

Abstract
Inflammation is implicated in metabolic abnormalities in obesity and type 2 diabetes. Because θ-defensins have anti-inflammatory activities, we tested whether RTD-1, a θ-defensin, improves metabolic conditions in diet-induced obesity (DIO). DIO was induced by high-fat feeding in obese-prone CD rats from 4 wk of age. Starting at age 10 wk, the DIO rats were treated with saline or RTD-1 for 4 or 8 wk. DIO rats gained more weight than low-fat-fed controls. RTD-1 treatment did not alter body weight or calorie intake in DIO rats. Plasma glucose, FFA, triglyceride (TG), and insulin levels increased in DIO rats; RTD-1 normalized plasma glucose and FFA levels and showed tendencies to lower plasma insulin and TG levels. Hepatic and skeletal muscle TG contents increased in DIO rats; RTD-1 decreased muscle, but not hepatic, TG content. Insulin sensitivity, estimated using homeostasis model assessment of insulin resistance and the glucose clamp technique, decreased in DIO rats, but this change was markedly reversed by RTD-1. RTD-1 had no significant effects on plasma cytokine/chemokine levels or IL-1β and TNF-α expression in liver or adipose tissues. RTD-1 treatment decreased hepatic expression of phosphoenolpyruvate carboxykinase and glucose-6-phosphatase, suggesting that the effect of RTD-1 on plasma glucose (or insulin action) might be mediated by its effect to decrease hepatic gluconeogenesis. Thus, RTD-1 ameliorated insulin resistance and normalized plasma glucose and FFA levels in DIO rats, supporting the potential of RTD-1 as a novel therapeutic agent for insulin resistance, metabolic syndrome, or type 2 diabetes.
Keywords: high-fat feeding, inflammation, insulin resistance, type 2 diabetes, gluconeogenesis
θ-defensins are naturally occurring, cyclic octadecapeptides expressed in Old World monkeys but not in great apes or humans (26, 30, 31). They are the only known class of cyclic peptides of animal origin (5). Selsted et al. (34) have shown that θ-defensins have potent anti-inflammatory effects in vitro and in vivo, in contrast to proinflammatory and immune-activating properties of mammalian α- and β-defensins, genetically related to θ-defensins (1, 11). The macrocyclic conformation of θ-defensins confers remarkable stability in vivo and in vitro (5). Preclinical studies have shown the peptides to be nontoxic, nonimmunogenic, and nonimmunosuppressive at doses that inhibit inflammation in vivo (25), providing strong support for their potential as anti-inflammatory therapeutics. Although the details of the mode of action are still being investigated, it is evident that the lead molecule rhesus θ-defensin 1 (RTD-1) suppresses Toll-like receptor (TLR)-mediated secretion of several pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6 (25).
Circulating levels of proinflammatory cytokines are increased in obesity (8). Inflammatory cytokines can induce insulin resistance (27) and may have direct effects on pancreatic β-cells (9, 35). Elevated levels of IL-1β, IL-6, and C-reactive protein are implicated in the pathogenesis of type 2 diabetes (8). Inflammatory cytokines are produced by macrophages that infiltrate adipose tissue in obesity (33, 38). This effect may be particularly prominent in visceral adipose tissue (3). Strategies to mitigate this inflammation include blockade of the cytokines/chemokines or their receptors that mediate macrophage infiltration [e.g., CC-chemokine ligand 2 (CCL2; also known as MCP1) and its receptor, CCR2 (3, 32)] and inhibition of cellular inflammatory responses [e.g., IKKβ and NF-κB by salsalate (10) or IL-1β by monoclonal antibodies or antagonists (8)]. To date, CCL2/CCR2 blockade has not resulted in significant effects to ameliorate insulin resistance (7), possibly due to the redundancy between multiple macrophage chemotactic factors (8, 14). Salsalate had very small effects on lowering A1C in people with type 2 diabetes (12). On the other hand, IL-1β blockade shows greater promise (18, 19). However, IL-1β blockade using monoclonal antibodies is likely to suffer from immunosuppressive side effects observed with TNF blockers (23). Thus, other anti-inflammatory approaches should be investigated. The present study was designed to test whether RTD-1 ameliorates insulin resistance or improves metabolic parameters in DIO.
MATERIALS AND METHODS
RTD-1.
The hydrochloride salt of synthetic RTD-1 (>98%) was prepared as described previously (30), dissolved in endotoxin-free diluent (0.9% NaCl) at 4 mg/ml, and stored at 4°C.
Induction of DIO and treatment with RTD-1.
