in kidney lysates from aldosterone-treated pendrin-null mice than in WT mice (15, 25). α-ENaC abundance could not be investigated by immunohistochemistry, however, due to the absence of a suitable antibody.
Fig. 4.
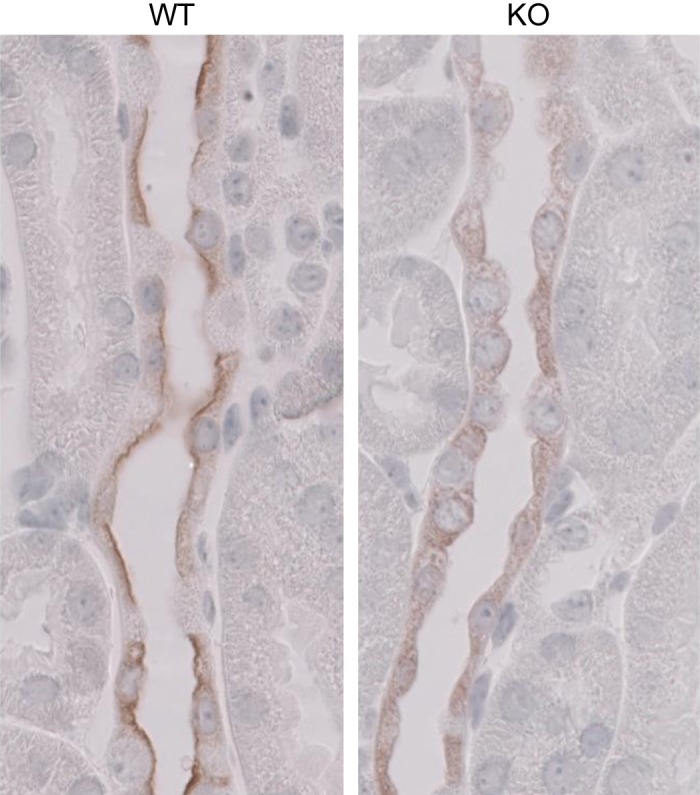
Pendrin gene ablation changes β-ENaC subcellular distribution. These images display β-ENaC labeling in a CCD from aldosterone-treated WT (left) and pendrin-null (KO) mice (right).
Fig. 5.

Pendrin gene ablation reduces γ-ENaC label in the region of the apical plasma membrane of mice harboring WT ENaC, whereas in mouse models of Liddle's syndrome, γ-ENaC is constitutively expressed on the apical plasma membrane, either with or without pendrin gene ablation. γ-ENaC immunolabel is shown in CCD principal cells from aldosterone-treated mice that fell into one of the following four groups: 1) mice with both WT ENaC and WT pendrin (ENaCWT, Pds+/+; WT mice), 2) homozygous pendrin-null mice that harbored WT ENaC (ENaCWT, Pds−/−; KO mice), 3) mice homozygous Liddle's mutation but harboring WT pendrin (ENaCLL, Pds +/+; LL/WT mice), and 4) homozogous pendrin-null mice that were also homozygous for Liddle's mutation (ENaCLL, Pds−/−; LL/KO mice). γ-ENaC immunolabel was more diffuse and less intense in CCDs from KO mice than from WT mice. However, with introduction of Liddle's mutation, γ-ENaC immunolabel appeared intense and discrete in the region of the apical plasma membrane whether mice harbored WT pendrin or were pendrin null.
Table 2.
Effect of pendrin gene ablation on epithelial Na channel subunit protein abundance and subcellular distribution
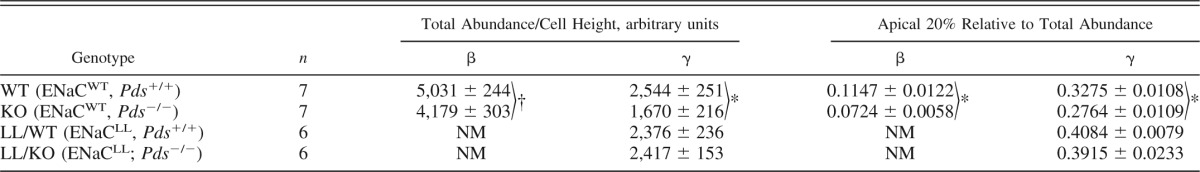
Values are means ± SE. NM, not measured.