Obese-prone CD rats were purchased from Charles River at the age of 4 wk. The animals were housed under controlled temperature (22 ± 2°C) and lighting (12-h light, 6 AM–6 PM; 12-h dark, 6 PM–6 AM) with free access to water and food. The animals were fed either a high-fat (60%) diet (HFD; Harlan Teklad, TD.06414) or normal rat chow [low-fat (12%) diet; LFD] for up to 14 wk. The animals on HFD received either RTD-1 [5 mg/kg sc (25) injected twice per week as 4 mg/ml in normal saline] or normal saline (in matched volume and frequency) treatments after 6 wk of high-fat feeding (n = 8 for each treatment). The RTD-1 and saline treatments were continued for 4 or 8 wk while animals were maintained on HFD. The animals on LFD (n = 8) received neither saline nor RTD-1 injections. All procedures involving animals were approved by the Institutional Animal Care and Use Committee at the University of Southern California.
Tail catheterization for blood sampling and hyperinsulinemic-euglycemic clamps.
At least 4 days before blood sampling and euglycemic clamps (see Experimental protocols), animals were placed in individual cages and accommodated to tail restraints, as previously described (16, 17), required to protect tail blood vessel catheters during the experiments. The animals were free to move about and were allowed unrestricted access to food and water. One tail vein infusion catheter was placed the day before the experiment, and one tail artery blood sampling catheter was placed the morning of the experiment (∼7 AM).
Experimental protocols.
Experiments were conducted in conscious states, starting at ∼1 PM, after a 6-h fast. Blood samples were obtained using the tail artery catheter for basal plasma levels of glucose, insulin, FFA, TG, and cytokines/chemokines. Blood samples were rapidly spun, and plasma samples were aliquoted, frozen immediately in liquid N2, and stored at −70°C for later analysis. Subsequently, a 2-h hyperinsulinemic-euglycemic clamp was conducted by infusing human insulin (5 mU·kg−1·min−1 Novolin; Novo Nordisk, Princeton, NJ) to raise plasma insulin in physiological ranges (16, 17). Plasma glucose was monitored at 10- to 20-min intervals, and 20% dextrose solution was infused to maintain plasma glucose at basal levels (∼140 mg/dl). Glucose infusion rates required during the clamp represent insulin's action to inhibit hepatic glucose output and increase peripheral glucose uptake. At the end of the clamp periods, the animals were euthanized by an intravenous injection of pentobarbital sodium, and tissue samples (liver, epididymal fat, and gastrocnemius muscle) were excised, freeze-clamped, and stored at −70°C for later analysis.
Assays.
Plasma glucose was analyzed using a glucose oxidase method on a Beckman Glucose Analyzer II (Beckman, Fullerton, CA). Plasma insulin was determined using a Rat Ultrasensitive Insulin ELISA kit from ALPCO (Salem, NH). Plasma FFA levels were measured using an acyl-CoA oxidase-based colorimetric kit from Wako Chemicals (Richmond, VA). Plasma TG levels were analyzed using a Ponte Scientific TG reagent (Thermo Fisher Scientific, Waltham, MA). Plasma cytokines were analyzed using a Rat Cytokine/Chemokine Multiplex kit from Millipore (RECYMAG65K27PMX) in the Metabolic Core of the USC Diabetes Obesity Research Institute. Liver and muscle TG were analyzed by homogenizing liver and muscle samples in cell lysis buffer (50 mM Tris·HCl, 5 mM EDTA and 1% Triton X-100, pH 7.4, 10% wt/vol) and measuring TG in the homogenates using the Ponte Scientific TG reagent.
Homeostasis model assessment of insulin resistance.
Insulin sensitivity was estimated based on basal plasma glucose and insulin levels using the homeostasis model assessment (HOMA). HOMA-IR, an index of insulin resistance, was calculated as (plasma insulin) × (plasma glucose) ÷ 405 (22).
Quantitative RT-PCR.
Tissue cytokine expression and hepatic expression of gluconeogenic genes [i.e., phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase)] were determined using quantitative (q)RT-PCR. Total RNA was isolated from individual tissues using TRI Reagent (Sigma-Aldrich, St. Louis, MO). Reverse transcription was performed using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems Life Technologies, Grand Island, NY). Real-time PCR was performed using a B-R SYBR Green SuperMix for iQ (Quanta Biosciences, Gaithersburg, MD) on a CFX 96 Real-Time System (Bio-Rad Laboratories). Primer sequences used were as follows: IL-1β, F 5′-TCT CCA CCT CAA TGG ACA GAA C-3′, R 5′-CCC AAG GCC ACA GGG ATT T-3′; IL-6, F 5′-TGT TCT CAG GGA GAT CTT GGA AA-3′, R 5′-CAG TGC ATC ATC GCT GTT CAT AC-3′; TNF-α, F 5′-GGG CTC CCT CTC ATC AGT TC-3′, R 5′-GTG GGC TAC GGG CTT GTC-3′; PEPCK (Pck1), F 5′-GCC GCT GGA TGT CAG AAG AG-3′, R 5′-ACA TGG TGC GGC CTT TCA TG-3′; G6Pase (G6pc), F 5′-GGC TCA CTT TCC CCA TCA GG-3′, R 5′-ATC CAA GTG CGA AAC CAA ACA G-3′; GAPDH, F 5′-GCC AAG GTC ATC CAT GAC AAC-3′, R 5′-GGG CCA TCC ACA GTC TTC TG-3′. Thermal cycling was carried out with a denaturation phase of 3 min at 95°C, followed by 45 cycles of 30 s at 95°C and 30 s at 60°C.