P < 0.05;
P = 0.07 (WT vs. KO or LL/WT vs. LL/KO).
To quantify the change in ENaC subcellular distribution that follows pendrin gene ablation, we used quantitative immunohistochemistry to measure the ratio of β- and γ-ENaC expression in the most apical 20% relative to total subunit abundance in the same cell. As shown in Table 2, pendrin gene ablation reduced the proportion of β- and γ-ENaC expressed in the apical 20% of the cell relative to total cell abundance. We conclude that pendrin gene ablation reduces the relative abundance of β- and γ-ENaC subunits in the apical region of principal cells and also reduces β- and γ-ENaC total protein abundance.
Introduction of Liddle's mutation blunts pendrin-dependent changes in ENaC subunit total protein abundance and subcellular distribution.
Further experiments explored whether pendrin modulates ENaC function in the absence of changes in ENaC subunit total protein abundance or subcellular distribution. To answer this question, we used a mouse model of Liddle's syndrome (26). Liddle's syndrome results from a deletion mutation in either the β- or γ-ENaC subunit, which prevents subunit association with the ubiquitin ligase NEDD4-2. As such, reduced internalization and degradation of ENaC is observed, which increases ENaC channel density (7, 26).
In the first series of experiments, we used quantitative immunohistochemistry to examine the effect of pendrin gene ablation on ENaC subunit abundance and subcellular distribution in mice harboring the Liddle's mutation. Figure 5 and Table 2 show that in mice harboring WT ENaC, pendrin gene ablation reduced both total γ-ENaC abundance per cell and the relative abundance of γ-ENaC in the apical region of principal cells. However, the Liddle's mutation blunted both the change in γ-ENaC subunit protein abundance and the change in γ-ENaC subcellular distribution observed with pendrin gene ablation (Table 2). Moreover, the results shown in Fig. 6 demonstrate that while pendrin gene ablation reduces the abundance of all three ENaC subunits in whole kidney lysates from mice with WT ENaC, pendrin-dependent changes in subunit abundance were blunted in kidney lysates from mice with the Liddle's mutation. We could not quantify β-ENaC abundance in mice with Liddle's mutation because the β-ENaC antibody used in this study recognizes a β-ENaC epitope that is clipped off with the Liddle's mutation (35). We conclude that Liddle's mutation blunts the change in ENaC subunit protein abundance and subcellular distribution observed with pendrin gene ablation.
Fig. 6.

Pendrin gene ablation reduces ENaC subunit abundance more in mice harboring WT ENaC than those with the Liddle's ENaC mutation. A: representative blots showing ENaC subunit abundance in kidney lysates taken from mice in each group described in Fig. 5. B: relative band density of α-, β-, and γ-subunits of ENaC in kidney lysates of mice taken from each group. ENaC subunit abundance was compared in aldosterone-treated WT and KO kidneys. For each ENaC subunit, abundance was expressed relative to that observed in the aldosterone-treated WT kidney. Lysates from aldosterone-treated LL/KO and LL/WT mice were also compared. ENaC subunit abundance was expressed relative to that of LL/WT mice. *P < 0.05.
Pendrin gene ablation reduces ENaC-mediated transport even when total ENaC subunit abundance and subcellular distribution do not change.
Since the introduction of Liddle's mutation blunts the change in ENaC subunit abundance and subcellular distribution observed with pendrin gene ablation, we hypothesized that introducing the Liddle's mutation will blunt or eliminate the fall in ENaC activity observed with pendrin gene ablation. To test this hypothesis, we measured the benzamil-sensitive change in voltage as an indicator of ENaC channel activity in CCDs from pendrin-null (KO), WT, LL/KO, and LL/WT mice. As shown in Fig. 7, in mice expressing WT ENaC, pendrin gene ablation reduced total VT. Benzamil-sensitive VT was also numerically lower in CCDs from pendrin-null relative to WT mice, although differences did not reach statistical significance. Nevertheless, these data are consistent with the observed fall in benzamil-sensitive Na+ absorption shown in Fig. 1. When Liddle's mutation was introduced into pendrin-null mice, total and benzamil-sensitive VT rose (LL/KO vs. KO mice), as expected with increased apical plasma membrane ENaC expression (Fig. 5 and Table 2). However, pendrin gene ablation appeared to reduce benzamil-sensitive VT more in mice with Liddle's mutation than in mice harboring WT ENaC. Pendrin gene ablation reduced benzamil-sensitive VT by ∼21 mV in mouse models of Liddle's syndrome (LL/WT vs. LL/KO mice, P < 0.05). In contrast, benzamil-sensitive VT was 9 mV lower in CCDs from pendrin-null than WT mice, which represents a difference not reaching statistical significance. We conclude that while Liddle's mutation blunts pendrin-dependent changes in ENaC subunit protein abundance and subcellular distribution, Liddle's mutation did not reduce pendrin-dependent changes in ENaC activity. Accordingly, pendrin gene ablation in mice with the Liddle's mutation reduces channel activity through a mechanism other than through changes in subunit total protein abundance or subunit subcellular distribution.
Fig. 7.
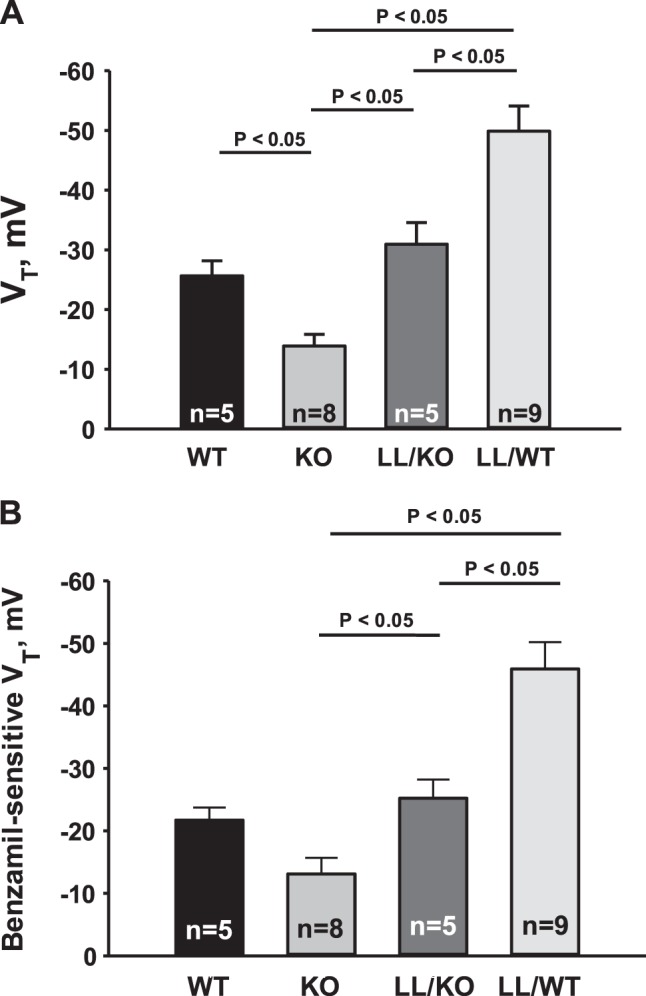
Pendrin gene ablation reduces ENaC activity even when ENaC is constitutively upregulated. A and B: VT (A) and the benzamil-sensitive component of VT (B) in CCDs perfused in vitro that were taken from mice in each of the groups described in Fig. 6.
Pendrin gene ablation reduces ENaC open probability in mouse models of Liddle's syndrome.