Statistical analysis.
All data are expressed as means ± SE. The significance of differences in the mean value was assessed by one-way ANOVA followed by ad hoc analysis using the Bonferonni method for multiple comparisons. A P value < 0.05 was considered to be statistically significant.
RESULTS
Food intake and body weight.
High-fat feeding in obese-prone CD rats resulted in expected increases in body weight compared with those fed LFD (Fig. 1A). RTD-1 treatment did not significantly alter body weight in HFD-fed rats compared with the saline-treated group (P > 0.05; Fig. 1, A and B). HFD markedly increased fat tissue in the abdominal cavity, and this was not significantly reduced by RTD-1 treatment. RTD-1 treatment showed a tendency to decrease calorie intake during high-fat feeding, but this effect did not gain statistical significance (P = 0.102; Fig. 1, C and D).
Fig. 1.

Changes in body weight (A and B) and calorie intake (C and D) during feeding and drug treatments. B and D: data for week 14. Data are means ± SE for 4 or 8 rats.
Plasma glucose, FFA, TG, and insulin levels.
At the end of 4-wk or 8-wk treatments with RTD-1 or saline, changes in plasma glucose, TG, FFA, and insulin (and other data), induced by high-fat feeding or RTD-1 treatment, were identical between the 4-wk- and 8-wk-treated groups, and the two sets of data were combined for analysis. Compared with LFD, HFD significantly increased basal plasma glucose in the absence of RTD-1, but this increase was completely prevented by RTD-1 treatment in the parallel HFD group (Fig. 2A). High-fat feeding showed a tendency to increase basal plasma FFA levels (by 20%); this increase was completely prevented by RTD-1 treatment (Fig. 2B). Basal plasma TG and insulin levels were significantly raised by high-fat feeding (Fig. 2, C and D). RTD-1 decreased plasma TG and insulin levels in HFD-fed rats, but these effects did not gain statistical significance.
Fig. 2.

Plasma glucose (A), FFA (B), TG (C), and insulin (D) levels in the 3 experimental groups. Blood samples were collected after a 6-h fast. Data are means ± SE for 7 or 8 rats.
Liver and muscle TG levels.
HFD increased hepatic TG levels ninefold compared with rats maintained on LFD (Fig. 3A). RTD-1 treatment had no effect on hepatic steatosis in HFD-fed rats. High-fat feeding also increased muscle TG levels, and RTD-1 treatment decreased muscle TG levels with a marginal statistical significance (Fig. 3B).
Fig. 3.
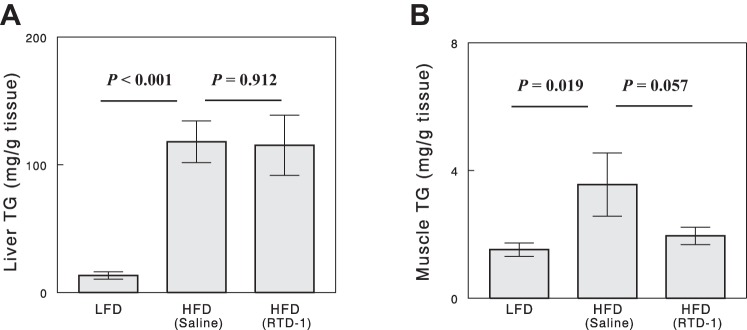
Liver (A) and muscle (B) TG levels in the 3 experimental groups. Data are means ± SE for 7 or 8 rats.
HOMA-IR.
Insulin resistance estimated by HOMA-IR was significantly increased by high-fat feeding (Fig. 4). The increase in HOMA-IR was markedly (59%) reversed by RTD-1 treatment (P = 0.04), indicating a significant insulin-sensitizing effect.
Fig. 4.
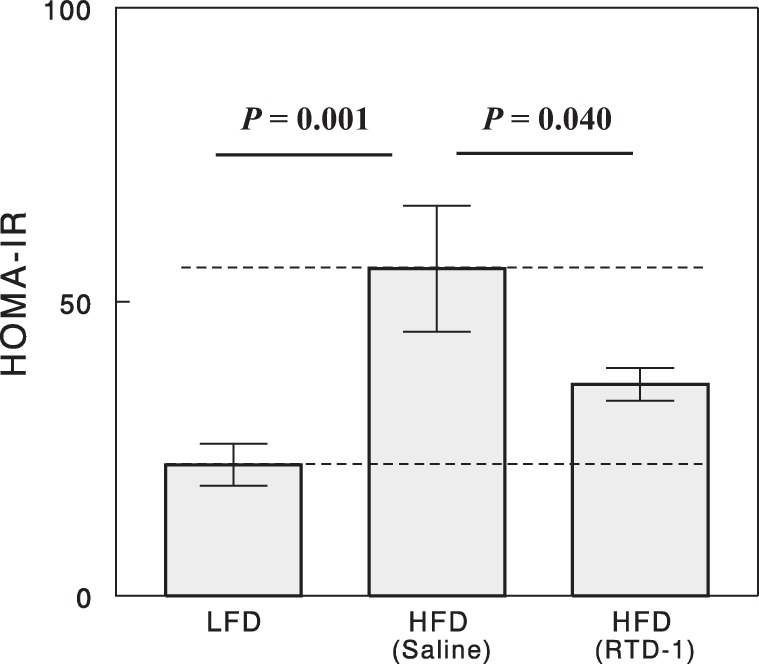
Homeostasis model of assessment of insulin resistance (HOMA-IR) for the 3 experimental groups. Data are means ± SE for 7 or 8 rats.
Insulin sensitivity measured by the glucose clamp.
During glucose clamp studies, plasma glucose concentrations (Fig. 5A) were well matched among the three study groups. Plasma insulin concentrations (Fig. 5B) were raised to similar levels in HFD-fed rats with vs. without RTD-1; these level were higher than those in LFD-fed rats, but increases from baseline were not statistically different among the groups, due in part to the higher fasting insulin levels in HFD groups. Glucose infusion rates (GINF) required during the clamp (i.e., during insulin infusion) represent insulin's action to inhibit hepatic glucose output and increase peripheral glucose uptake. GINF was substantially lower in the HFD group than in the LFD group (Fig. 5, C and D). RTD-1 treatment increased GINF by 56% in HFD rats, reversing insulin resistance by 46%, consistent with the HOMA data.
Fig. 5.

Plasma glucose (A), insulin (B), and glucose infusion rate (GINF; C) during a hyperinsulinemic-euglycemic clamp. D: steady-state (final 30 min) GINF. Data are means ± SE for 7 or 8 rats.
Plasma and adipose tissue cytokine levels.
Of 27 cytokines that can be detected by the Rat Cytokine/Chemokine Multiplex Kit from Millipore, only 10 were measured reliably (i.e., SE < 20% in all groups; Table 1). Plasma levels of leptin, IL-4, VEGF, and CCL11 (eotaxin) were significantly increased by high-fat feeding; RTD-1 treatment did not reverse these changes. Other cytokines, such as CCL5 (RANTES), CXCL5 (LIX), CX3CL1 (fractalkine), IL-5, IL-12 p70, and MCP-1, were affected neither by high-fat feeding nor by RTD-1. The kit did not have sufficient sensitivity to detect TNF-α or IL-6 in our basal plasma samples and produced too much variation for IL-1β, so we do not know whether plasma levels of these cytokines were affected by HFD or RTD-1. We next measured the expressions of IL-1β, IL-6, and TNF-α in epididymal adipose tissue and liver using qRT-PCR. Figure 6 shows that neither IL-1β nor TNF-α showed changes in gene expression in liver or adipose tissue with HFD or RTD-1 treatment (P > 0.05). IL-6 expression levels were too low to be accurately quantified (data not shown).
Table. 1.