Since pendrin gene ablation reduces ENaC function in mouse models of Liddle's syndrome without changing either ENaC subunit total protein abundance or subcellular distribution, we hypothesized that pendrin gene ablation in a mouse model of Liddle's syndrome lowers ENaC channel activity by reducing open probability instead of channel density. To test this hypothesis, we examined the effect of pendrin gene ablation on channel activity, channel density, and open probability in split-open CCDs from aldosterone-treated mice with the Liddle's mutation. Typical channel recordings are shown in Fig. 8A. As shown in Fig. 8B, channel activity (NPo) was 2.71 ± 0.31 in LL/WT mice, which compares with a NPo of 0.894 ± 0.123 observed in WT mice (Fig. 2). While these values were not directly compared, they are consistent with previous observations that Liddle's mutation increases channel activity (2, 7). Figure 8B shows that in a mouse model of Liddle's syndrome, pendrin gene ablation reduced channel activity, which is consistent with the fall in benzamil-sensitive voltage observed in these mutant mice (Fig. 7). Figure 8, C and D, shows that in a mouse model of Liddle's syndrome, pendrin gene ablation reduces channel activity through changes in open probability rather than channel density. Pendrin gene ablation did not change ENaC channel density in mice with Liddle's syndrome, as expected, since pendrin gene ablation did not change ENaC subunit abundance or subcellular distribution in mice with the Liddle's mutation (Figs. 5 and 6 and Table 2). However, in a mouse model of Liddle's syndrome, pendrin gene ablation markedly reduced channel open probability. We conclude that constitutively expressing ENaC on the apical membrane does not eliminate pendrin-induced changes in ENaC function since a critical component of the fall in ENaC channel activity and ENaC-mediated Na+ absorption observed with pendrin gene ablation occurs from changes in channel open probability.
Fig. 8.

Pendrin gene ablation reduces ENaC open probability (Po) in mouse models of Liddle's syndrome. A: typical single channel recordings of principal cell apical membranes taken from CCDs of aldosterone-treated Liddle's mice harboring WT pendrin and in Liddle's mice that were pendrin null. Representative records were selected from patches that appeared to have only one channel so that differences in open probability were more easily recognized. As shown, ENaC channel activity (NPo) (B) was much lower in LL/KO mice relative to LL/WT mice. However, channel density (N) (C) was similar in LL/WT and LL/KO mice. In contrast, channel open probability (D) was much lower in LL/KO than LL/WT mice. Numbers in each bar are the number of individual patches from 6 tubules from LL/WT mice and 4 tubules from LL/KO mice. *P < 0.05.
DISCUSSION
In people and in rodent models of salt-sensitive hypertension (18, 19), blood pressure rises with increased NaCl intake. While salt-sensitive hypertension is generally attributed to Na+ intake, blood pressure can vary more with intake of Cl− than with Na+ (18, 19), suggesting that the coordinated regulation of renal Na+ and Cl− absorption is important in blood pressure regulation. In the CCD, Na+ and Cl− absorption occur across different cell types. Whereas Na+ is absorbed primarily by principal cells, Cl− is absorbed across intercalated cells (29), in large part through pendrin-mediated Cl−/HCO3− exchange. While principal cell Na+ transport has been well studied, less is known about intercalated cell Cl− transport mechanisms and how these Cl− transporters impact blood pressure.