Changes in plasma cytokines with HFD or RTD-1 treatment
|
P Values: HFD (Saline) |
|||||
|---|---|---|---|---|---|
| Cytokine | LFD | HFD (Saline) | HFD (RTD-1) | vs. LFD | vs. RTD-1 |
| Leptin | 17.5 ± 2.0 | 43.1 ± 5.7 | 48.0 ± 6.1 | 0.001* | 0.495 |
| IL-4 | 3.8 ± 0.4 | 7.7 ± 0.9 | 7.1 ± 1.0 | 0.003* | 0.562 |
| CCL11 | 9.9 ± 0.8 | 13.1 ± 1.2 | 11.5 ± 0.8 | 0.029* | 0.249 |
| VEGF | 67 ± 6 | 89 ± 9 | 81 ± 5 | 0.032* | 0.416 |
| CCL5 | 487 ± 39 | 570 ± 27 | 630 ± 66 | 0.245 | 0.397 |
| CXCL5 | 298 ± 52 | 379 ± 63 | 306 ± 45 | 0.302 | 0.345 |
| CX3CL1 | 40 ± 4 | 47 ± 5 | 43 ± 3 | 0.309 | 0.505 |
| IL-5 | 38 ± 3 | 44 ± 5 | 49 ± 5 | 0.326 | 0.400 |
| IL-12 (p70) | 24.0 ± 4.7 | 28.7 ± 2.7 | 35.0 ± 4.5 | 0.441 | 0.305 |
| MCP-1 | 468 ± 51 | 624 ± 109 | 772 ± 143 | 0.512 | 0.348 |
Data are means ± SE for 7 or 8 rats. Units are ng/ml for leptin and pg/ml for all others. LFD, low-fat diet; HFD, high-fat diet; RTD-1, rhesus θ-desmin 1.
P < 0.05.
Fig. 6.
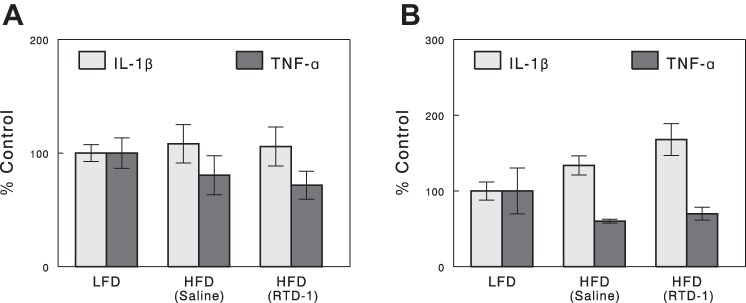
IL-1β and TNF-α expression in adipose (A) and liver (B) tissues determined by qRT-PCR. Data are means ± SE for 7 or 8 rats.
Hepatic gene expression of gluconeogenic enzymes.
G6Pase and PEPCK are rate-limiting enzymes for gluconeogenesis. Liver mRNA expression of G6Pase showed tendencies to increase with HFD (P = 0.15 vs. LFD; Fig. 7) and to decrease with RTD-1 treatment (P = 0.07 vs. saline-treated, HFD-fed rats). PEPCK expression was not altered by high-fat feeding but was decreased by RTD-1 with a marginal statistical significance (P = 0.05).
Fig. 7.
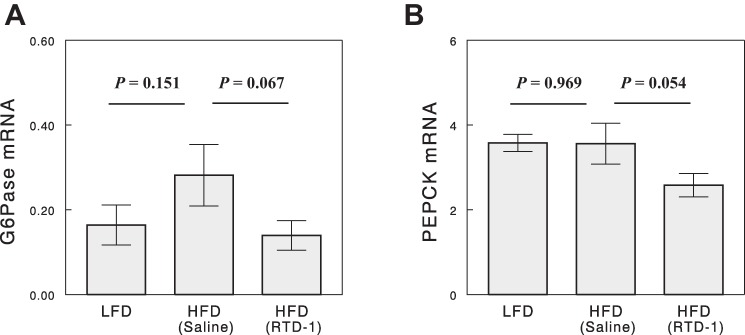
Hepatic glucose-6-phosphatase (G6Pase; A) and phosphoenolpyruvate carboxykinase (PEPCK; B) mRNA expression (in arbitrary units) determined by qRT-PCR. Data are means ± SE for 6–8 rats.
DISCUSSION
The present study demonstrates that RTD-1, a θ-defensin, improves insulin action and normalizes plasma glucose and FFA levels in DIO rats. These effects occurred without altering body weight, food intake, or fat accumulation in the liver. Interestingly, RTD-1 treatment altered neither plasma cytokine/chemokine levels nor adipose or hepatic cytokine expression, and it is unclear whether the RTD-1 effects on insulin action, plasma glucose, and plasma FFA relate to its anti-inflammatory properties (see below). RTD-1 did reduce fat in skeletal muscle and lowered liver expression of two important enzymes for gluconeogenesis. Although the contribution of these changes to the effects of RTD-1 are unclear, our results support the potential of RTD-1 (and possibly other θ-defensins) as a new class of therapeutics for insulin resistance and related disorders such as type 2 diabetes.
Human studies on the natural history of type 2 diabetes showed that insulin resistance occurs long before the development of hyperglycemia (2). Normally, euglycemia can be maintained as β-cells secrete more insulin to compensate for insulin resistance. Blood glucose increases when β-cell function (or compensation) becomes impaired (2, 37). Amelioration of insulin resistance with insulin-sensitizing thiazolidinedione drugs can arrest β-cell decline (4, 36), suggesting that insulin resistance is a cause of falling β-cell function that leads to hyperglycemia. Increased fasting glucose in HFD rats treated with saline suggests inadequate compensation for insulin resistance in those animals. The full reversal of fasting hyperglycemia by RTD-1 despite only partial amelioration of insulin resistance in HFD rats suggests additional mechanisms that cannot be determined from the present study. However, the reduction in insulin levels observed following RTD-1 may have biological importance. The strongest predictor of prevention of diabetes with thiazolidinediones was an initial reduction in insulin levels (4, 36), reflecting reduced secretory demands on β-cells. To the extent that RTD-1 reduces secretory demands on β-cells, it holds promise as an intervention that can alter the natural history of the β-cell disease that leads to hyperglycemia in type 2 diabetes.
Insulin sensitivity was assessed in the present study by HOMA-IR, which is based on basal glucose and insulin levels, and GINF required during a glucose clamp. The two independent methods agreed well to indicate that insulin resistance in DIO rats was ∼50% reversed by RTD-1 treatment. However, these methods do not disclose whether insulin action was improved in the liver (to decrease hepatic glucose production), peripheral tissues (to increase glucose uptake), or both. Regarding this issue, muscle TG was significantly increased by high-fat feeding, but this change was prevented by RTD-1 treatment. It is well established that accumulation of TG in muscle fibers is associated with peripheral insulin resistance (15, 28). Therefore, RTD-1-mediated improvement of whole body insulin action may be mediated, at least in part, by normalizing TG content (and thus insulin action) in skeletal muscle. In addition, hepatic expression of major gluconeogenic genes was decreased by RTD-1 treatment (Fig. 7), which would lead to improved insulin action to suppress hepatic glucose production. Thus, RTD-1 might improve insulin action both in the liver and in peripheral tissues in DIO rats. The mechanisms by which RTD-1 treatment decreased muscle TG content are unclear; this effect may relate to RTD-1's effects to lower plasma FFA levels, but the possibility that RTD-1 increased lipid oxidation in skeletal muscle cannot be excluded. RTD-1 treatment showed a tendency to decrease calorie intake, and this effect may play a role in lowering plasma FFA or muscle TG levels. The effects of RTD-1 to decrease hepatic expression of gluconeogenic genes were associated with no effect on fatty liver. These data are consistent with previous suggestions that there is a strong dissociation between fatty liver and hepatic glucose production or insulin resistance (29), and inhibition of hepatic gluconeogenesis can improve hepatic insulin resistance but also lead to fatty liver (13).
Our data show impressive effects of RTD-1 to completely normalize basal blood glucose and FFA levels in DIO rats. This is in contrast to only a partial reversal of insulin resistance with RTD-1 treatment. The effects on blood glucose and FFA levels might arise, at least in part, from hormonal changes in the basal state. There is a set of hormones, such as catecholamines, glucagon, and cortisol (corticosterone in rodents), that increase both glucose and FFA levels by stimulating hepatic glucose production and lipolysis, respectively. If these hormone levels are decreased in DIO rats by RTD-1, such effects would explain normalization of plasma glucose and FFA. The finding that RTD-1 decreased hepatic expression of gluconeogenic genes supports this idea. In addition, because these hormones are known to induce insulin resistance (6, 24), such effects could also explain the improvement of insulin resistance by RTD-1 treatment.
Schaal et al. (25) demonstrated anti-inflammatory effects of θ-defensins in vitro and in vivo. θ-Defensins suppressed proinflammatory cytokine/chemokine release in vitro in peripheral blood leukocytes and cultured THP-1 monocytes stimulated by various TLR agonists. In addition, RTD-1 treatment suppressed rises of proinflammatory cytokines in vivo in mice challenged with bacterial infection. Thus, anti-inflammatory properties of θ-defensins were demonstrated with inflammatory stimuli or bacterial infections. In contrast, RTD-1 treatment did not alter cytokine/chemokine levels in uninfected (i.e., unstimulated) animals. In the present study, RTD-1 treatment did not significantly alter the plasma cytokine/chemokine levels that we measured in DIO rats, as the animals were studied in basal (i.e., unstimulated) states, and inflammation in these animals, if any, was very mild, indicated by the changes in plasma cytokine/chemokine levels (Table 1).