We have observed previously that pendrin gene ablation produces natriuresis and chloriuresis after dietary NaCl restriction, which contributes to the reduced blood pressure observed in pendrin-null mice (15). This natriuresis occurs in part from reduced ENaC subunit abundance and function. In particular, we observed that pendrin gene ablation reduces the abundance of the 70-kDa fragment of γ-ENaC, which is thought to be the functionally active γ-ENaC fragment. These data led us to explore whether pendrin gene ablation interferes with the normal ENaC processing that occurs with aldosterone administration, such as ENaC cleavage. We observed that administration of an ENaC-cleaving protease increased the lumen-negative VT in CCDs from aldosterone-treated pendrin-null mice but not in WT mice. One interpretation of these results is that pendrin gene ablation interferes with aldosterone-induced ENaC cleavage, and that this defect in channel processing is rescued in part with the application of an ENaC-cleaving protease. The absence of an effect of trypsin on VT in CCDs from aldosterone-treated WT mice may occur because ENaC subunits are already fully cleaved. How pendrin regulates luminal protease activity is beyond the scope of this report but might involve changes in luminal pH (25). Moreover, the trypsin-induced increment in VT observed in CCDs from pendrin-null mice was relatively small and did not eliminate the fall in VT observed with pendrin gene ablation. Therefore, changes in ENaC cleavage events do not fully explain the pendrin-dependent change in ENaC function.
Aldosterone regulates renal Na+ and Cl− absorption largely by changing the number of functional transporters in the cell membrane. This steroid hormone produces salt-sensitive hypertension partly by stimulating renal Na+ and Cl− transporters such as ENaC and pendrin (12, 22, 24, 36). Aldosterone increases ENaC surface expression largely through a mechanism that involves the ubiquitin ligase NEDD4-2 (20, 33). After binding to NEDD4-2, transporters such as ENaC are ubiquitinated and then endocytosed and degraded (10, 20, 27). With aldosterone administration, NEDD4-2 is phosphorylated, which prevents the association of this ubiquitin ligase with ENaC (8). Thus, plasma membrane ENaC abundance increases, which increases renal Na+ absorption thereby increasing blood pressure (27, 33). Liddle's syndrome results from truncation mutations in either the β- or γ-subunit of ENaC, which prevents NEDD4-2 from associating with ENaC (7), thereby increasing channel activity by reducing ENaC endocytosis and degradation. While we did not compare channel density in aldosterone-treated WT mice and mice with Liddle's mutation directly, we observed channel number per principal cell apical membrane patch to be 2.75 ± 0.30 in aldosterone-treated WT mice and 5.75 ± 0.51 in aldosterone-treated mice with the Liddle's mutation, which is consistent with previous observations demonstrating that the Liddle's mutation increases ENaC channel density (7).
We exploited mouse models of Liddle's syndrome to study the effect of pendrin gene ablation on ENaC activity when total and apical plasma membrane ENaC abundance are constitutively upregulated. These experiments demonstrated that while pendrin gene ablation modulates ENaC activity through changes in total protein abundance and subcellular distribution, pendrin also strongly modulates intrinsic ENaC channel properties (open probability).
A previous study (25) by our laboratory has shown that pendrin gene ablation reduces ENaC subunit protein abundance, at least in part through changes in luminal pH or HCO3− concentration. A similar mechanism may contribute to pendrin-dependent changes in ENaC open probability (Po) or subcellular distribution, since channel activity (or NPo) in cultured A6 Xenopus cells is virtually eliminated when the HCO3− concentration in culture media is reduced from 25 to 5 mM (25). However, the effect of apical HCO3− concentration (or pH) on ENaC open probability and subcellular distribution remains to be determined.
Intercalated cells also modulate ENaC through ATP-triggered PGE2 release into the luminal fluid (11). In this model, pendrin gene ablation greatly reduces the abundance of H+-ATPase in type B intercalated cells. In so doing, ATP secretion into the luminal fluid is increased, which acts through purinergic receptors on the apical plasma membrane of principal cells to stimulate PGE2 release, thereby inhibiting ENaC function and reducing ENaC subunit abundance (11). In addition to reducing ENaC subunit abundance, ATP is well known to reduce ENaC open probability (21). Therefore, the fall in ENaC open probability observed in the pendrin-null mouse may occur through increased secretion of ATP. How pendrin gene ablation regulates ENaC channel activity therefore requires further study.