A previous study by Cao et al. (21) showed that human neutrophil α-defensins (HNP-1) reduced hepatic expression of gluconeogenic enzymes (i.e., PEPCK and G6Pase), lowered blood glucose, and improved insulin action, similar to the RTD-1 effects observed in the present study. Whereas θ-defensins have anti-inflammatory properties, several α-defensins, including HNP-1, have proinflammatory properties (1). Similar effects of RTD-1 and HNP-1 suggest the intriguing possibility that the RTD-1 effects arise from structural features shared by α- and θ-defensins [e.g., six conserved cysteines, three intramolecular disulfide bonds, β-sheet regions, etc. (20)] rather than RTD-1's anti-inflammatory properties. This is supported by the finding that RTD-1 had no significant effects on plasma cytokine levels or IL-1β and TNF-α expression in liver and adipose tissue. Future studies are warranted to address this important issue.
GRANTS
This work was supported by ADA Basic Science Award No. 7-12-BS-214 and NIH CTSI Pilot grant (to J. H. Youn), AI-022931, DE-021341, and Arthritis Foundation No. 5997 (to M. Selsted), and UL1 TR-000130 (to T. A. Buchanan).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Y.T.O., T.A.B., M.E.S., and J.H.Y. conception and design of research; Y.T.O. and D.T. performed experiments; Y.T.O. and J.H.Y. analyzed data; Y.T.O., T.A.B., M.E.S., and J.H.Y. interpreted results of experiments; Y.T.O. prepared figures; Y.T.O. and J.H.Y. drafted manuscript; Y.T.O., D.T., T.A.B., M.E.S., and J.H.Y. edited and revised manuscript; Y.T.O., D.T., T.A.B., M.E.S., and J.H.Y. approved final version of manuscript.
ACKNOWLEDGMENTS
J. Youn is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
REFERENCES
- 1.Ahn JK, Huang B, Bae EK, Park EJ, Hwang JW, Lee J, Koh EM, Cha HS. The role of α-defensin-1 and related signal transduction mechanisms in the production of IL-6, IL-8 and MMPs in rheumatoid fibroblast-like synoviocytes. Rheumatology (Oxford) 52: 1368–1376, 2013. [DOI] [PubMed] [Google Scholar]
- 2.Bogardus C. Insulin resistance in the pathogenesis of NIDDM in Pima Indians. Diabetes Care 16: 228–231, 1993. [DOI] [PubMed] [Google Scholar]
- 3.Bruun JM, Lihn AS, Pedersen SB, Richelsen B. Monocyte chemoattractant protein-1 release is higher in visceral than subcutaneous human adipose tissue (AT): implication of macrophages resident in the AT. J Clin Endocrinol Metab 90: 2282–2289, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Buchanan TA, Xiang AH, Peters RK, Kjos SL, Marroquin A, Goico J, Ochoa C, Tan S, Berkowitz K, Hodis HN, Azen SP. Preservation of pancreatic β-cell function and prevention of type 2 diabetes by pharmacological treatment of insulin resistance in high-risk Hispanic women. Diabetes 51: 2796–2803, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Conibear AC, Craik DJ. The chemistry and biology of theta defensins. Angew Chem Int Ed 53: 10612–23, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deibert DC, DeFronzo RA. Epinephrine-induced insulin resistance in man. J Clin Invest 65: 717–21, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Prospero NA, Artis E, Andrade-Gordon P, Johnson DL, Vaccaro N, Xi L, Rothenberg P. CCR2 antagonism in patients with type 2 diabetes mellitus: a randomized, placebo-controlled study. Diabetes Obes Metab 16: 1055–1064, 2014. [DOI] [PubMed] [Google Scholar]
- 8.Donath MY, Shoelson SE. Type 2 diabetes and inflammatory disease. Nat Rev Immunol 11: 98–107, 2011. [DOI] [PubMed] [Google Scholar]
- 9.Donath MY, Storling J, Berchtold LA, Billestrup N, Mandrup-Poulsen T. Cytokines and beta cell biology: from concept to clinical translation. Endocrinol Rev 29: 334–350, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Fleischman A, Shoelson SE, Bernier R, Goldfine AB. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care 31: 289–294, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Froy O. Regulation of mammalian defensin expression by Toll-like receptor-dependent and independent signalling pathways. Cell Microbiol 7: 1387–1397, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE. The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med 152: 346–357, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gómez-Valadés AG, Méndez-Lucas A, Vidal-Alabró A, Blasco FX, Chillon M, Bartrons R, Bermúdez J, Perales JC. Pck1 gene silencing in the liver improves glycemia control, insulin sensitivity, and dyslipidemia in db/db mice. Diabetes 57: 2199–2210, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiao P, Chen Q, Shah S, Du J, Tao B, Tzameli I, Yan W, Xu H. Obesity-related upregulation of monocyte chemotactic factors in adipocytes: involvement of nuclear factor-kappaB and c-Jun NH2-terminal kinase pathways. Diabetes 58: 104–115, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelley DE, Goodpaster BH, Storlien L. Muscle triglyceride and insulin resistance. Annu Rev Nutr 22: 325–346, 2002. [DOI] [PubMed] [Google Scholar]
- 16.Kim JK, Wi JK, Youn JH. Metabolic impairment precedes insulin resistance in skeletal muscle during high-fat feeding in rats. Diabetes 45: 651–658, 1996. [DOI] [PubMed] [Google Scholar]
- 17.Kim JK, Youn JH. Prolonged suppression of glucose metabolism causes insulin resistance in rat skeletal muscle. Am J Physiol Endocrinol Metab 272: E288–E296, 1997. [DOI] [PubMed] [Google Scholar]
- 18.Larsen CM, Faulenbach M, Vaag A, Ehses JA, Donath MY, Mandrup-Poulsen T. Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care 32: 1663–1668, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356: 1517–1526, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Lehrer RI, Cole AM, Selsted ME. θ-Defensins: cyclic peptides with endless potential. J Biol Chem 287: 27014–27019, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu HY, Collins QF, Moukdar F, Zhuo D, Han J, Hong T, Collins S, Cao W. Suppression of hepatic glucose production by human neutrophil alpha-defensins through a signaling pathway distinct from insulin. J Biol Chem 283: 12056–12063, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28: 412–419, 1985. [DOI] [PubMed] [Google Scholar]
- 23.Nanau RM, Neuman MG. Safety of anti-tumor necrosis factor therapies in arthritis patients. J Pharm Pharm Sci 17: 324–361, 2014. [DOI] [PubMed] [Google Scholar]
- 24.Purnell JQ, Kahn SE, Samuels MH, Brandon D, Loriaux DL, Brunzell JD. Enhanced cortisol production rates, free cortisol, and 11β-HSD-1 expression correlate with visceral fat and insulin resistance in men: effect of weight loss. Am J Physiol Endocrinol Metab 296: E351–E357, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaal JB, Tran D, Tran P, Ösapay G, Trinh K, Roberts KD, Brasky KM, Tongaonkar P, Ouellette AJ, Selsted ME. Rhesus macaque theta defensins suppress inflammatory cytokines and enhance survival in mouse models of bacteremic sepsis. PLoS One 7: e51337, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selsted ME. Theta-defensins: cyclic antimicrobial peptides produced by binary ligation of truncated alpha-defensins. Curr Protein Pept Sci 5: 365–371, 2004. [DOI] [PubMed] [Google Scholar]
- 27.Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology 132: 2169–2180, 2007. [DOI] [PubMed] [Google Scholar]
- 28.Stannard SR, Johnson NA. Insulin resistance and elevated triglyceride in muscle: more important for survival than “thrifty” genes? J Physiol 554: 595–607, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun Z, Lazar MA. Dissociating fatty liver and diabetes. Trends Endocrinol Metab 24: 4–12, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang YQ, Yuan J, Osapay G, Osapay K, Tran D, Miller CJ, Ouellette AJ, Selsted ME. A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated alpha-defensins. Science 286: 498–502, 1999. [DOI] [PubMed] [Google Scholar]
- 31.Tongaonkar P, Tran P, Roberts K, Schaal J, Osapay G, Tran D, Ouellette AJ, Selsted ME. Rhesus macaque theta-defensin isoforms: expression, antimicrobial activities, and demonstration of a prominent role in neutrophil granule microbicidal activities. J Leukoc Biol 89: 283–290, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Liebel RL, Ferrante AW Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 116: 115–124, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wohlford-Lenane CL, Meyerholz DK, Perlman S, Zhou H, Tran D, Selsted ME, McCray PB Jr. Rhesus theta-defensin prevents death in a mouse model of severe acute respiratory syndrome coronavirus pulmonary disease. J Virol 83: 11385–11390, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiang AH, Kawakubo M, Trigo E, Kjos SL, Buchanan TA. Declining beta-cell compensation for insulin resistance in Hispanic women with recent gestational diabetes mellitus: association with changes in weight, adiponectin, and C-reactive protein. Diabetes Care 33: 396–401, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiang AH, Peters RK, Kjos SL, Marroquin M, Goico J, Ochoa C, Kawakubo M, Buchanan TA. Effect of pioglitazone on pancreatic β-cell function and diabetes risk in Hispanic women with prior gestational diabetes. Diabetes 55: 517–522, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiang AH, Wang C, Peters RK, Trigo E, Kjos SL, Buchanan TA. Coordinate changes in plasma glucose and pancreatic β-cell function in Latino women at high risk for type 2 diabetes. Diabetes 55: 1074–1079, 2006. [DOI] [PubMed] [Google Scholar]
- 38.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112: 1821–1830, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]