In conclusion, pendrin gene ablation reduces ENaC function in aldosterone-treated mice, in part, by inhibiting subunit processing, such as subunit cleavage, which contributes to the fall in channel open probability observed in pendrin-null mice. Pendrin gene ablation also reduces membrane channel density by reducing subunit total protein abundance and by reducing the relative abundance of subunits in the region of the apical membrane.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-46493 (to S. M. Wall), DK-03793 (to D. C. Eaton), and T32-DK-07656 (to Y. Lazo-Fernandez) and by ASN Career Development Grant 145596 (to V. Pech).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: V.P., S.M.W., D.C.E., and J.W.V. conception and design of research; V.P., M.N., H.-F.B., Y.-H.K., Y.L.-F., Q.Y., T.D.P., and J.W.V. performed experiments; V.P., S.M.W., M.N., H.-F.B., Y.-H.K., Y.L.-F., T.D.P., D.C.E., and J.W.V. analyzed data; V.P., S.M.W., Y.-H.K., D.C.E., and J.W.V. interpreted results of experiments; V.P. and S.M.W. drafted manuscript; V.P., S.M.W., Y.-H.K., D.C.E., and J.W.V. edited and revised manuscript; V.P., S.M.W., M.N., H.-F.B., Y.-H.K., Y.L.-F., Q.Y., T.D.P., D.C.E., and J.W.V. approved final version of manuscript; S.M.W., Y.-H.K., T.D.P., D.C.E., and J.W.V. prepared figures.
ACKNOWLEDGMENTS
The authors thank Dr. Bernard Rossier and Dr. Tom Kleyman for helpful suggestions. The authors thank Dr. Edith Hummler for providing the mouse models of Liddle's syndrome. We thank Dr. Sharon W. Matthews, Tanisha Thomas, and Denise Klein for technical assistance.
REFERENCES
- 1.Amlal H, Petrovic S, Xu J, Wang Z, Sun X, Barone S, Soleimani M. Deletion of the anion exchanger Slc26a4 (pendrin) decreases the apical Cl−/HCO3− exchanger activity and imipairs bicarbonate secretion in the kidney collecting duct. Am J Physiol Cell Physiol 299: C33–C41, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anantharam A, Tian Y, Palmer LG. Open probability of the epithelial sodium channel is regulated by intracellular sodium. J Physiol 574: 333–347, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bao HF, Thai TL, Yue Q, Ma HP, Eaton AF, Cai H, Klein JD, Sands JM, Eaton DC. ENaC activity is increased in isolated, split-open cortical collecting ducts from protein kinase Cα knockout mice. Am J Physiol Renal Physiol 306: F309–F320, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bourdeau JE, Hellstrom-Stein RJ. Voltage-dependent calcium movement across the cortical collecting duct. Am J Physiol Renal Fluid Electrolyte Physiol 242: F285–F292, 1982. [DOI] [PubMed] [Google Scholar]
- 5.Bruns JB, Carattino MD, Sheng S, Maarouf A, Weisz OA, Pilewski JM, Hughey RP, Kleyman TR. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the g-subunit. J Biol Chem 282: 6153–6160, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Carattino MD, Sheng S, Bruns JB, Pilewski JM, Hughey RP, Kleyman TR. The epithelial Na+ channel is inhibited by a peptide derived from proteolytic processing of its a subunit. J Biol Chem 281: 18901–18907, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Dahlmann A, Pradervand S, Hummler E, Rossier BC, Frindt G, Palmer LG. Mineralocorticoid regulation of epithelial Na+ channels is maintained in a mouse model of Liddle's syndrome. Am J Physiol Renal Physiol 285: F310–F318, 2003. [DOI] [PubMed] [Google Scholar]
- 8.Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J 20: 7052–7059, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, Hoogstraten-Miller SL, Kachar B, Wu DK, Green ED. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet 10: 153–161, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Flores SY, Loffing-Cueni D, Kamynina E, Daidie D, Gerbex C, Chabanel S, Dudler J, Loffing J, Staub O. Aldosterone-induced serum and glucocorticoid-induced kinase 1 expression is accompanied by Nedd4-2 phosphorylation and increased Na+ transport in cortical collecting duct cells. J Am Soc Nephrol 16: 2279–2287, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Gueutin V, Vallet M, Jayat M, Peti-Peterdi J, Corniere N, Leviel F, Sohet F, Wagner CA, Eladari D, Chambrey R. Renal β-intercalated cells maintain body fluid and electrolyte balance. J Clin Invest 123: 4219–4231, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim GH, Masilamani S, Turner R, Mitchell C, Wade JB, Knepper MA. The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci USA 95: 14552–14557, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim HY, Baylis C, Verlander JW, Han KH, Reungjui S, Handlogten ME, Weiner ID. Effect of reduced renal mass on renal ammonia transporter famiily, Rh C glycoprotein and Rh B glycoprotein, expression. Am J Physiol Renal Physiol 293: F1238–F1247, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Kim YH, Kwon TH, Frische S, Kim J, Tisher CC, Madsen KM, Nielsen S. Immunocytochemical localization of pendrin in intercalated cell subtypes in rat and mouse kidney. Am J Physiol Renal Physiol 283: F744–F754, 2002. [DOI] [PubMed] [Google Scholar]
- 15.Kim YH, Pech V, Spencer KB, Beierwaltes WH, Everett LA, Green ED, Shin WK, Verlander JW, Sutliff RL, Wall SM. Reduced ENaC expression contributes to the lower blood pressure observed in pendrin null mice. Am J Physiol Renal Physiol 293: F1314–F1324, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Knepper MA, Good DW, Burg MB. Ammonia and bicarbonate transport by rat cortical collecting ducts perfused in vitro. Am J Physiol Renal Fluid Electrolyte Physiol 249: F870–F877, 1985. [DOI] [PubMed] [Google Scholar]
- 17.Knepper MA, Good DW, Burg MB. Mechanism of ammonia secretion by cortical collecting ducts of rabbits. Am J Physiol Renal Fluid Electrolyte Physiol 247: F729–F738, 1984. [DOI] [PubMed] [Google Scholar]
- 18.Kurtz TW, Hamoudi AAB, Morris RC. “Salt-sensitive” essential hypertension in men. N Engl J Med 317: 1043–1048, 1987. [DOI] [PubMed] [Google Scholar]
- 19.Kurtz TW, Morris RC. Dietary chloride as a determinant of “sodium-dependent” hypertension. Science 222: 1139–1141, 1983. [DOI] [PubMed] [Google Scholar]
- 20.Loffing-Cueni D, Flores SY, Sauter D, Daidie D, Siegrist N, Meneton P, Staub O, Loffing J. Dietary sodium intake regulates the ubiquitin-proteinligase Nedd4-2 in the renal collecting system. J Am Soc Nephrol 17: 1264–1274, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Ma HP, Li L, Zhou ZH, Eaton DC, Warnock DG. ATP masks stretch activation of epithelial sodium channels in A6 distal nephron cells. Am J Physiol Renal Physiol 282: F501–F505, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC α, β and γ subunit proteins in rat kidney. J Clin Invest 104: R19–R23, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morimoto T, Liu W, Woda C, Carattino MD, Wei Y, Hughey RP, Apodaca G, Satlin LM, Kleyman TR. Mechanism underlying flow stimulation of sodium absorption in the mammalian collecting duct. Am J Physiol Renal Physiol 291: F663–F669, 2006. [DOI] [PubMed] [Google Scholar]
- 24.O'Shaughnessy KM, Karet FE. Salt handling and hypertension. J Clin Invest 113: 1075–1081, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pech V, Pham TD, Hong S, Weinstein AM, Spencer KB, Duke BJ, Walp E, Kim YH, Sutliff RL, Bao HF, Eaton DC, Wall SM. Pendrin modulates ENaC function by changing luminal HCO3−. J Am Soc Nephrol 21: 1928–1941, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pradervand S, Wang Q, Burnier M, Beermann F, Horisberger JD, Hummler E, Rossier BC. A mouse model for Liddle's syndrome. J Am Soc Nephrol 10: 2527–2533, 1999. [DOI] [PubMed] [Google Scholar]
- 27.Rotin D, Staub O. Role of the ubiquitin system in regulating ion transport. Pflügers Arch 461: 1–21, 2011. [DOI] [PubMed] [Google Scholar]
- 28.Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green ED. Pendrin, encoded by the pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci USA 98: 4221–4226, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schlatter E, Greger R, Schafer JA. Principal cells of cortical collecting ducts of the rat are not a route of transepithelial Cl− transport. Pflügers Arch 417: 317–323, 1990. [DOI] [PubMed] [Google Scholar]
- 30.Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP. The pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet 21: 440–443, 1999. [DOI] [PubMed] [Google Scholar]
- 31.Shcheynikov N, Yang D, Wang Y, Zeng W, Karniski LP, So I, Wall SM, Muallem S. Slc26a4 functions as an electroneutral Cl−/I−/HCO3− exchanger: role of Slc26a4 and Slc26a6 in I− and HCO3− secretion and in regulation of CFTR in the parotid duct. J Physiol 586: 3814–3824, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheng S, Carattino MD, Bruns JB, Hughey RP, Kleyman TR. Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am J Physiol Renal Physiol 290: F1488–F1496, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Shi PP, Cao XR, Sweezer EM, Kinney TS, Williams NR, Husted RF, Nair R, Weiss RM, Williamson RA, Sigmund CD, Snyder PM, Staub O, Stokes JB, Yang B. Salt-sensitive hypertension and cardiac hypertrophy in mice deficient in the ubiquitin ligase Nedd4-2. Am J Physiol Renal Physiol 295: F462–F470, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Snyder PM. The epithelial Na+ channel: cell surface insertion and retrieval in Na+ homeostasis and hypertension. Endocr Rev 23: 258–275, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Vallet M, Picard N, Loffing-Cueni D, Fysekidis M, Bloch-Faure M, Deschenes G, Breton S, Meneton P, Loffing J, Aronson PS, Chambrey R, Eladari D. Pendrin regulation in mouse kidney primarily is chloride-dependent. J Am Soc Nephrol 17: 2153–2163, 2006. [DOI] [PubMed] [Google Scholar]
- 36.Verlander JW, Hassell KA, Royaux IE, Glapion DM, Wang ME, Everett LA, Green ED, Wall SM. Deoxycorticosterone upregulates Pds (Slc26a4) in mouse kidney: role of pendrin in mineralocorticoid-induced hypertension. Hypertension 42: 356–362, 2003. [DOI] [PubMed] [Google Scholar]
- 37.Verlander JW, Kim YH, Shin WK, Pham TD, Hassell KA, Beierwaltes WH, Green ED, Everett LA, Matthews SW, Wall SM. Dietary Cl− restriction upregulates pendrin expression within the apical plasma membrane of type B intercalated cells. Am J Physiol Renal Physiol 291: F833–F839, 2006. [DOI] [PubMed] [Google Scholar]
- 38.Wall SM. NH4+ augments net acid secretion by a ouabain-sensitive mechanism in isolated perfused inner medullary collecting ducts. Am J Physiol Renal Fluid Electrolyte Physiol 270: F432–F439, 1996. [DOI] [PubMed] [Google Scholar]
- 39.Wall SM, Hassell KA, Royaux IE, Green ED, Chang JY, Shipley GL, Verlander JW. Localization of pendrin in mouse kidney. Am J Physiol Renal Physiol 284: F229–F241, 2003. [DOI] [PubMed] [Google Scholar]
- 40.Wall SM, Kim YH, Stanley L, Glapion DM, Everett LA, Green ED, Verlander JW. NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl− conservation. Hypertension 44: 1–6, 2004. [DOI] [PubMed] [Google Scholar]
- 41.Zhelyaskov VR, Liu SY, Broderick MP. Analysis of nanoliter samples of electrolytes using a flow-through microfluorometer. Kidney Int 57: 1764–1769, 2000. [DOI] [PubMed] [Google Scholar]